Metal-free synthesis of nitrile based partially reduced thia-and oxa-thia[5]helicenes: conformation and dynamics†
Ramendra
Pratap
*a,
Abhinav
Kumar
b,
Rigoberg
Pick
c,
Volker
Hüch
d and
Vishnu Ji
Ram
*b
aDepartment of Chemistry, University of Delhi, North Campus, Delhi, 110007, India. E-mail: ramendrapratap@gmail.com; Tel: +919899637945
bDepartment of Chemistry, Lucknow University, Lucknow, 226007, India. E-mail: vjiram@yahoo.com
cUniversität des Saarlandes, Institut für Organische Chemie, Im Stadtwald, Geb. C4.2, D-66123, Saarbrücken, Germany
dUniversität des Saarlandes, Institut für Anorganische Chemie, Im Stadtwald, Geb. C4.1, D-66123, Saarbrücken, Germany
First published on 22nd December 2011
Abstract
An expeditious, metal free, simple and convenient synthesis of partially reduced thia[5]helicenes and oxa-thia[5]helicenes, appended with nitrile and amino functionalities such as 3-sec.amino-5,6-dihydro-2H-1-thia-dibenzo[c,g]phenanthrene-4-carbonitriles 7 and (Z)-2-(5,6-dihydrobenzo[f]thiochromeno[3,4-c]-3-(2H)-ylidene)acetonitriles 6 has been delineated through base-catalyzed ring transformation of 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4 by 2-tetralones 5. The molecular structure of one of the partially reduced thia[5]helicenes has been determined by X-ray crystallographic analysis. In order to calculate the inversion barrier of the helimeric enantiomers P-7a and M-7a, the molecular geometries of both, the ground states and the transition states have been optimized by the density functional theory (DFT) using B3LYP/6-311G.** Based on these structures, helimerization barrier of 44.36 K cal mol−1 has been predicted using MP2/6-311G** single-point energy calculations.
Introduction
Helicenes are thermally stable, non-planar helical molecules endowed with inherent chirality,1 comprised of ortho-annulated aromatic and or heteroaromatic rings. The unique supramolecular architecture2 and extraordinary optical,3 electronic4 and chelating5 properties and their potential applications to liquid crystals,6 sensors,7dyes,7 asymmetric synthesis,8,9 molecular recognition,8,9polymer synthesis,10 electrostatic switches11 and circularly polarized luminescent11 and non-linear optical (NLO) materials for the development of ultra-fast large scale optical data processing devices12,13 made helicenes a constant subject of investigation. Very little has been studied on the NLO properties of heterohelicenes14 though the synthesis was first reported14b in the year 1903. Recently, heterohelicenes have emerged as extremely attractive molecules.14c–l The presence of a heteroatom in helicenes not only imparts helicity but also affects the racemisation barrier (RB). The racemisation barrier of heterohelicenes depends on several factors such as the number of ortho-annulated rings, substitution at various sites of the helix, nature and site of the heteroatom and lastly steric crowding on the terminal positions. The introduction of a sulfur atom in a helicene is extremely useful to the electronic, optical and photorefractive properties of a thiohelicene based material.3d The wide ranging applications of thiohelicenes in material science, made imperative the development of an efficient synthesis of partially reduced thia-, and oxa-thiahelicenes endowed with various functionalities for their broad applications in material science.This class of molecules have classically been synthesized by oxidative photocyclization of bis(stilbenes)15 with poor regiocontrol.16 Despite significant progress in helicene chemistry in the past decade,17 various synthetic strategies based on Diels–Alder cycloadditions,18 transition metal mediated cyclotrimerization of triynes or dienetriynes,19,20olefin metathesis,21 intramolecular Pd-catalyzed cyclization,22 ring transformation strategy22 and Friedel-Crafts cyclization22 emerged. Stemming from the Katz methodology,1b Carreno and Urbano developed23 cleverly its asymmetric version, enabling the enantioselective access to helicene quinones with high optical purity. The remarkable high stereocontrol in the synthesis of [5]helicenes has been achieved by Karikomi who used the completely diastereoselective aromatic oxy-Cope rearrangement as a key step.24 A different concept of obtaining non-racemic helicenes was published by Genet25 who took advantage of chirality transfer from an enantiopure tether to helicene backbone under thermal conditions, making configurationally locked. The nature of substituents, their presence and pattern on the helicene core are highly dependent on the synthetic strategy. There are some examples where variable substituents are introduced through transformation of groups present on preformed helicenes.26,27 Despite remarkable progress, development of an efficient economical and concise route for highly functionalized heterohelicenes is highly demanding and remains a challenge to the state-of-art synthesis.
Results and discussion
The elegance of the helicene architecture and its unexplored potential of envisioned applications stimulated us to devise an efficient and short synthesis through base catalyzed ring transformations of suitably functionalized 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[3,4-c]pyran-3-carbonitriles by 2-tetralones, a feasible concept exhibiting a high degree of synthetic flexibility for the construction of partially reduced thia- and oxa-thia[5]helicenes, not reported so far. We have non-catalytically synthesized various partially reduced thia- and oxa-thia[5]helicenes endowed with halo, amino and nitrile functionalities, transformable to some other groups either catalytically or non-catalytically. The beauty of the synthetic strategy lies in obtaining partially reduced helicenes, not easy obtainable either chemically or through catalytic regioselective hydrogenation. It has been observed that an increase in the non-planarity in polycyclic arenes reduces their ability to be metabolically activated to form DNA-damaging adducts,28 responsible for carcinogenic properties. Thus, we developed a synthetic strategy to induce the distortion in the co-planarity in the molecule through partial reduction.As it is apparent from the retrosynthetic path way outlined in Scheme 1, the primary precursor, 4-methylthio-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 3 can be obtained from the reaction of thiochroman-4-one 2 and methyl 2-cyano-3,3-dimethylthioacrylate291 which on amination30 with sec.amine in boiling ethanol yielded 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4, as useful precursor for the ring transformation reactions (Scheme 2).
![Retrosynthetic pathway for the synthesis of [5]helicenes](/image/article/2012/RA/c1ra00824b/c1ra00824b-s1.gif) | ||
| Scheme 1 Retrosynthetic pathway for the synthesis of [5]helicenes | ||
![Synthesis of 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4](/image/article/2012/RA/c1ra00824b/c1ra00824b-s2.gif) | ||
| Scheme 2 Synthesis of 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4 | ||
The synthetic potential of lactone 4 is enormous due to the uniqueness in their structural features and functionalities present for the construction of a variety of arenes and heteroarenes.
As evident from the topography of the thiochromeno[4,3-b]pyran-3-carbonitrile 4, the positions C2, C4, and C10b are electron deficient centers in which the latter is highly electrophilic and prone to nucleophilic attack because of an extended conjugation and the presence of an electron-withdrawing CN substituent at position 3 of the thiochromene ring 4.
Our synthetic strategy is based on the easy availability of starting materials, or readily obtainable through synthesis, so that a variety of functionalized thia[5]helicenes and oxa-thia[5]helicenes could be obtained by exploiting the topography and the presence of electrophilic center C10b in 4 by the attack of a carbanion for the ring transformation.
Thus, an equimolar mixture of the 2H-benzo[h]chromene-3-carbonitrile 4a or 4c, 2-tetralone 5a, and NaH as a base was stirred for 3–4 h in dry THF with continuous monitoring of the reaction progress by TLC. After completion of the reaction, the product was isolated by column chromatography on neutral alumina, giving the respective oxa-thiahelicene 6 and thiahelicene 7, as characterized by spectroscopic analyses (Scheme 3). When we ran this reaction using KOH as a base and DMF as a solvent, only product 6 was obtained in low yield. (Table 1).
![Synthesis of the (Z)-2-(5,6-dihydrobenzo[f]thiochromeno[3,4-c]-3(2H)-ylidene)-acetonitriles 6 and 3-sec.amino-5,6-dihydro-2H-1-thia-dibenzo[c,g]phenanthrene-4-carbonitriles 7](/image/article/2012/RA/c1ra00824b/c1ra00824b-s3.gif) | ||
| Scheme 3 Synthesis of the (Z)-2-(5,6-dihydrobenzo[f]thiochromeno[3,4-c]-3(2H)-ylidene)-acetonitriles 6 and 3-sec.amino-5,6-dihydro-2H-1-thia-dibenzo[c,g]phenanthrene-4-carbonitriles 7 | ||
In order to assess the suitability of lactones 4 with pyrrolidine, piperidine and morpholine substituted for the preparation of 8-oxa-thia[5]helicenes 6 and thia[5]helicenes 7, various ring transformations were carried out by 2-tetralones but only 4-(morpholin-4-yl)-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles (4c, d) were found comparatively better for the synthesis of oxa-thiahelicenes (6a–m) possibly due to an inductive effect of the oxygen in the morpholine ring. However in only two cases we were able to isolate thiahelicenes 7a and 7b. During the course of the reaction it was observed that the use of 7-methoxy-2-tetralone as a source of carbanion always gave comparatively a better yield of 7. However, the ring transformation of 4 with 6-methoxy-2-tetralone yield of product 6 was predominant. As depicted in Scheme 4, the reaction of 4 and 5 possibly initiates through a base catalyzed Michael addition of a 2-tetralone at C10b of the 4-sec.amino-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitrile 4 and thereafter the reaction may follow path A and/or B. In the case of path A, the Michael adduct P may undergo an intramolecular enolate addition to the enamine followed by elimination of carbon dioxide and the respective amine (piperidine or morpholine) to yield the 8-oxa-thia[5]helicenes 6. However, if the reaction follows path B, the Michael adduct intermediate P may undergo an intramolecular C–C bond formation by addition of the enamine to the carbonyl function followed by elimination of carbon dioxide and water to yield the thia[5]helicenes 7.
![A plausible mechanism for the formation of the partially reduced oxa-thia[5]helicene 6 and thia[5]helicene 7.](/image/article/2012/RA/c1ra00824b/c1ra00824b-s4.gif) | ||
| Scheme 4 A plausible mechanism for the formation of the partially reduced oxa-thia[5]helicene 6 and thia[5]helicene 7. | ||
Thus, the formation of two products from a single reaction is an indication of competition between paths A and B leading to the formation of products, 8-oxa-thia[5]helicenes 6 and thia[5]helicenes 7.
Crystal structure analysis of 7
Crystals of X-ray quality were obtained by slow evaporation of the solution of compound in acetone at room temperature. The conformation of the compound with arbitrary numbering is shown as an ORTEP diagram (Fig. 1), illustrating the non-planar, helically distorted conformation of the molecule. The compound crystallized in the P2[5]2(1)2(1) space group with four molecules in the orthorhombic unit cell. The least-square plane calculation from X-ray crystallographic data of the compound indicates that the fully unsaturated rings A, C, and E are as such nearly planar, while the rings B and D adopt half-chair conformation. The average mean plane angle for the twist between the rings A and C is significant (36.69°), while between the rings C and E is 34.58°. The helical distortion between the terminal rings, A and E, is even 52.20°. This large helical distortion can be due to the presence of sulfur atom S1 in the ring D. The short contact between the two protons at C5 and C18 (3.09 Å) also causes strain, which is relieved either through stretching and bending of the chemical bonds or buckling of the aromatic rings. The torsion angles (C5-C6-C7-C17, C6-C7-C17-C16, and C7-C17-C16-C18) of the compound are 35.78°, 19.52°, and 33.38°, respectively.![The conformation of the compound with arbitrary numbering is Shown as an ORTEP diagram, illustrating the non-planar, helically distorted conformation of the 9-methoxy-3-(piperidin-1-yl)-5,6-dihydro-2H-thia-dibenzo[c,g] phenanthrene-4-carbonitrile 7a.](/image/article/2012/RA/c1ra00824b/c1ra00824b-f1.gif) | ||
| Fig. 1 The conformation of the compound with arbitrary numbering is Shown as an ORTEP diagram, illustrating the non-planar, helically distorted conformation of the 9-methoxy-3-(piperidin-1-yl)-5,6-dihydro-2H-thia-dibenzo[c,g] phenanthrene-4-carbonitrile 7a. | ||
There are a number of weak intermolecular van der Waal interactions present in the crystal structure which stabilizes solid state structure of the molecule. However, the most important is the weak intermolecular C–H π interaction31 having a dimension of 2.759 Å operating between the centroid of ring A and the H2 atom of ring E to form a dimer (Fig. 2).
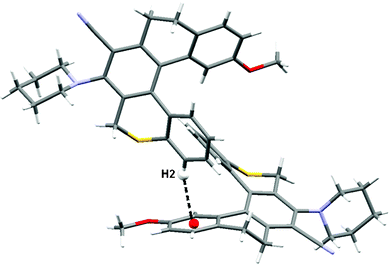 | ||
| Fig. 2 Dimeric structure for 7a displaying weak intermolecular C–H π interaction. | ||
Analysis of the configurational stability of 7
In order to assess the influence of orientation of the methyl in the methoxy group present on the ring A on the degree of helicity, quantum chemical calculations were performed using X-ray structure having C28-O1-C19-C18 torsion angle 180° and 0°. However, the alteration of this torsion angle from 180° to 0°causes no change in energy (the energy difference is of only −1.725 × 10−18 kcal mol−1). Hence, it was concluded that the orientation of the methoxy group does not influence the helicity of the molecule.In order to gain a deeper insight into the stereochemical behavior of 7, the helimeric inversion barrier for this compound was calculated using the quantum chemical methodology.
The key optimized structural parameters for the ground as well as the transition states are presented in Table S1 (ESI†). Calculations revealed that the helimerization barrier for 7 is ΔE = 44.36 kJ mol−1. The calculated value of the inversion barrier indicates a rapid interconversion of P and M isomers of 7a at room temperature.
Conclusions
In summary, we have developed a metal free, efficient, economical and concise route for the construction of partially reduced thia-, and oxa-thiahelicenes through base catalyzed ring transformation of 4-sec.amino-2-oxo-2,5-dihydrothiochromeno- [4,3-b]pyran-3-carbonitriles (4) by 2-tetralones(5) only in two steps. The beauty of protocol lies in the synthesis of partially reduced heterohelicenes without chemical or catalytic reduction. The synthetic methodology offers a high flexibility for the construction of partially reduced [5]heterohelicenes with halo, sec.amino and nitrile functionalities on the outer core of the frame-work and can be exploited for further ring formation. The exemplary conformational studies of 7a by X-ray and computational analysis revealed good agreement with theoretical and experimental values of torsional angles and molecular geometries of synthesized heterohelicenes.Experimental section
General remarks: Commercially available reagents were used without purification. 1H and 13C NMR spectra were taken on a Bruker 400 MHz and 300 MHz NMR spectrometer. CDCl3 and DMSO-d6 was used as solvent. Chemical shift are reported in parts per million shift (δ-value) from Me4Si (δ 0 ppm for 1H) or based on the middle peak of the solvent (CDCl3) (δ 77.00 ppm for 13C NMR) as an internal standard. Signal pattern are indicate as s, singlet; d, doublet; dd, double doublet; t, triplet; m, multiplet; bs, broad singlet; bm, broad multiplet. Coupling constant (J) are given in hertz (Hz). Infrared (IR) spectra were recorded on a Perkin-Elmer AX-1 spectrophotometer in KBr disc and reported in wave number (cm−1). ESIMS spectrometer were used for mass spectra analysis.General procedure for the synthesis of 4-methylthio-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles (3a,b): A mixture of methyl 2-cyano-3,3-dimethylthioacrylate (1.0 mmol), thiochroman-4-one (1.0 mmol) and powdered KOH (1.5 mmol) in dry DMF was stirred for 4-5 h at room temperature. The progress of the reaction was monitored by TLC. After completion, the reaction mixture was poured onto the crushed ice with vigorous stirring for 1–2 h. The resulted yellow precipitate was filtered, washed with distilled water several times and dried. The crude product was crystallized with ethanol for purification.
(3a) 1-Methylsulfanyl-3-oxo-3H,10H-4-oxa-9-thiaphenanthr- ene-2-carbonitrile: Yellow amorphous solid, mp: 180 °C, 1HNMR (400 MHz, DMSO-d6): δ 2.95 (s, 3H, SCH3), 4.06 (s, 2H, CH2), 7.36 (dd, J = 3.6 Hz, 1H, Ar-H) , 7.47 (d, J = 3.6 Hz, 2H, Ar-H), 7.8 (d, J = 8.0 Hz, 1H, Ar-H); 13C (100 MHz, DMSO-d6): δ 17.3, 23.1, 94.5, 109.2, 114.4, 125.4, 126.2, 127.2, 132.0, 135.9, 153.4, 156.8, 166.7. MS (CI) m/z 287 (M+); HRMS (CI) calcd for C14H9NO2S2 287.0075 found 287.0084.
(3b) 6-Chloro-1-methylsulfanyl-3-oxo-3H,10H-4-oxa-9-thiaphenanthrene-2-carbonitrile: Yellow amorphous solid, mp: 193 °C, 1HNMR (400 MHz, DMSO-d6): δ 2.94 (S, 3H, SCH3), 4.07 (s, 2H, CH2), 7.39–7.50 (m, 2H, Ar-H), 7.72 (s, 1H, Ar-H); 13C (100 MHz, DMSO-d6): δ 17.8, 23.5, 95.4, 110.2, 114.8, 125.9, 127.3, 129.2, 130.9, 131.9, 135.4, 152.3, 157.0, 166.9 MS (CI) m/z 321 (M+). HRMS (CI) calcd for C14H8ClNO2S2 320.9685 found 320.9704.
General procedure for the synthesis of 4-sec.amino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles (4): To a suspension of 4-methylthio-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitrile (3, 1.0 mmol) in ethanol, sec.amine (1.5 mmol) was added and the resulting mixture was refluxed for 5–6 h. Thereafter, reaction mixture was cooled and the precipitate was filtered and washed with cold ethanol to remove any unreacted amine. Finally, the crude product was crystallized from ethanol and characterized by spectroscopic techniques.
2-Oxo-4-(piperidin-1-yl)-2,5-dihydrothiochromeno[4,3-b] pyran-3-carbonitrile (4a): Orange colored crystalline solid; mp 183–185 °C; IR (KBr): 2202 (CN), 1716 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1HNMR: (400 MHz, DMSO-d6): δ 1.69 (bs, 6H, 3xCH2), 3.51 (m, 4H, 2xNCH2), 3.87 (s, 2H, SCH2), 7.33 (m, 1H, Ar-H), 7.745 (m, 2H, Ar-H), 7.74 (d, 1H, J = 7.8 Hz); 13CNMR (100 MHz, DMSO-d6): δ 22.9, 24.7, 25.9, 52.7, 79.6, 106.8, 116.6, 126.0, 126.4, 127.2, 127.5, 131.7, 135.8, 155.9, 160.2, 165.5; MS (CI): m/z 324 (M+); HRMS (CI): calcd for C18H16N2O2S: 324.0932 (M+); found: 324.0939.
O); 1HNMR: (400 MHz, DMSO-d6): δ 1.69 (bs, 6H, 3xCH2), 3.51 (m, 4H, 2xNCH2), 3.87 (s, 2H, SCH2), 7.33 (m, 1H, Ar-H), 7.745 (m, 2H, Ar-H), 7.74 (d, 1H, J = 7.8 Hz); 13CNMR (100 MHz, DMSO-d6): δ 22.9, 24.7, 25.9, 52.7, 79.6, 106.8, 116.6, 126.0, 126.4, 127.2, 127.5, 131.7, 135.8, 155.9, 160.2, 165.5; MS (CI): m/z 324 (M+); HRMS (CI): calcd for C18H16N2O2S: 324.0932 (M+); found: 324.0939.
9-Chloro-2-oxo-4-(piperidin-1-yl)-2,5-dihydrothiochromeno [4,3-b]pyran-3-carbonitrile (4b): Orange colored crystalline solid; mp 240 °C; IR (KBr): 2211 (CN), 1712 (CO); 1HNMR: (300 MHz, DMSO-d6): δ 1.68 (bs, 6H, 3xCH2), 3.51 (bs, 4H, 2xNCH2), 3.89 (s, 2H, SCH2), 7.49 (s, 2H, Ar-H), 7.79 (s, 1H, Ar-H); 13CNMR (75 MHz, DMSO-d6): δ 22.9, 24.7, 25.9, 52.8, 79.9, 107.5, 116.5, 125.3, 128.8, 129.2, 130.8, 131.3, 134.9, 154.6, 159.9, 165.2; MS (CI): m/z 324 (M+); HRMS (ESI): calcd for C18H15ClN2O2S: 359.0621 (M+); found: 359.0618.
4-Morpholino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitrile (4c): Orange colored crystalline solid; mp 183 °C; IR (KBr): 2212 (CN), 1714 (CO); 1HNMR: (300 MHz, DMSO-d6): δ 3.58-3.68 (m, 4H, 2xOCH2), 3.59–3.77 (m, 4H, 2xNCH2), 3.89 (s, 2H, SCH2), 7.32 (m, 1H, Ar-H), 7.743 (m, 2H, Ar-H), 7.74 (d, 1H, J = 7.8 Hz, Ar-H); 13CNMR (75 MHz, DMSO-d6): δ 24.6, 51.8, 66.3, 79.9, 106.6, 116.6, 126.0, 126.5, 127.1, 127.5, 131.8, 135.8, 156.2, 160.1, 165.0; MS (CI): m/z 326 (M+); HRMS (CI): calcd for C17H14N2O3S: 326.0759 (M+); found: 326.0756.
9-Chloro-4-morpholino-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitrile (4d): Orange colored crystalline solid; IR (KBr): 2213 (CN), 1717 (CO); 1H NMR: (400 MHz, DMSO-d6): δ 3.60 (t, 4H, J = 4.4 Hz, 2xNCH2), 3.78 (t, 4H, J = 4.4 Hz, 2xOCH2), 3.93 (s, 2H, SCH2), 7.51 (d, 2H, J = 1.2 Hz, Ar-H), 7.71 (t, 1H, J = 1.2 Hz, Ar-H); 13C NMR (100 MHz, DMSO-d6): δ 24.2, 51.4, 65.9, 79.9, 106.9, 116.1, 124.9, 128.3, 128.8, 130.4, 130.9, 134.5, 154.4, 159.4, 164.4, 164.8; MS (CI): m/z 360 (M+); HRMS (CI): calcd for C17H13ClN2O3S2: 360.0335 (M+); found: 360.0332.
2-Oxo-4-(pyrrolidin-1-yl)-2,5-dihydrothiochromeno[4,3-b] pyran-3-carbonitrile (4e): Orange colored crystalline solid; mp 230–231 °C; IR (KBr): 2197 (CN), 1689 (CO); 1HNMR: (400 MHz, DMSO-d6): δ 1.91 (t, J = 6.4 Hz, 4H, 2xCH2), 3.88 (t, J = 6.4 Hz, 4H, 2xNCH2), 4.06 (s, 2H, SCH2), 7.33–7.35 (m, 1H, Ar-H), 7.43–7.46 (m, 2H, Ar-H), 7.74 (d, 1H, J = 7.6 Hz, Ar-H); 13CNMR (100 MHz, DMSO-d6): δ 25.0, 26.7, 53.9, 70.9, 104.3, 118.2, 125.9, 126.2, 127.3, 131.2, 135.5, 153.8, 159.2, 131.1; MS (CI): m/z 310 (M+); HRMS (CI): calcd for C17H14N2O2S2: 310.0776 (M+); found: 310.0775.
General procedure for the synthesis of (Z)-2-(5,6-dihydrobenzo[f]thiochromeno[3,4-c]chromen-3(2H)ylidene) acetonitriles (6) and 3-sec. amino-5,6-dihydro-2H-1-thia-dibenzo[c,g] phenanthrene-4-carbonitriles (7):
Procedure A: A mixture of 4-(piperidin-1-yl)-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4 (1 mmol), 2-tetralone 2 (1.2 mmol) and NaH (1.5 mmol) in THF (6.0 mL) was stirred for 3-4 h at ambient temperature. Completion of the reaction was monitored by TLC. The excess of THF was removed under reduced pressure and the reaction mixture was poured onto crushed ice with vigorous stirring followed by neutralization with 10% HCl. The precipitate obtained was filtered, washed with water and dried. TLC of the crude product showed two main spots and were separated on neutral alumina column using 25% ethyl acetate in n-hexane as the eluent. The structure of the product with high Rf was assigned as (Z)-2-(5,6-dihydrobenzo[f]thiochromeno[3,4-c]chromen-3-(2H)-ylidene)acetonitriles 6 while compound isolated with low Rf was characterized as 3-(piperidin-1-yl)-5,6-dihydro-2H-1-thia-dibenzo[c,g]phenanthrene-4-carbonitriles 7.
Procedure B: A reaction mixture of 4-(piperidin-1-yl)-2-oxo-2,5-dihydrothiochromeno[4,3-b]pyran-3-carbonitriles 4 (1 mmol), 2-tetralone 2 (1.2 mmol) in DMF (6 mL) was stirred with powdered KOH (1.5 mmol) for 5–6 h at room temperature. The progress of the reaction mixture was monitored with TLC. After the completion of reaction the content was poured onto crushed ice with vigorous stirring followed by neutralization with 10% HCl. The resulted precipitate was filtered, washed with water and finally dried. The crude product on TLC showed one major spot which was isolated by column chromatography using neutral alumina as solid phase and 25% ethyl acetate in hexane as eluent. The isolated product was identical to the product 6.
(Z)-2-(5,6-Dihydrobenzo[f]thiochromeno[3,4-c]chromen-3(2H)-ylidene)acetonitrile (6a): Red-orange amorphous solid from acetone-hexane; mp 169 °C; 1HNMR (400 MHz, CDCl3): δ 2.77 (bd, 2H, J = 4.8 Hz, CH2), 2.87–3.11 (bm, 2H, CH2), 3.21–3.46 (bm, 2H, SCH2), 4.53 (s, 1H, CH), 6.40 (d, 1H, J = 7.8 Hz, Ar-H), 6.87–6.90 (m, 1H, Ar-H), 6.94–7.01 (m, 2H, Ar-H), 7.05–7.09 (m, 1H, Ar-H), 7.20–7.25 (m, 2H, Ar-H), 7.50 (d, 1H, J = 7.8 Hz, Ar-H); 13CNMR (100 MHz, CDCl3): δ 25.3, 27.4, 28.2, 66.2, 111.2, 117.8, 125.1, 125.3, 125.7, 126.3, 127.2, 127.5, 129.1, 129.4,130.0, 131.1, 131.2, 134.0, 134.2, 136.0, 159., 163.1; MS (CI): m/z 341 (M+); HRMS (CI): calcd for C22H15NOS: 341.0874 (M+); found: 341.0881.
(Z)-2-(12-Chloro-5,6-dihydrobenzo[f]thiochromeno[3,4-c]chromen-3(2H)-ylidene)acetonitrile (6b): Red amorphous solid from acetone-hexane; mp 165–167 °C; 1HNMR (400 MHz, CDCl3): δ: 2.77–2.79 (m, 2H, CH2), 2.99 (bs, 2H, CH2), 3.35 (bs, 2H, SCH2), 4.57 (s, 1H, CH), 6.39 (d, 1H, J = 7.6 Hz, Ar-H), 6.95–6.98 (m, 2H, Ar-H), 7.02–7.14 (m, 1H, Ar-H), 7.22–7.24 (m, 2H, Ar-H), 7.45 (d, 1H, J = 7.6 Hz, Ar-H); 13CNMR (100 MHz, CDCl3): δ 25.3, 27.3, 28.1, 67.1, 110.8, 117.5, 125.4, 125.8, 126.6, 126.8, 127.7, 129.2, 129.7, 130.0, 130.4, 131.1, 132.4, 132.6,134.0, 135.2, 160.0,162.7; MS (CI): m/z 375 (M+); HRMS (CI): calcd for C22H14ClNOS: 375.0455 (M+); found: 375.0458.
(Z)-2-(8-Methoxy-5,6-dihydrobenzo[f]thiochromeno[3,4-c] chromen-3(2H)-ylidene)acetonitrile (6c): Red amorphous solid from acetone-hexane; Rf in CH2Cl2 0.76; 1HNMR (400 MHz, CDCl3): δ: 2.74–2.75 (m, 2H, CH2), 2.94 (bs, 2H, CH2), 3.33 (bs, 2H, SCH2), 3.77 (s, 3H, OCH3), 4.50 (s, 1H, CH), 6.31 (d, 1H, J = 7.6 Hz, Ar-H), 6.43–6.45 (m, 1H, Ar-H), 6.77 (s, 1H, Ar-H), 6.97–7.05 (m, 2H, Ar-H), 7.22–7.25 (m, 1H, Ar-H), 7.50 (d, 1H, J = 7.6 Hz, Ar-H); 13CNMR (100 MHz, CDCl3): δ 25.3, 27.3, 28.7, 55.2, 65.7, 101.8, 110.9, 113.4, 117.9, 123.7, 124.8, 125.3, 128.4, 129.1, 129.3, 129.9, 131.2, 134.3, 135.9, 136.3, 157.9, 158.4, 163.2; MS (CI): m/z 372 (M++1), 371 (M+); HRMS (CI): calcd for C23H17NO2S: 371.0980 (M+); found: 371.0965
(Z)-2-(12-Chloro-8-methoxy-5,6-dihydrobenzo[f] thiochromeno[3,4-c]chromen-3(2H)-ylidene)acetonitrile (6d): Red amorphous solid from acetone-hexane; mp 120–125 °C; 1HNMR (400 MHz, CDCl3): δ: 2.73 (bm, 2H, CH2), 2.88 (bs, 1H, CH), 3.01 (bs, 1H, CH), 3.32 (bs, 1H, SCH2), 3.75 (s, 3H, SCH3), 4.49 (s, 1H, CH), 6.26 (d, 1H, J = 8.8 Hz, Ar-H), 6.44–6.47 (dd, 1H, J = 8.8 Hz, Ar-H), 6.74 (d, 1H, J = 2.4 Hz, Ar-H), 6.96 (d, 1H, J = 2.4 Hz, Ar-H), 7.17–7.19 (m, 1H, Ar-H), 7.40 (d, 1H, J = 8.0 Hz, Ar-H); 13C NMR (100 MHz, CDCl3): δ 25.4, 27.3, 28.6, 55.3, 66.6, 110.6, 110.9, 113.7, 117.7, 123.0, 125.3, 128.1, 129.2, 129.6, 130.0, 131.2, 132.7, 132.5, 132.7, 135.9, 158.1, 158.6, 162.9; MS (CI): m/z 405 (M+); HRMS (CI): calcd for C23H16ClNO2S: 405.0590 (M+); found: 405.0595.
(Z)-2-(9-Methoxy-5,6-dihydrobenzo[f]thiochromeno[3,4-c] chromen-3(2H)-ylidene)acetonitrile (6e): Red amorphous solid from acetone-hexane; Rf 0.33 in CH2Cl2, mp 165 °C; 1HNMR (400 MHz, CDCl3): δ 2.75 (bs, 2H, CH2), 2.85 (bs, 1H, CH2), 2.99 (bs, 1H, CH2), 3.36 (bs, 1H, SCH2), 3.37 (s, 3H, OCH3), 3.38 (bs, 1H, SCH2), 4.53 (s, 1H, CH), 5.93 (d, 1H, J = 2.8 Hz, Ar-H), 6.63 (dd, 1H, J = 8.0 and 2.8 Hz, Ar-H), 7.01–7.09 (m, 2H, Ar-H), 7.11 (d, 1H, J = 8.4 Hz, Ar-H), 7.23–7.27 (m, 1H, Ar-H), 7.49–7.70 (d, 1H, J = 8.4 Hz, Ar-H); 13CNMR (100 MHz, CDCl3): δ 25.3, 27.3, 27.7, 54.9, 66.3, 111.2, 112.3, 112.9, 117.8, 124.8, 125.3, 126.1, 128.2, 129.1, 129.3, 130.2, 130.9, 132.0, 134.4, 136.2, 157.4, 160.3, 163.0; MS (CI): m/z 371 (M+); HRMS (CI): calcd for C23H17NO2S: 371.0980 found: 371.0987.
(Z)-2-(12-Chloro-9-methoxy-5,6-dihydrobenzo[f] thiochromeno[3,4-c]chromen-3(2H)-ylidene)acetonitrile (6f): Red amorphous solid from acetone-hexane; mp 101–103 °C; 1HNMR (400 MHz, CDCl3): δ 2.75 (bs, 2H, CH2), 2.79 (bs, 1H, CH2), 2.87 (bs, 1H, CH2), 3.32 (bs, 1H, SCH2), 3.36 (bs, 1H, SCH2), 3.46 (s, 3H, OCH3), 4.56 (s, 1H, CH), 5.93 (d, 1H, J = 2.8 Hz, Ar-H), 6.67 (dd, 1H, J = 8 Hz, Ar-H), 6.99 (d, 1H, J = 2 Hz, Ar-H), 7.13 (d, 1H, J = 8 Hz, Ar-H), 7.23 (dd, 1H, J = 8.0 and 2.0 Hz, Ar-H), 7.44 (d, 1H, J = 8.4 Hz, Ar-H); 13CNMR (100 MHz, CDCl3): δ 25.3, 27.2, 27.7, 55.2, 67.2, 110.8, 112.6, 112.9, 117.5, 125.3, 126.3, 128.5, 129.1, 129.9,130.1, 131.1, 131.4, 132.2, 132.9, 135.2, 157.6, 160.6, 162.7; MS (CI): m/z 405 (M+); HRMS (CI): calcd for C23H16ClNO2S: 405.0590 (M+); found: 405.0597.
9-Methoxy-3-(piperidin-1-yl)-5,6-dihydro-2H-phenanthro [3,4-c]thiochromene-4-carbonitrile (7a): It was prepared from the reaction of 4a (1 mmol) and 5c (1 mmol) in THF (5.0 mL) in the presence of NaH (1.5 mmol) as a base. The progress of the reaction was monitored with TLC. The reaction mixture was worked up as usual and purified on Si gel column chromatography. The upper spot isolated as an oil which solidified after 48 h. The solid was crystallized with little acetone and characterized by spectroscopic analyses. It was finally identified by single crystal X-ray diffraction. White crystalline solid, mp 212 °C, 1HNMR (400 MHz, CDCl3): δ 1.74 (bs, 4H, CH2), 1.83 (bs, 2H, CH2), 2.64 (bs,1H, CH2), 2.84–2.86 (m, 2H, CH2), 3.01 (bs, 1H, CH2), 3.22 (bs, 2H, CH2), 3.31 (s, 3H, OCH3), 3.34 (bs, 1H, CH2), 3.55(bs, 1H, CH2), 3.76 (bs, 2H, CH2), 6.18-(d, 1H, J = 2.8 Hz, Ar-H), 6.66 (m, 1H, Ar-H), 6.94–6.96 (m, 1H, Ar-H), 7.11–7.19 (m, 3H, Ar-H), 7.58 (d, 1H, J = 8 Hz, ArH). 13C NMR (100 MHz, CDCl3): δ 24.0, 28.0, 29.0, 54.7, 107.1, 114.6, 117.9, 125.4, 128.1, 129.3, 130.3, 130.9, 132.0, 134.2, 135.4, 136.3, 137.2, 138.9, 144.9, 150.7, 157.2. MS (CI): m/z 438 (M+); HRMS (CI): calcd for C28H26N2OS: 438.1766 (M+); found: 438.1770.
9-Methoxy-3-(morpholin-4-yl)-5,6-dihydro-2H-phenanthro [3,4-c]thiochromene-4-carbonitrile (7b): It was obtained by stirring a mixture of 4c (1.0 mmol) and 5c (1.0 mmol) in THF (5.0 mL) using NaH (60%, 1.5 mmol)) as a base at room temperature. The usual workup and purification on Si gel column using ethyl acetate and hexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) as an eluent gave title compound as a viscous oil. The other compound isolated as upper spot was identified as 6e.
:3) as an eluent gave title compound as a viscous oil. The other compound isolated as upper spot was identified as 6e.
1HNMR (400 MHz, CDCl3): δ 2.82–2.85 (m, 1H, CH2), 3.16–3.27 (m, 2H, CH2), 3.40–3.44 (m, 5H, CH2), 3.68–3.69 (m, 3H, OCH3), 3.69 (s, 1H, CH2), 3.72(s, 1H, CH2), 3.80–3.83 (m, 4H, CH2), 6.29 (d, J = 2.8 Hz, 1H, Ar-H), 6.49 (dd, J = 8.8 and 2.8 Hz, 1H, Ar-H), 6.79 (d, J = 0.8 and 2.8 Hz, 1H, ArH), 6.88 (ddd, J = 7.6 and 1.2 Hz, 1H, ArH), 7.05 (ddd, J = 7.6 and 1.2 Hz, 1H, ArH), 7.19 (d, 1H, J = 7.6 Hz, ArH), 7.28 (d, J = 1.2 Hz, 1H, ArH). 13C NMR (100 MHz, CDCl3): δ 23.6, 27.7, 39.4, 50.1, 54.9, 111.8, 113.1, 118.0, 125.2, 125.6, 127.1, 127.2, 128.7, 129.0, 129.7, 132.7, 133.2, 134.9, 151.6, 158.6, 162.7; MS (CI): m/z 440 (M+); HRMS (CI): calcd for C27H24N2O2S: 440.1558 (M+); found: 440.1610.
Crystallographic details: Intensity data for the colorless crystal were collected at 293(2) K on a Bruker APEX-II CCD diffractometer system equipped with graphite monochromated Cu-Kα radiation λ = 1.54178 Å. The final unit cell determination, scaling of the data, and corrections for Lorentz and polarization effects were performed with Bruker SAINT.32 A symmetry-related numerical absorption correction had been applied. The structures were solved by direct methods (SHELXS-97)33 and refined by a full-matrix least-squares procedure based on F2.34 All non-hydrogen atoms were refined anisotropically; hydrogen atoms were located at calculated positions and refined using a riding model with isotropic thermal parameters fixed at 1.2 times the Ueq value of the appropriate carrier atom. Figure for the compound 7a was prepared using ORTEP.35
Crystal data for 7a: C28H26N2OS, formula mass 438.57, orthorhombic space groupP212121, a = 10.313(2), b = 13.170(3), c = 16.748(8) Å, V = 2274.7(8) Å3, Z = 4, dcalcd = 1.281 mg m−3, linear absorption coefficient 1.434 mm−1, F(000) = 928, crystal size 0.26 × 0.20 × 0.16 mm, reflections collected 1505, independent reflections 1505. Final indices [I > 2σ(I)] R1 = 0.0470 wR2 = 0.0979, R indices (all data) R1 = 0.0711, wR2 = 0.1138, gof 1.123, Largest difference peak and hole 0.189 and −0.249 e Å−3.
Computational details
The geometries for both the ground state and transition state were calculated using hybrid Lee–Yang–Parr functional36 using 6-311G** basis set for all the atoms. For the ground state geometry optimization stable keyword was employed, while for both the structures GDIIS algorithm used. Harmonic vibrational frequencies were calculated for all of the stationary points. For the optimized ground state the frequency analysis verified the absence of any imaginary frequencies, whereas the transition state showed only one single imaginary frequency. For the optimization of both the ground as well as the transition state single crystal X-ray coordinates was the starting point. Both the ground and transition state structures were subjected to MP2/6-311G** single-point energy calculations to provide more reliable values for the enthalpy of formation. All the calculations were performed using G03 program.37Acknowledgements
VJR is thankful to AvH Foundation, Germany for financial support and Prof. Uli Kazmaier, Universität des Saarlandes, Saarbrucken, Germany for providing laboratory facilities. UGC support for Emeritus Fellowship is gratefully acknowledged. RP is thankful to University of Delhi to provide fund under R & D grant for research.References
- (a) For reviews, see: H. Osuga and K. Tanaka, J. Synth. Org. Chem. Jpn., 2002, 60, 593 CrossRef CAS; (b) K. Sato and S. AraiIn Cyclophane Chemistry for the 21th centuary (Ed.: H. Takemura). Research Signpost, Kerala, 2002 chap. 8, p. 173 Search PubMed; (c) H. Hopf, Classics in Hydrocarbon Chemistry: Syntheses, Concept Perspectives, Wiley-VCH, Weinheim, 2000, chap. 12. p. 321 Search PubMed; (d) K. P. Meurer and F. Vögtle, Top. Curr. Chem., 1985, 127, 1 CrossRef CAS; (e) W. H. Laarhoven and W. J. C. Prinsen, Top. Curr. Chem., 1984, 125, 63 CAS; (f) R. H. Martin, Angew. Chem., Int. Ed. Engl., 1974, 13, 649 CrossRef; (g) I. G. Stara and I. Stary, Science of Synthesis, 2010, 45b, 885–953 Search PubMed; (h) I. G. Stara and I. Stary, Strained Hydrocarbons (H. Dodziuk, Ed.), Wiley-VCH 2009, Chapter 4.3, 166–176 Search PubMed; (i) A. Rajca and M. Miyasaka, Syntheses and Strategies; T. J. J. Mueller, U. H. F. Bunz, ed.; Wiley-VCH: Weinheim, 2007, 543–577 Search PubMed.
- K. Nakano, Y. Hidechira, K. Takahashi, T. Hiyama and K. Nozaki, Angew. Chem., Int. Ed., 2005, 44, 7136–7138 CrossRef CAS.
- (a) T. J. Wigglesworth, D. Sud, T. B. Norsten, V. S. Lekhi and N. R. Branda, J. Am. Chem. Soc., 2005, 127, 7272–7273 CrossRef CAS; (b) E. Botek, B. Champagne, M. Turki and J. -M. Andre, J. Chem. Phys., 2004, 120, 2042–2048 CrossRef CAS; (c) C. Nuckolls, T. J. Katz, T. Verbiest, S. Van Elshocht, H.–G. Kuball, S. Kiesewalter, A. J. Lovinger and A. Persoons, J. Am. Chem. Soc., 1998, 120, 8656–8660 CrossRef CAS; (d) S. Maiorana, A. Papagni, E. Licandro, R. Annunziata, P. Paravidino, D. Perdicchia, C. Giannini, M. Bencini, K. Clays and A. Persoons, Tetrahedron, 2003, 59, 6481–6488 CrossRef CAS.
- (a) F. Furche, R. Ahrlichs, C. Wachsmann, E. Weber, A. Sobanski, F. Vögtle and S. Grimme, J. Am. Chem. Soc., 2000, 122, 1717–1724 CrossRef CAS; (b) G. Treboux, P. Lapstun, Z. Wu and K. Silverbrook, Chem. Phys. Lett., 1999, 301, 493–497 CrossRef CAS; (c) D. Beljonne, Z. Shuai, J. L. Bredas, M. Kaurranen, T. Verbiest and A. Persoons, J. Chem. Phys., 1998, 108, 1301–1304 CrossRef CAS.
- (a) Y. Xu, Y. X. Zhang, H. Sugiyama, T. Umano, H. Osuga and K. Tanaka, J. Am. Chem. Soc., 2004, 126, 6566–6567 CrossRef CAS; (b) I. Sato, R. Yamashima, K. Kadowaki, J. Yamamoto, T. Shibata and K. Soai, Angew. Chem., Int. Ed., 2001, 40, 1096–1098 CrossRef CAS.
- C. Nuckolls, R. Shao, W. -G. Jang, N. A. Clark, D. M. Welba and T. J. Katz, Chem. Mater., 2002, 14, 773–776 CrossRef CAS.
- (a) B. Laleu, P. Mobian, C. Herse, B. W. Laursen, G. Hopfgartner, G. Bernardinelli and J. Lacour, Angew. Chem., Int. Ed., 2005, 44, 1879–1883 CrossRef CAS; (b) M. T. Reetz and S. Sostmann, Tetrahedron, 2001, 57, 2515–2520 CrossRef CAS.
- (a) S. D. Dreher, T. J. Katz, K.–C. Lam and A. L. Rheingold, J. Org. Chem., 2000, 65, 815–822 CrossRef CAS; (b) M. T. Reetz and S. Sostmann, J. Organomet. Chem., 2000, 603, 105–109 CrossRef CAS; (c) M. T. Reetz, E. W. Beuttenmü and R. Goddard, Tetrahedron Lett., 1997, 38, 3211–3214 CrossRef CAS.
- (a) H. Sugiura, Y. Takahira and M. Yamaguchi, J. Org. Chem., 2005, 70, 5698–5708 CrossRef CAS; (b) O. Ermer and J. Neudoerfl, Helv. Chim. Acta, 2001, 84, 1268–1313 CrossRef CAS; (c) E. Murguly, R. McDonald and N. R. Branda, Org. Lett., 2000, 2, 3169–3172 CrossRef CAS; (d) K. Deshayes, R. D. Broene, I. Chao, C. B. Knobler and F. Diedrich, J. Org. Chem., 1991, 56, 6787–6795 CrossRef CAS; (e) L. Owens, C. Thilgen, F. Diederich and C. B. Knobler, Helv. Chim. Acta, 1993, 76, 2757–2774 CrossRef CAS.
- (a) A. J. Lovinger, C. Nuckolls and T. J. Katz, J. Am. Chem. Soc., 1998, 120, 264–268 CrossRef CAS; (b) T. P. Bender, Y. Qi, J. P. Gao and Z. Y. Wang, Macromolecules, 1997, 30, 60001–60006 CrossRef; (c) Y. J. Dai and T. J. Katz, J. Org. Chem., 1997, 62, 1274–1285 CrossRef CAS; (d) J. M. Fox, D. Lin, Y. Itagaki and T. Fujita, J. Org. Chem., 1998, 63, 2031–2038 CrossRef CAS.
- (a) T. J. Katz, Angew. Chem., Int. Ed., 2000, 39, 1921 CrossRef CAS; (b) C. Nuckolls, T. Z. Katz, G. Katz, P. J. Collings and L. Castellanos, J. Am. Chem. Soc., 1999, 121, 79 CrossRef CAS; (c) C. Nuckolls, T. J. Katz, G. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef; (d) C. Nuckolls and T. J. Katz, J. Am. Chem. Soc., 1998, 120, 9541 CrossRef CAS.
- (a) In The Principles of Non-linear Optics; Y. R. Shen, Ed.; Wiley: New York, 1984 Search PubMed; (b) In Introduction to Non- linear Optical Effects in Molecules and Polymers; P. N. Prasad, D. J. Williams, ed.; Wiley: New York, 1991 Search PubMed; (c) In Principles and Applications of Non-linear Optical Materials; R. W. Munn, C. N. Ironside, ed.; Chapman and Hall: London, 1993 Search PubMed; (d) In Molecular Non-linear Optics; J. Zyss, Ed.; Academic: London, 1994 Search PubMed; (e) In Photonic Polymer Systems: Fundamentals, Methods and Applications; D. L. Wise, G. E. Wnek, D. J. Trantolo, T. M. Cooper, J. D. Gresser, ed.; Marcel Dekker: New York, 1998 Search PubMed; (f) In Non-linear Effects and Materials; P. Gunter, Ed.; Springer: Berlin, 2000 Search PubMed.
- (a) S. Sioncke, S. Van Elshocht, T. Verbiest, A. Persoons, M. Kauranen, K. E. S. Phillips and T. J. Katz, Chem. Phys., 2000, 113, 7578–7581 CAS; (b) D. Belijonne, Z. Shuai, J. L. Brédas, M. Kauranen, T. Verbiest and A. Persoons, J. Chem. Phys., 1998, 104, 1301–1304 CrossRef; (c) L. Minuti, A. Taticchi, A. Marrocchi, E. Gacs-Baitz and R. Galeazzi, Eur. J. Org. Chem., 1999, 3155–3163 CrossRef CAS; (d) H. Soon Choi and K. S. Kim, J. Phys. Chem. B, 2000, 104, 11006–11009 CrossRef.
- (a) H. Ashitaka and H. Sasabe, Non-linear Opt., 1995, 14, 81–89 CAS; (b) J. Meisenheimer and K. White, Chem. Ber., 1903, 36, 4153 CrossRef CAS; (c) For recent review, see: F. Dumitrascu, D. G. Dumitrescu and I. Aron, ARKIVOC, 2010, 1, 1–32 Search PubMed; (d) A. Rajca, M. Pink, S. Xiao, M. Miyasaka, S. Rajca, K. Das and K. Plessel, J. Org. Chem., 2009, 74, 7504–7513 CrossRef CAS; (e) M. Miyasaka, M. Pink, S. Rajca and A. Rajca, Angew. Chem., Int. Ed., 2009, 48, 5954–5957 CrossRef CAS; (f) K. Tanaka, N. Fukawa, T. Suda and K. Noguchi, Angew. Chem., Int. Ed., 2009, 48, 5470–5473 CrossRef CAS; (g) S. Graule, M. Rudolph, N. Vanthuyne, J. Autschbach, Ch. Roussel, J. Crassous and R. Reau, J. Am. Chem. Soc., 2009, 131, 3183–3185 CrossRef CAS; (h) L. Adriaenssens, L. Severa, T. Sālovā, I. Císarovā, R. Pohl, D. Saman, S. V. Rocha, N. S. Finney, L. Pospísil, P. Slavícek and F. Teply, Chem.–Eur. J., 2009, 15, 1072–1076 CrossRef CAS; (i) Ch. Li, J. Shi, L. Xu, Y. Wang, Y. Cheng and H. Wang, J. Org. Chem., 2009, 74, 408–411 CrossRef CAS; (j) S. K. Collins and M. P. Vachon, Org. Biomol. Chem., 2006, 4, 2518–2524 RSC; (k) K. Sato, Y. Katayama, T. Yamagishi and S. Arai, J. Heterocycl. Chem., 2006, 43, 177–181 CrossRef CAS; (l) K. Nakano, Y. Hidehira, K. Takahashi, T. Hiyama and K. Nozaki, Angew. Chem., 2005, 117, 7298–7300 CrossRef; (m) T. Kaseyama, S. Furumi, X. Zhang, K. Tanaka and M. Takeuchi, Angew. Chem., Int. Ed., 2011, 50, 3684–3687 CrossRef CAS.
- (a) F. Aloui, R. El Abed, A. Marinetti and B. Ben Hassine, Tetrahedron Lett., 2008, 76, 1439 Search PubMed; (b) M. Reetz and S. Sostmann, Tetrahedron, 2001, 57, 2515 CrossRef CAS; (c) L. Liu, B. Yang, T. J. Katz and M. K. Pointdexter, J. Org. Chem., 1991, 56, 3769 CrossRef CAS; (d) H. R. Talele, A. R. Chaudhary, P. R. Patel and A. V. Bedekar, ARKIVOC, 2011, 9, 15–37 Search PubMed; (e) N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef CAS.
- (a) K. Yano, M. Osatani, K. Tani, T. Adachi, K. Yamamoto and H. Matsubara, Bull. Chem. Soc. Jpn., 2000, 73, 185–189 CrossRef CAS; (b) C. Stammel, R. Froehlich, C. Wolff, H. Weneck, A. De Meijere and J. Mattay, Eur. J. Org. Chem., 1999, 1709–1718 CrossRef CAS; (c) A. Terfort, H. Goerls and H. Brunner, Synthesis, 1997, 79–86 CrossRef CAS; (d) W. H. Laarhoven and W. J. C. Prinsen, Top. Curr. Chem., 1984, 125, 63–130 CAS.
- (a) For recent nonphotochemical syntheses, see: M. C. Carreno, S. Garcia-Cerrada and A. Urbano, J. Am. Chem. Soc., 2001, 123, 7929 CrossRef CAS; (b) M. C. Carreno, S. Garcia-Cerrada, M. J. Sanz-Cuesta and A. Urbano, Chem. Commun., 2001, 1452 RSC; (c) H. Okubo, D. Nakano, S. Anzai and M. Yamaguchi, J. Org. Chem., 2001, 66, 557 CrossRef CAS; (d) J. Eskildsen, F. C. Krebs, A. Faldt, P. Sommer-Larsen and K. Bechgaard, J. Org. Chem., 2001, 66, 200 CrossRef CAS; (e) E. Murguly, R. McDonald and N. R. Branda, Org. Lett., 2000, 2, 3169 CrossRef CAS; (f) H. Okubo, D. Nakano, M. Yamaguchi and C. Kabuto, Chem. Lett., 2000, 1316 CrossRef CAS; (g) A. Modlerspreitzer, R. Fritsch and A. Mannschreck, Collect. Czech. Chem. Commun., 2000, 65, 555 CrossRef CAS; (h) L. Minuti, A. Taticchi, A. Marrocchi, E. Gacs- Baitz and R. Galeazzi, Eur. J. Org. Chem., 1999, 3155 CrossRef CAS; (i) M. C. Carreno, R. Hernández-Sánchez, J. Mahugo and A. Urbano, J. Org. Chem., 1999, 64, 1387 CrossRef CAS; (j) I. G. Stará, I. Starỳ, A. Kollárovič, F. Teplỳ, Sÿ. Vyskočil and D. Sÿaman, Tetrahedron Lett., 1999, 40, 1993 CrossRef; (k) M. Gingras and F. Dubois, Tetrahedron Lett., 1999, 40, 1309 CrossRef CAS; (l) F. Dubois and M. Gingras, Tetrahedron Lett., 1998, 39, 5039 CrossRef CAS; (m) H. Okubo, M. Yamaguchi and C. Kabuto, J. Org. Chem., 1998, 63, 9500 CrossRef CAS; (n) L. Minuti, A. Taticchi and A. Marrocchi, Synth. Commun., 1998, 28, 2181 CrossRef CAS; (o) K. Tanaka, H. Suzuki and H. Osuga, J. Org. Chem., 1997, 62, 4465 CrossRef CAS; (p) K. Tanaka, H. Suzuki and H. Osuga, Tetrahedron Lett., 1997, 38, 457 CrossRef CAS; (q) L. Minuti, A. Taticchi, A. Marrocchi and E. Gacs-Baitz, Tetrahedron, 1997, 53, 6873 CrossRef CAS; (r) S. Cossu, O. De Lucchi, D. Fabbri, G. Valle, G. F. Painter and A. J. Smith, Tetrahedron, 1997, 53, 6073 CrossRef CAS; (s) I. Pischel, S. Grimme, S. Kotila, M. Nieger and F. Vögtle, Tetrahedron: Asymmetry, 1996, 7, 109 CrossRef CAS; (t) J. Larsen and K. Bechgaard, J. Org. Chem., 1996, 61, 1151 CrossRef CAS; (u) M. Yamaguchi, H. Okubo and M. Hirama, Chem. Commun., 1996, 1771 RSC; (v) A. Dore, D. Fabbri, S. Gladiali and G. Valle, Tetrahedron: Asymmetry, 1995, 6, 779 CrossRef CAS; (w) I. G. Stará, I. Starỳ, M. Tichỳ, J. Závada and V. Hanuš, J. Am. Chem. Soc., 1994, 116, 5084 CrossRef; (x) A. Urbano, Angew. Chem., Int. Ed., 2003, 42, 3986–3989 CrossRef CAS.
- (a) M. C. Carreno, S. Garcia-Cerrada and A. Urbano, Chem.–Eur. J., 2003, 9, 4118–4131 CrossRef CAS; (b) M. del Mar Real, J. P. Sestelo and L. A. Sarandeses, Tetrahedron Lett., 2002, 43, 9111–9114 CrossRef.
- (a) I. G. Stara, I. Stray, A. Kollarovic, F. Teply, D. Saman and P. Fiedler, Collect. Czech. Chem. Commun., 2003, 68, 917–930 CrossRef CAS; (b) F. Teply, I. G. Stara, I. Stary, A. Kollarovic, D. Saman, L. Rulisek and P. Fiedler, J. Am. Chem. Soc., 2002, 124, 9175–9180 CrossRef CAS; (c) I. G. Stara, Z. Alexandrova, F. Teply, P. Sehnal, I. Stary, D. Saman, M. Budesinsky and J. Cvacka, Org. Lett., 2005, 7, 2547–2550 CrossRef CAS; (d) D. C. Haarowven, M. I. T. Nunn and D. R. Fenwick, Tetrahedron Lett., 2002, 43, 3189–3191 CrossRef; (e) J. Misek, F. Teply, I. G. Stara, M. Tichy, D. Saman, I. Cisarova, P. Vojtisek and I. Stary, Angew. Chem., Int. Ed., 2008, 47, 3188–3191 CrossRef CAS; (f) P. Sehnal, I. G. Stara, D. Saman, M. Tichy, J. Misek, J. Cvacka, L. Rulisek, J. Chocholousova, J. Vacek, G. Goryl, M. Szymonski, I. Cisarova and I. Stary, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13169–13174 CrossRef CAS.
- (a) S. K. Collins, A. Grandbois, M. P. Vachon and J. Gote, Angew. Chem., Int. Ed., 2006, 45, 2923 CrossRef CAS; (b) A. Grandbois and S. K. Collins, Chem.–Eur. J., 2008, 14, 9323–9329 CrossRef CAS.
- K. Kamikawa, I. Takemoto, S. Takemoto and H. Matsuzaka, J. Org. Chem., 2007, 72, 7406 CrossRef CAS.
- (a) J. Ichikawa, M. Yokota, T. Kudo and S. Umezaki, Angew. Chem., Int. Ed., 2008, 47, 4870 CrossRef CAS; (b) A. Goel, D. Verma, R. Pratap, G. Taneja, Y. Hemmberger, M. Knauer, R. Raghunandan, P. R. Maulik, V. J. Ram and G. Bringmann, Eur. J. Org. Chem., 2011, 2940–2947 CrossRef CAS.
- (a) M. C. Carreno, M. S. Gonzales-Lopez and A. Urbano, Chem. Commun., 2005, 611 RSC; (b) M. Carmen Carreno, A. Enriquez, S. Garcia-Cerrada, M. J. Sanz-Cuesta, A. Urbano, F. Maseras and A. Nonell-Canals, Chem.–Eur. J., 2008, 14, 603–620 CrossRef; (c) A. Latorre, A. Urbano and M. C. Carreno, Chem. Commun., 2009, 6652–6654 RSC; (d) A. Latorre, A. Urbano and M. C. Carreno, Chem. Commun., 2011, 47, 8103–8105 RSC.
- Y. Ogawa, M. Toyama, M. Karikomi, K. Seki, K. Haga and T. Uyehara, Tetrahedron Lett., 2003, 44, 2167 CrossRef CAS.
- E. R. Abed, B. B. Hassine, G. P. Genet, M. Gorsane, J. Madec, L. Richard and A. Marinetti, Synthesis, 2004, 2515 Search PubMed.
- (a) A. Aloui, E. R. Abed, A. Marinetti and B. Ben Hassine, Tetrahedron Lett., 2008, 76, 1439 Search PubMed; (b) L. Liu, B. Yang, T. J. Katz and M. K. Pointdexter, J. Org. Chem., 1991, 56, 3769 CrossRef CAS.
- F. Teply, I. G. Stara, I. Stary, A. Kollarovic, D. Saman, S. Vyskocil and P. Fiedler, J. Org. Chem., 2003, 68, 5193 CrossRef CAS.
- M. K. Lakshman, P. L. Kole, S. Chaturvedi, J. H. Saugier, H. J. C. Yeh, J. P.. Glusker, H. L. Carrell, A. K. Katz, C. E. Afshar, W-M. Dashwood, G. Kenniston and W. M. Baird, J. Am. Chem. Soc., 2000, 122, 12629–12636 CrossRef CAS.
- R. Gompper and W. Topfl, Chem. Ber., 1962, 95, 2861 CrossRef CAS.
- V. J. Ram, M. Nath, P. Srivastava, S. Sarkhel and P. R. Maulik, J. Chem. Soc., Perkin Trans. 1, 2000, 3719–3723 RSC.
- V. J. Ram, A. Goel, S. Sarkhel and P. R. Maulik, Bioorg. Med. Chem., 2002, 10, 1275–1280 CrossRef CAS.
- SAINT+ 6.02 ed.; Bruker AXS; Madison, WI, 1999.
- G. M. Sheldrick, SHELXS97, Program for Crystal Structure Solution, University of Göttingen, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, 1997 Search PubMed.
- M. N. Burnett and C. K. Johnson, ORTEP-III, Oak Ridge Thermal Ellipsoid Plot Program for Crystal Structure Illustrations, Report ORNL-6895, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1996.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 1133 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, T. Vreven Jr., K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. D. Dapprich, A. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, W. M. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D. 01) Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: All the NMR spectra are given in this section. See DOI: 10.1039/c1ra00824b/ |
| This journal is © The Royal Society of Chemistry 2012 |