Investigation of heterodimeric and homodimeric radical cations of the series: [F2O2]+, [F2Cl2]+, [Cl2O2]+, [F4]+, and [Cl4]+†
Tobias
Schlöder
and
Sebastian
Riedel
*
Albert-Ludwigs Universität Freiburg, Institut für Analytische und Anorganische Chemie, Albertstrasse, 21, 79104, Freiburg, Germany. E-mail: sebastian.riedel@psichem.de, www.psichem.de; Fax: 0049 761 203 6001; Tel: 0049 761 203 8717
First published on 22nd November 2011
Abstract
In this state-of-the-art quantum-chemical investigation we report structures, thermochemical stabilities, Born-Fajans-Haber cycles as well as vibrational data of heterodimeric and homodimeric radical cations of the series [F2O2]+, [F2Cl2]+, [Cl2O2]+, [F4]+, and [Cl4]+. The so far experimentally unknown species [F4]+, [F2O2]+ and [F2Cl2]+ are predicted to be thermochemically stable and could be possible targets for gas-phase or matrix-isolation experiments. Furthermore, their stabilities as homodimeric [X4]+ or heterodimeric [X2Y2]+ radical cation salts in the solid state have been estimated by Born-Fajans-Haber cycles.
Introduction
Almost ten years ago the cationic species [Cl4]+ and [Cl2O2]+ were prepared and structurally characterized.1,2[Cl4]+ was synthesized outgoing from IrF6 and Cl2 forming the ion-pair complex [Cl4]+[IrF6]−.1 The crystal structure contains a rectangular [Cl4]+ ion with no significant contacts to the counter ion. A detailed estimation of the thermochemical stability of this species has to the best of our knowledge not been published. Nevertheless, the stability of the theoretical fluorine homologue [F4]+ was computationally investigated at ab initio level3 indicating computational difficulties of the Møller–Plesset theory which could be overcome by higher level calculations like CCSD(T).3,4 A qualitative analysis of the bonding situation was also performed at various levels of theory describing the bonding as a π*–π* interaction.1–3This type of association is also observed for the [Cl2O2]+ cation which was synthesized in anhydrous HF as [Cl2O2]+[SbF6]−, [Cl2O2]+[Sb2F11]− and [Cl2O2]+[HIr2F12]− ion-pair complexes in which the cations show similar trapezoidal structures. Even the dissociation energy of this [Cl2O2]+ molecule cation was measured by Sunderlin and co-workers using energy resolved collision-induced dissociation experiments in a flowing afterglow-tandem mass spectrometer.5 Applying the same technique the same group has also attempted to prepare the analogous fluorine compound [F2O2]+ but without any success.5
Herein, we report structures, thermochemical stabilities, and vibrational frequencies for [F2O2]+ and [F2Cl2]+ obtained by state-of-the-art quantum-chemical calculations. To put these data into perspective, we have also investigated the stability of [F4]+, [Cl4]+ and [Cl2O2]+ in the gas-phase as well as bulk material.
Computational methods
As previous analysis have shown that a careful treatment of electron correlation is crucial for the balanced description of the molecules in question we have performed state-of-the-art quantum-chemical calculations up to CCSD(T) level. The coupled-cluster calculations with single and double substitutions as well as perturbative triple excitations [CCSD(T) level] were carried out with MOLPRO 20066 using an ROHF reference wavefunction. All species were fully optimized at a given computational level. Several Dunning correlation consistent basis sets [double-ζ (aug-cc-pVDZ), triple-ζ (aug-cc-pVTZ), and quadruple-ζ (aug-cc-pVQZ); for brevity the basis sets are denoted as aVXZ] were used to evaluate the basis-set effects. Corrections for the incompleteness of the basis-set size have been adjusted by using the correlation-consistent basis sets which can be extrapolated to the complete basis set limit (CBS) using equation E(lmax) = ECBS + B/lmax3 (B are values of the three highest lmax).7 Anharmonic corrections and isotopic shifts of the vibrational spectra have been calculated at the DFT (B3LYP8/aVTZ) level using the program package Gaussian 09.9 Some properties have been investigated using the BP8610,11 functional. Scalar and spin–orbit relativistic effects have not been considered in this study. All investigated species have shown normal T1-diagnostic values and can be treated by single reference methods.Results and discussion
Structures
As expected, rectangular structures (D2h symmetry) are observed for the two homodimeric systems [F4]+ and [Cl4]+, see Fig. 1.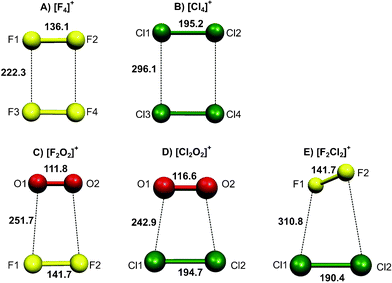 | ||
| Fig. 1 Optimized structures at CCSD(T)/aVTZ level. | ||
In the case of [F4]+ the intrafragmental bond length (dF1–F2 = dF3–F4 = 136.1 pm) lies between the bond lengths of the two separated fragments F2 and [F2]+, the bond being 5.2 pm shorter than the F–F bond in F2 (dF–F = 141.3 pm) and 5.6 pm longer than the one in [F2]+ (dF–F = 130.7 pm), all values computed at CCSD(T)/aVQZ level. The distance between the two fragments in [F4]+ has been computed to be dF1–F3 = dF2–F4 = 222.3 pm which is significantly shorter than the sum of the van der Waals radii (2⋯rF = 280 pm)12 and in good agreement with the known literature values of ranging between 220.7 and 221.8 pm.3
The picture is the same for the heavier homologue [Cl4]+ as the length of the intrafragmental bond (dCl1–Cl2 = dCl3–Cl4 = 195.2 pm) is intermediate between the bond lengths in two free fragments (Cl2: dCl–Cl = 200.3 pm, [Cl2]+: dCl–Cl = 190.5 pm) at CCSD(T)/aVQZ level; the distance between the two fragments is calculated to be 296.1 pm. All these values are in good agreement with experimental findings, see Table 1.
| Species | aVDZ | aVTZ | aVQZ | Exp. | QC–Ref. |
|---|---|---|---|---|---|
| a Distances in pm, angles in degree. b UMP2 optimized structure.3 c UQCISD(T) optimized structure.3 d UCCSD(T) optimized structure.3 e RCCSD(T) optimized structure.3 f Optimized structure at MP2/6311++G(3df,3pd) see ref. 1. g Optimized structure at B3LYP/6311++G(3df,3pd).1 h [Cl2O2]+ [HIr2F12]− see ref. 1. i [Cl2O2]+[SbF6]− see ref. 2. j [Cl2O2]+[Sb2F11]− see ref. 2. k [Cl2O2]+ B3LYP/aVTZ see ref. 5. l [Cl2O2]+ optimized at B3LYP/6311++G(3df).2 m [Cl2O2]+ optimized at CASPT2 level.2 | |||||
| [F4]+ | |||||
| F1–F1 distance | 139.3 | 136.6 | 136.1 | 142.9b 139.9c 139.7d 139.8e | |
| F1–F2 distance | 221.7 | 223.0 | 222.3 | 221.8b 220.8c 220.7d 220.9e | |
| [Cl4]+ | |||||
| Cl1–Cl1 distance | 200.8 | 196.7 | 195.2 | 194.11 | 194.0f 195.6g |
| Cl1–Cl2 distance | 301.9 | 298.1 | 296.1 | 293.71 | 297.5f 334.3g,f |
| [F2O2]+ | |||||
| F–F distance | 145.5 | 142.2 | 141.7 | ||
| O–O distance | 113.0 | 112.2 | 111.8 | ||
| F–O distance | 275.2 | 251.9 | 251.7 | ||
| O–O–F angle | 93.4 | 93.4 | 93.4 | ||
| [Cl2O2]+ | |||||
| Cl–Cl distance | 200.0 | 195.6 | 194.7 | 188.8h 191.6i 190.9j | 196k 194.5l 190.6m |
| O–O distance | 118.0 | 117.3 | 116.6 | 116h 118.5i 120.7j | 116.2k 116.1l 122m |
| Cl–O distance | 246.6 | 244.2 | 242.9 | 244h 242.5i 241.4j | 267k 264.7l 242m |
| O–O–Cl angle | 99.6 | 99.2 | 99.3 | ||
| [F2Cl2]+ | |||||
| F–F distance | 145.5 | 142.2 | 141.7 | ||
| Cl–Cl distance | 195.8 | 191.9 | 190.4 | ||
| F–Cl distance | 323.1 | 311.8 | 310.8 | ||
| F–F–Cl angle | 74.7 | 89.7 | 90.4 | ||
| F2–Cl2 tor. angle | 80.2 | 43.0 | 40.0 | ||
| X-X distance | |||||
| F2 | 145.0 | 141.8 | 141.3 | 141.213 | |
| [F2]+ | 133.6 | 131.3 | 130.7 | 130.517 | |
| Cl2 | 206.2 | 201.9 | 200.3 | 198.713 | |
| [Cl2]+ | 196.0 | 192.1 | 190.5 | 189.113 | |
| O2 | 122.0 | 121.3 | 120.7 | 120.8 13 | |
| [O2]+ | 113.1 | 112.1 | 111.7 | 111.6 13 |
The two oxygen-containing heterodimeric cations [F2O2]+ and [Cl2O2]+ both showed similar C2v-symmetrical trapezoidal structures (see Fig. 1).
For [F2O2]+ the calculations at CCSD(T)/aVQZ level yielded a fluorine–fluorine distance of dF–F = 141.7 pm which is almost identical to the value for free F2 and an oxygen–oxygen bond length that corresponds to the one found for the dioxygenyl cation ([F2O2]+: dO–O = 111.8 pm, [O2]+: 111.7 pm [calc.] and 111.6 pm [exp.13]); thus the molecule can be regarded as a complex between [O2]+ and F2, the interfragmental bond length (dF–O = 251.7 pm) being shorter than the sum of the van der Waals radii (rF + rO = 280 pm)12 but much longer than the analogous bond length in [F4]+, where the positive charge is delocalized over the whole molecule.
The structural parameters of [Cl2O2]+ computed at CCSD(T)/aVQZ level hint a delocalization of the positive charge which can be understood by the similar electronegativities of chlorine and oxygen: both intrafragmental bond lengths lie between the bond lengths found for the neutral and monocationic monomers, respectively (dCl–Cl = 200.3, 194.7 and 190.5 pm; dO–O = 120.7, 116.6 and 111.7 pm in Cl2, [Cl2O2]+ and [Cl2]+). The interfragmental bond length of dO–Cl = 242.9 is significantly shorter than the sum of the van der Waals radii (rO + rCl = 320 pm12). Again, the calculated values are in very good agreement with the ones found experimentally.2 Note, that the weakly bound neutral Cl2O2 species was predicted to show a “hockey stick” structure like for the recently predicted [F5]−14 and no trapezoidal one like for the cation.15,16
Contrary to the structures of the [X4]+ and [X2O2]+ (X = F, Cl) cations, the homodimeric [F2Cl2]+ cation is not planar. Instead it has a twisted structure of only C2 symmetry where the F2 and Cl2 units are skewly arranged; the C2v-symmetrical trapezoidal structure analogous to the [X2O2]+ cases showed an imaginary frequency of 58.5 cm−1 at CCSD(T)/aVTZ level.
As for [F2O2]+ the calculated bond lengths (CCSD(T)/aVQZ level) in both fragments (dF–F = 141.7 pm and dCl–Cl = 190.4 pm) hint at a positive charge localized at the Cl2–fragment; the long distance between the two fragment (dF–Cl = 310.8 pm) which is only slightly smaller than the sum of the van der Waals radii (rF + rCl = 320 pm)12 also points at an only weak interaction. Note however, that a structure optimization on DFT level using either the B3LYP or BP86 functional could not reproduce the distortion obtained at coupled-cluster-level. A scan of the potential energy surface was performed at CCSD(T)/aVQZ level where the F2–Cl2–torsional angle was varied at the otherwise unchanged optimized structure. The result (see Fig. 2) shows a very flat potential with a maximum at an angle of 90° and only about 0.2 kJ mol−1 higher in energy. A possible explanation for the deviation from planarity might be a quadrupole–quadrupole interaction between the two fragments.
![Potential energy scan for [F2Cl2]+ calculated at CCSD(T)/aVQZ level; the energy is plotted as function of the F2–Cl2 torsional angle.](/image/article/2012/RA/c1ra00804h/c1ra00804h-f2.gif) | ||
| Fig. 2 Potential energy scan for [F2Cl2]+ calculated at CCSD(T)/aVQZ level; the energy is plotted as function of the F2–Cl2 torsional angle. | ||
Electronic structures
In a molecular orbital picture the bonding situation in [F4]+, [Cl4]+ and [Cl2O2]+ can be described as π*–π* interaction leading to a delocalization of the positive charge. The π* orbitals of both constituent fragments in the plane of the dimer interact to give a bonding and an anti-bonding combination. In the case of these two homodimeric cations the two orbitals contain three electrons resulting in a filled bonding orbital and a half-filled anti-bonding orbital. For [Cl2O2]+ only two electrons occupy the bonding π*–π* orbital and the unpaired electron occupies the π* (O2) orbital perpendicular to the molecular plane. Fig. 3 shows the singly occupied molecular orbital (SOMO) and the bonding π*–π* orbital of [F4]+ and [Cl2O2]+ calculated at CCSD(T)/aVTZ level; the corresponding orbitals for [Cl4]+ are analogous to those of [F4]+. In the two [F2X2]+ (X = Cl, O) cations the charge remains localized on the [X2]+ fragment and consequently no significant overlap between the molecular orbitals of each fragment can be observed.![Selected molecular orbitals for [F4]+ and [Cl2O2]+ calculated at CCSD(T)/aVTZ level.](/image/article/2012/RA/c1ra00804h/c1ra00804h-f3.gif) | ||
| Fig. 3 Selected molecular orbitals for [F4]+ and [Cl2O2]+ calculated at CCSD(T)/aVTZ level. | ||
Gas phase thermochemistry
In order to evaluate the stability of the investigated molecules different decomposition reactions were considered for each. All given reaction energies have been calculated on CCSD(T)/aVXZ (X = D, T, Q) level and were extrapolated to the CBS limit.As reference system for reliable decomposition energies we have used the [Cl2O2]+ molecule due experimental determined values for the reaction to [Cl2]+ and O2. This is the only observed decomposition channel in an energy resolved collision-induced dissociation experiment in a flowing afterglow-tandem mass spectrometer at 0 K,5 leading to a dissociation energy of 57 ± 7 kJ mol−1.
This value agrees very well with our computed dissociation energy of 69.6 kJ mol−1(see Table 2). The second possible dissociation of [Cl2O2]+ into [Cl2] and [O2]+ is calculated to be much more endothermic (121.4 kJ mol−1) and thus not observed in the experiment. This shows that the chosen protocol of CCSD(T)/aVXZ (X = D, T, Q) calculations is working fine and should be reliable for the stability prediction of the other species in question.
| Species | aVDZ | aVTZ | aVQZ | CBSb | QC-Ref. |
|---|---|---|---|---|---|
| a Values in parentheses are ZPE contributions. b Estimation of the complete-basis set limit (CBS) by using equation E(lmax) = ECBS + B/lmax3 (B are values of the three highest lmax).7 c Values computer at UQCISD(T), UCCSD, UCCSD(T), RCCSD, RCCSD(T) level using the 6-31G(d) basis set, respectively.3 d Experimental dissociation energies using: neon 58 ± 6 kJ mol−1, and argon 55 ± 7 kJ mol−1 as collision gas and average value 57 ± 7 kJ mol−1. e CASPT2 value with ZPE correction from B3LYP/aVTZ frequencies.2 f Values at B3LYP/6311++G(3df,3pd).2 g Values at B3LYP/aVTZ.5 h Computed value at CASPT2 level.2 | |||||
| [F4]+ → F2 + [F2]+ | 58.1 (−6.4) | 55.7 (−6.5) | 54.3 (−6.8) | 54.1 | 67.4, 54.4, 67.4, 53.6, 67.4c |
| [F4]+ → F + [F3]+ | 124.8 (−7.3) | 136.2 (−7.2) | 137.8 (−7.5) | 140.2 | |
| [Cl4]+ → Cl2 + [Cl2]+ | 75.2 (−5.8) | 76.2 (−4.6) | 78.2 (−4.1) | 77.9 | |
| [Cl4]+ → Cl + [Cl3]+ | 127.1 (−5.8) | 132.6 (−4.5) | 137.5 (−4.0) | 137.5 | |
| [F2O2]+ → F2 + [O2]+ | 21.7 (−0.9) | 20.3 (−0.8) | 19.1 (−0.8) | 19.1 | |
| [F2O2]+ → O2 + [F2]+ | 380.4 (−1.7) | 371.0 (−1.6) | 373.4 (−1.6) | 370.4 | |
| [Cl2O2]+ → Cl2 + [O2]+ | 118.6 (−10.2) | 121.7 (−6.2) | 120.2 (−5.7) | 121.4 | |
| [Cl2O2]+ → O2 + [Cl2]+d | 68.5 (−11.8) | 67.4 (−7.7) | 70.8 (−7.2) | 69.6 | 49.1e, 77.4f, 69.9g, 53.9h |
| [F2Cl2]+ → F2 + [Cl2]+ | 11.7 (0.0) | 11.2 (0.0) | 10.8 | 10.8 | |
| [F2Cl2]+ → Cl2 + [F2]+ | 420.7 (0.7) | 416.3 (0.8) | 414.5 | 414.0 |
For the lighter homologue [F2O2]+ our calculation show that the preferred decomposition channel, the dissociation into F2 and [O2]+, is still endothermic by 19.1 kJ mol−1; as expected the other fragmentation yielding [F2]+ and O2 is computed to be even less favorable (see Table 2). Also, an elimination reaction of [F2O2]+ → OF2 + O+ type is very unlikely to proceed due to an enormous endothermic energy of 770.6 kJ mol−1. However, [F2O2]+ is about 50.1 kJ mol−1 less stable than the experimentally known [Cl2O2]+ which is mainly due to the larger difference in the electron affinities between fluorine and oxygen. Nevertheless it shows a similar stability like the known HgF4 species which was synthesized at cryogenic temperature in a matrix-isolation experiment.18,19
After the consideration of these two molecules we have also investigated the stability of the interhalogen compound [F2Cl2]+ to complete the series. Because of the similarity in ionization energies between oxygen (12.08 eV)20 and chlorine (11.48 eV)20 a thermochemical stability comparable to that of [F2O2]+ is expected.
Indeed the most probable decomposition channel is the fragmentation into F2[Cl2]+; the computed reaction energy is 10.8 kJ mol−1 indicating that this species might also be stable at low temperatures.
For the two homodimeric cations, [F4]+ and [Cl4]+, of which only the latter is experimentally known and stabilized as the hexafluoroiridate(V), two decomposition pathways, the elimination of atomic and molecular halogen, have been investigated. For [Cl4]+ the decomposition channel leading to Cl2 and [Cl2]+ is calculated to be endothermic by 77.9 kJ mol−1 (Table 2). The second possible decomposition would lead to a [Cl3]+ cation and a chlorine atom and is computed to be endothermic by 137.5 kJ mol−1. Note, that the [Cl4]+ cation is gaining stability as ion-pair complex by considering lattice energies, see below.
The radical cation [F4]+ is calculated to show a slightly less endothermic elimination reaction [F4]+ → [F2]+ + F2 (54.1 kJ mol−1) than the corresponding fragmentation of [Cl4]+. The elimination of a fluorine atom is similarly endothermic (140.2 kJ mol−1) as the analogous decomposition of the heavier homologue. This outcome is in agreement with a previous investigation of the [F4]+ cation where the concerted F2 elimination was computed to be 67.4 kJ mol−1 at CCSD(T)/6-31G(d) level.3 The computed difference of 13.0 kJ mol−1 between our CBS extrapolation and the previous value is due to basis-set deficiencies in the used 6-31G(d) basis set. Nevertheless, both results indicate that [F4]+ is predicted to be surprisingly stable against decomposition in either [F2]+ or [F3]+.
The different stabilities of the [X4]+ and [X2Cl2]+ cations can be explained by valence bond theory where the 3 electron bond is described in terms of valence bond structures which lead to stability by resonance energy.3 The largest gain of resonance energy is achieved if the valence bond structures show equal (or similar) weights (corresponding to a charge delocalization), which is the case for the homodimeric radical cations and for [Cl2O2]+, but not for the heterodimeric radical cations [F2X2]+ (X = Cl, O) where the charge is localized on the X2 side.
Born-Fajans-Haber cycles
In this section the possibility of stabilizing the homodimeric [X4]+ or heterodimeric [X2Y2]+ radical cation salts (compared to the corresponding [X2]+ salts and gaseous X2 or Y2) in the solid state is investigated. We constructed Born–Fajans–Haber cycles with [SbF6]− as the counter ion which was successfully used to stabilize [Cl2O2]+ (see Scheme 1).![Representative thermochemical (Born-Fajans-Haber) cycles for the [X2O2]+ [SbF6]− complexes (X = F, Cl) at 298.15 K; values in kJ mol−1.](/image/article/2012/RA/c1ra00804h/c1ra00804h-s1.gif) | ||
| Scheme 1 Representative thermochemical (Born-Fajans-Haber) cycles for the [X2O2]+ [SbF6]− complexes (X = F, Cl) at 298.15 K; values in kJ mol−1. | ||
Thermal corrections to energies as well as entropy values have been calculated using the statistical thermodynamics tool implemented in the Gaussian09 package21 based on optimized structures and harmonic frequencies at B3LYP/aug-cc-pVTZ level. The results (Table S1†) show that the two decompositions of the fluorine-containing heterodimeric cations [F2Y2]+ (Y = Cl, O) yielding elemental fluorine and the corresponding monomeric cation become exothermic when subjected to standard conditions whereas they are endothermic at 0 K.
Lattice enthalpies have been estimated using the generalized Kapustinskii equation and lattice entropies by an empirical correlation, where both formulas have been derived by Jenkins et al.22,23. The ionic volumes have been either taken from literature1,2,22 or estimated on the base of cations judged to have a sufficiently similar volume (Table S2†).
As expected, the constructed Born–Haber cycles (Table 3) show that the addition of neutral Y2 to [X2]+[SbF6]− is less favourable than the corresponding gas phase reaction. This is due to the decrease of lattice enthalpy caused by the increasing cationic volumes. Thus, the two known cations [Cl2O2]+ and [Cl4]+ become unstable with respect to elimination of O2 and Cl2, respectively. Although we estimate the error of this kind of lattice enthalpy as about 30 kJ mol−1, these results are in good agreement with the reported thermal instability of the two salts which decompose at temperatures of above −78 °C ([Cl4]+[IrF6]− (the ionic volume of the two anions are comparable)1 and well below 0 °C ([Cl2O2]+[SbF6]−)2. By contrast, the [F4]+ salt is computed to be stable at room temperature, but still, its synthesis will probably not be possible for the want of [F2]+-donating reactants.
| [X2]+/Y2 | ΔrG° ([X2]+(g) +Y2(g) → [X2Y2]+(g)) | − ΔlatG ([X2]+[SbF6]−) | ΔlatG ([X2Y2]+[SbF6]−) | ΔrG° ([X2]+[SbF6]−(s) + Y2(g) → [X2Y2]+[SbF6]−(s)) |
|---|---|---|---|---|
| [F2]+/F2 | −24.8 | 461.2 | −424.0 | 12.4 |
| [F2]+/O2 | −335.1 | 461.2 | −426.8 | −300.6 |
| [F2]+/Cl2 | −392.0 | 461.2 | −388.4 | −319.2 |
| [Cl2]+/F2 | +13.6 | 435.9 | −388.4 | 61.1 |
| [Cl2]+/O2 | −30.6 | 435.9 | −398.6 | 6.7 |
| [Cl2]+/Cl2 | −44.6 | 435.9 | −385.2 | 6.1 |
| [O2]+/F2 | + 11.5 | 463.1 | −426.8 | 47.8 |
| [O2]+/Cl2 | − 89.7 | 463.1 | −398.6 | −25.2 |
Vibrational frequencies
Time-of-flight mass spectrometry, IR photodissociation spectroscopy and matrix-isolation spectroscopy are ideal experimental techniques for the characterization of the species in question. These techniques were successfully used for the experimental verification e.g. of the [O4]+ radical cation and its larger homologues.24–29 To allow an adequate identification for future experimental attempts, we report state-of-the-art vibrational frequencies up to CCSD(T) level summarized in Table S3.†Unfortunately, only experimental Raman measurements of the reference systems [Cl4]+ and [Cl2O2]+ are available for comparison. For [Cl4]+ the calculated wavenumber for symmetrical chlorine–chlorine stretch mode (Ag) at B3LYP/aVTZ level is in surprisingly good agreement with the experimentally determined one [Δνs(Cl–Cl) = 6 cm−1]. The agreement can be increased to Δνs(Cl–Cl) = 2 cm−1 if one considers anharmonicity effects. (Table S3†)
Coupled-cluster calculations in the CCSD(T)/aVXZ (X = D,T,Q) series show a decline in agreement with increasing basis set size, leading to Δνs(Cl–Cl) = 24 cm−1 for the aVQZ basis set. The good performance of the density functional theory is probably due to error compensation. An adequate treatment of anharmonic corrections at coupled-cluster level should compensate the overshooting of the harmonic frequencies. A comparison of the experimental and calculated wavenumbers for the B3g mode (δs (Cl2⋯Cl2)) shows that all used computational levels fail by more than 96 cm−1. The agreement of the CCSD(T)/aug-cc-pVQZ calculation with the experimental band for the symmetric Cl2⋯Cl2 stretch (Ag) at 175 cm−1 is instead good.
Our computed O–O stretching mode in [Cl2O2]+ at CCSD(T)/aVQZ level (1618 cm−1) shows an hypsochromic shift of 84 cm−1 when compared with the experimental band at 1534 cm−1; the agreement should be increased by 20–30 cm−1 when taking anharmonic corrections into account. At DFT level (B3LYP) the calculated wavenumber is by 148 cm−1 to high and the anharmonic correction amounts to 25 cm−1. However, for this band, the 16O/18O is experimentally known (86 cm−1) and be compared to our calculations: at B3LYP/aVTZ the computed shift value of 96 cm−1 agrees well with the experiment. For the Cl–Cl stretching mode the difference between the experimental and calculated wavenumber (CCSD(T)/aVQZ level) is only Δν(Cl–Cl) = 14 cm−1 (see Table S3†).
Conclusions
Our present state-of-the-art quantum chemical calculations suggest that the homodimeric and heterodimeric radical cations [F4]+ and [F2O2]+ are thermochemically stable molecules and thus these systems have a realistic chance of experimental observation in matrix-isolation spectroscopy or in gas-phase experiments. The good agreement between experiment and theoretical prediction is demonstrated for the case of [Cl2O2]+ where experimental reported value of 57 ± 7 kJ mol−1 is nicely reproduced in silico (69.2 kJ mol−1). Nevertheless this outcome only considers gas-phase chemistry and not stabilization in bulk compounds. Calculations by Born-Haber cycles confirm the stability of the already known compounds [Cl4]+[IrF6]− and [Cl2O2]+[SbF6]−. They also show that the predicted [F4]+ cation could be stabilized as ion-pair complex; nevertheless, this stability is likely to be hypothetical due to the lack of experimental [F2]+[A]− reactants.In conclusion, our calculations predict the [F4]+, [F2Cl2]+ and [F2O2]+ as possible target species in gas-phase experiments. We hope that the computed harmonic and anharmonic frequencies together with predicted isotopic shifts allow an adequate characterization of these possible compounds.
Acknowledgements
The authors are grateful to M. Kaupp for kindly providing computational resources and to Carsten Knapp for stimulating discussions. We thank the Fonds der Chemischen Industrie, the DFG, the Alexander von Humboldt Foundation and the Institut für Anorganische und Analytische Chemie of the Universität Freiburg for financial support.References
- S. Seidel and K. Seppelt, Angew. Chem., Int. Ed., 2000, 39, 3923–3925 CrossRef CAS.
- T. Drews, W. Koch and K. Seppelt, J. Am. Chem. Soc., 1999, 121, 4379–4384 CrossRef CAS.
- P. C. Hiberty and N. Berthe-Gaujac, J. Phys. Chem. A, 1998, 102, 3169–3174 Search PubMed.
- R. D. Cohen and C. D. Sherrill, J. Chem. Phys., 2001, 114, 8257–8269 CrossRef CAS.
- J. M. Bailey, C. Hao, B. J. Johnson and L. S. Sunderlin, Int. J. Mass Spectrom., 2005, 241, 133–138 Search PubMed.
- H.-J. Werner, P. J. Knowles, R. Lindh, F. R. Manby, M. Schütz, P. Celani, T. Korona, G. Rauhut, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, C. Hampel, G. Hetzer, A. W. Lloyd, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklaß, P. Palmieri, R. Pitzer, U. Schumann, H. Stoll, A. J. Stone, R. Tarroni and T. Thorsteinsson, MOLPRO 2006.1 a package of ab initio programs, Birmingham, UK, MOLPRO 2006.1 edn, 2006 Search PubMed.
- A. Halkier, T. Helgaker, P. Jorgensen, W. Klopper, H. Koch, J. Olsen and A. K. Wilson, Chem. Phys. Lett., 1998, 286, 243–252 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross. V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, Gaussian09, Revision A.1 edn , 2009 Search PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- E. Fluck and K. G. Heumann, Periodensystem der Elemente, Periodensystem der Elemente, Wiley-VCH, Weinheim 2002 Search PubMed.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure, 4: Constants of Diatomic Molecules, Van Nostrand, New York, 1979 Search PubMed.
- S. Riedel, T. Köchner, X. Wang and L. Andrews, Inorg. Chem., 2010, 49, 7156–7164 CrossRef CAS.
- I. V. Nikitin, Russ. Chem. Rev., 2002, 71, 85–97 RSC.
- R. S. Zhu and M. C. Lin, J. Chem. Phys., 2003, 118, 4094–4106 CrossRef CAS.
- R. P. Tuckett, A. R. Dale, D. M. Jaffey, P. S. Jarrett and T. Kelly, Mol. Phys., 1983, 49, 475–486 CAS.
- X. Wang, L. Andrews, S. Riedel and M. Kaupp, Angew. Chem., Int. Ed., 2007, 46, 8371–8375 CrossRef CAS.
- S. Riedel and M. Kaupp, Coord. Chem. Rev., 2009, 253, 606–624 CrossRef CAS.
- K. Watanabe, J. Chem. Phys., 1957, 26, 542–547 CrossRef CAS.
- M. J. T. Frisch, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Montgomery, Jr., J. A. Vreven, T. Kudin, K. N. Burant, J. C. Millam, J. M. Iyengar, S. S. Tomasi, J. Barone, V. Mennucci, B. Cossi, M. Scalmani, G. Rega, N. Petersson, G. A. Nakatsuji, H. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Klene, M. Li, X. Knox, J. E. Hratchian, H. P. Cross, J. B. Bakken, V. Adamo, C. Jaramillo, J. Gomperts, R. Stratmann, R. E. Yazyev, O. Austin, A. J. Cammi, R. Pomelli, C. Ochterski, J. W. Ayala, P. Y. Morokuma, K. Voth, G. A. Salvador, P. Dannenberg, J. J. Zakrzewski, V. G. Dapprich, S. Daniels, A. D. Strain, M. C. Farkas, O. Malick, D. K. Rabuck, A. D. Raghavachari, K. Foresman, J. B. Ortiz, J. V. Cui, Q. Baboul, A. G. Clifford, S. Cioslowski, J. Stefanov, B. B. Liu, G. Liashenko, A. Piskorz, P. Komaromi, I. Martin, R. L. Fox, D. J. Keith, T. Al-Laham, M. A. Peng, C. Y. Nanayakkara, A. Challacombe, M. Gill, P. M. W. Johnson, B. Chen, W. Wong, M. W. Gonzalez, C. Pople, J. A., Gaussian03 Revision C.02, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- H. D. B. Jenkins, H. K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef.
- H. D. B. Jenkins and L. Glasser, Inorg. Chem., 2003, 42, 8702–8708 CrossRef CAS.
- A. M. Ricks, G. E. Douberly and M. A. Duncan, Int. J. Mass Spectrom., 2009, 283, 69–76 Search PubMed.
- L. B. Knight, Jr., S. T. Cobranchi and J. Petty, J. Chem. Phys., 1989, 91, 4423–4424 CrossRef.
- W. E. Thompson and M. E. Jacox, J. Chem. Phys., 1989, 91, 3826–3837 CrossRef CAS.
- M. E. Jacox and W. E. Thompson, J. Chem. Phys., 1994, 100, 750–751 Search PubMed.
- C. L. Lugez, W. E. Thompson and M. E. Jacox, J. Chem. Phys., 1996, 105, 2153–2160 CrossRef CAS.
- M. Zhou, J. Hacaloglu and L. Andrews, J. Chem. Phys., 1999, 110, 9450–9456 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NPA charges, thermochemical data, vibrational data. See DOI: 10.1039/c1ra00804h |
| This journal is © The Royal Society of Chemistry 2012 |