Hydroamination of 1,1-dimethylallene with primary aryl amines under mild conditions: An atom-economical route to N-(1,1-dimethyl-2-propenyl)-anilines†
John F.
Beck
and
Joseph A.R.
Schmidt
*
Department of Chemistry, School for Green Chemistry and Engineering, College of Natural Sciences & Mathematics, The University of Toledo, 2801, W. Bancroft St. MS 602, Toledo, OH 43606-3390, USA. E-mail: Joseph.Schmidt@utoledo.edu
First published on 4th November 2011
Abstract
The synthesis of alicyclic 3-iminophosphine ligands was extended to include a new framework incorporating a cyclopentenyl backbone with a di-tert-butyl phosphine functionality (3IPtBu). The palladium complex [(3IPtBu)Pd(allyl)]OTf displayed excellent catalytic activity in the 100% atom-efficient hydroamination of 3-methyl-1,2-butadiene (1,1-dimethylallene) with primary aryl amines (anilines), selectively producing the branched allylic amine products (kinetic products) in high conversion at ambient temperature for non-halogenated substrates. Hydroamination using halogenated anilines was successful at 70 °C, providing moderate yields, with the formation of little or no linear product (thermodynamic product). Additionally, a subsequent aromatic amino Claisen rearrangement of selected allylic amine products, employing catalytic triflic acid, proved to be an effective atom-economical method for the production of ortho-allylic anilines in a high yielding two-step, one-pot synthesis.
We have previously demonstrated that alicyclic 3-iminophosphines are effective ligands for use in palladium-catalyzed hydroamination of allenes,1 1,3-dienes1,2 and alkynes.2 Our initial work detailed the hydroamination of 1,3-cyclohexadiene and phenyl acetylene in moderate yields using a diphenylphosphine-derived 3-iminophosphine precatalyst: [(3IP)Pd(allyl)]OTf.2 Recently, we reported the hydroamination of 3-methyl-1,2-butadiene (1,1-dimethylallene) and 2,3-dimethyl-1,3-butadiene with secondary amines using a related catalyst: [(3IPAr)Pd(allyl)]OTf.1 Inclusion of a six-membered alicyclic backbone and an aromatic imine substituent in this second-generation catalyst significantly improved catalysis. For example, the hydroamination of 3-methyl-1,2-butadiene with morpholine yielded the linear allylic amine in nearly quantitative yield at room temperature in only 4 h using [(3IPAr)Pd(allyl)]OTf, while the original catalyst, [(3IP)Pd(allyl)]OTf, yielded a mixture of the linear and branched products in a combined yield of only 89% under the same conditions.
In general, the synthesis of allylic aminesvia transition metal catalysis has long been of interest, with many allylic amination catalysts developed utilizing various metals, including iridium,3rhodium,4,5palladium6 and iron.7 The iridium, rhodium and iron catalysts often display a high degree of selectivity for the branched (kinetic) product, whereas most palladium catalysts favor the linear (thermodynamic) product.7–11 A more sustainable method to generate allylic amines involves the hydroamination of allenes or 1,3-dienes, which has the advantage of a higher degree of atom economy compared to allylic amination.12 There are several examples of gold-catalyzed intermolecular hydroamination of allenes yielding allylic amines, including those of Bertrand,13–16 Widenhoefer17,18 and Yamamoto,19–21 although most of these systems lack selectivity for the more desirable branched product. However, Widenhoefer22 has observed the opposite regioselectivity to that reported by Yamamoto21 and Bertrand,15 providing for isolation of the highly desirable branched allylic amine. This is likely due to the room temperature conditions that Widenhoefer utilized, compared to the higher temperatures required for the Bertrand and Yamamoto catalysts. In general, allylic amines are desirable because they can be further functionalized using transformations such as asymmetric hydroboration,23hydroformylation,24amino Claisen rearrangement,25 and alkene metathesis26 as part of the synthesis of value added molecules, such as natural products or pharmaceuticals.27
Aryl amino Claisen rearrangement is a useful reaction for the synthesis of ortho-allylic anilines, which have further synthetic utility in the construction of heterocycles.28–33 Despite the apparent utility of the aryl amino Claisen rearrangement, there are relatively few reports utilizing this reaction. This can be attributed to the slow reaction rates, low yields and high reaction temperatures generally required.34–36 Branched N-allylic anilines have proven cumbersome to synthesize via traditional organic techniques. In 2001, Ward and coworkers outlined a 4-step synthesis, using stoichiometric or greater amounts of copper reagents and nBuLi,25 analogous to that of Jolidon and Hansen from several decades before,37 to generate branched allylic amines (Scheme 1). Modern advances in catalysis now allow easier access to these compounds via allylic amination6 or the hydroamination of dienes18,38,39 in one step. Both hydroamination and allylic amination are more atom economical and reduce the number of steps required for the synthesis of branched allylic amines, as well as the amount of solvent used and energy required. Hydroamination is the most desirable of these reactions due to the 100% atom economy of the reaction (Scheme 1). To our knowledge, there has been no methodological report of the coupling of allylic amination or hydroamination of allenes with aryl amino Claisen rearrangement in one pot to generate ortho-allylic anilines. Kheinman and coworkers do postulate an aryl amino Claisen rearrangement as part of a thermal synthesis of 2-ethyl-2-methyl-2,3-dihydro-1H-indole, an insecticide, where their allylic amine starting material is synthesized via the hydroamination of isoprene with aniline.40
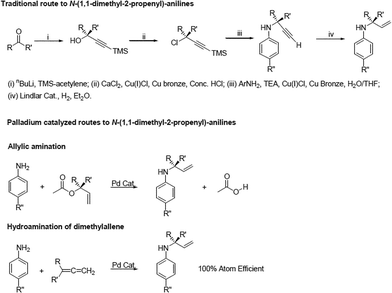 | ||
| Scheme 1 N-allylic anilines can be synthesized using a number of methods, including traditional stepwise synthesis and catalytic methods such as allylic amination and hydroamination of allenes. The hydroamination of allenes is the only method with 100% atom economy. | ||
Herein, we report the use of a new 3-iminophosphine catalyst, [(3IPtBu)Pd(allyl)]OTf, for the hydroamination of 3-methyl-1,2-butadiene at room temperature, generating the branched allylic amine product exclusively. Hydroamination of 3-methyl-1,2-butadiene was also coupled to an aryl amino Claisen rearrangement in a one-pot, two-step synthesis of 2-(3-methyl-2-butenyl)-anilines. Using this methodology, many new N-allylic and ortho-allylic anilines were produced and fully characterized for the first time, demonstrating the excellent utility of these new palladium catalysts.
The 3-iminophosphine (3IPtBu; 3) was synthesized in an analogous fashion to our previous ligands (Scheme 2)1,2 and isolated in moderate yield as a dark red oil. Its imine proton resonance appeared at 8.88 ppm, coupled to the phosphorus nucleus (4JPH = 6.0 Hz), and its 31P{1H} NMR spectrum showed a single resonance at 13.2 ppm. Reaction of 3 with [Pd(allyl)Cl]2 (0.5 equiv.) yielded (3IPtBu)Pd(allyl)Cl (4) in moderate yield (62%; Scheme 3). A single resonance at 42.7 ppm was observed in the 31P NMR spectrum of 4, indicating coordination of phosphorus to the palladium center while only broad resonances corresponding to the allylic protons were noted in its 1H NMR spectrum. Many of the resonances in the 1H, 13C{1H} and 31P{1H} NMR spectra of 4 were broad due to its fluxionality and cooling to –60 °C did not eliminate its fluxional behavior. Reaction of 4 with AgOTf afforded [(3IPtBu)Pd(allyl)]OTf (5) in good yield as a brown powder after recrystallization. A 31P resonance at 61.8 ppm and an upfield shift of 1.3 ppm for the imine proton indicated chelation of the ligand to the metal center in 5.
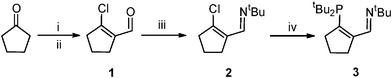 | ||
| Scheme 2 Synthesis of alicyclic 3IPtBu (3). Legend: (i) 2 eq. DMF, 1.6 eq. POCl3, 0 °C, 12 h; (ii) ice, NaHCO3; (iii) 1.5 eq. tert-butylamine, Et2O, molecular sieves, 0 °C, 12 h; (iv) 1.0 eq. LiPtBu2, Et2O, 0 °C, 2 h. | ||
![Metallation of 3 yielding the palladium complexes 4 and 5. Legend: (i) 0.5 eq. [Pd(allyl)Cl]2, CH2Cl2, 25 °C, 1 h; (ii) 1.0 eq. AgOTf, CH2Cl2/toluene (3 : 1), 25 °C, 4 h.](/image/article/2012/RA/c1ra00795e/c1ra00795e-s3.gif) | ||
Scheme 3
Metallation of 3 yielding the palladium complexes 4 and 5. Legend: (i) 0.5 eq. [Pd(allyl)Cl]2, CH2Cl2, 25 °C, 1 h; (ii) 1.0 eq. AgOTf, CH2Cl2/toluene (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 25 °C, 4 h. :1), 25 °C, 4 h. | ||
X-ray quality crystals of 5 were successfully grown, and its crystal structure (Fig. 1) revealed a palladium center supported by a chelating neutral 3-iminophosphine ligand with a bite angle of 92.52(6) degrees, an η3-allyl ligand and an outer sphere triflate anion. The ligand is puckered, rising above the coordination plane of the pseudo-square planar palladium center, resulting in a boat like conformation of this six-membered ring.
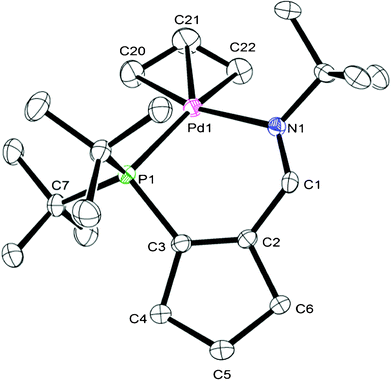 | ||
| Fig. 1 ORTEP diagram (50% thermal ellipsoids) of 5. Hydrogen atoms and triflate anion removed for clarity. Bond lengths (in Å): Pd1–N1 = 2.134(2), N1–C1 = 1.273(3), C1–C2 = 1.467(4), C2–C3 = 1.345(4), P1–C3 = 1.842(3), Pd1–P1 = 2.3356(8); angles (in °): N1–Pd1–P1 92.52(6), C1–N1–Pd1 = 123.3(2), C3–P1–Pd1 = 101.23(9). | ||
The hydroamination of 1,1-dimethylallene with anilines proceeded readily at room temperature in benzene, using catalytic [(3IPtBu)Pd(allyl)]OTf (5). Benzene was chosen as the solvent for initial catalytic screening on the NMR scale because other common NMR solvents, such as acetonitrile and THF, coordinate to the metal center and cause significant reductions in the activity of the catalyst. Production of greater than 98% conversion of the branched allylic amine was achieved in as little as 12-20 h for several substrates at ambient temperature (A; Table 1). We attribute this selectivity for the kinetic product to the low reaction temperatures available to catalyst 5, compared to the higher temperatures necessary for utilization of our previously reported hydroamination catalysts.1 Mild heating of the reaction (70 °C) increased the rate of catalysis and was necessary for halogenated anilines, which were found to be unreactive at room temperature. Heating at 70 °C for long periods of time (several days) resulted in formation of a palladium mirror, while heating to 115 °C resulted in rapid conversion to the linear (thermodynamic) product (B; Table 1) as part of a complex reaction mixture with conversions much lower than those observed via lower temperature methods. Moderate (alkyl) to strong (methoxy, dimethylamino) electron-donating groups enhanced catalysis significantly compared to unsubstituted aniline. In contrast, halogenated substrates (F, Cl, Br) showed lower conversion than aniline and required heating to 70 °C for productive catalysis to take place. Additionally, small amounts of the linear products were observed in the NMR spectra of the chlorinated anilines. 4-(Trifluoromethoxy)aniline proved to be completely unreactive, while 3-nitroaniline and 4-aminopyridine were not screened due to a lack of amine solubility. Steric effects also played an important role in this catalysis. Specifically, substituents at the ortho positions were found to virtually halt catalysis, even when the reaction was heated. The only exception to this trend was 2-methoxyaniline, which was observed to undergo hydroamination by 1H NMR spectroscopy, although conversion of the allylic amine was extremely low (10%) even after heating for 20 h at 70 °C.
|
|
|||
|---|---|---|---|
| Entry | Amine | Temp. | Conversion (A:B)a |
| a Catalytic procedure: amine (0.5 mmol) was added to a mixture of [(3IPtBu)Pd(allyl)]OTf (5 mol%) and benzene-d6 (0.8 ml). Subsequently, allene (1.0 mmol) was added and the reaction was sealed. Conversion and product ratios were determined viaNMR spectroscopy after 20 h. b Data reported at 12 h. | |||
| 1 | aniline | 25 °C | 60 (100:0) |
| 2 | 3-methylaniline | 25 °C | >98 (100:0) |
| 3 | 3-ethylaniline | 25 °C | 73 (100:0) |
| 4 | 4-methylaniline | 25 °C | >98 (100:0) |
| 5 | 3,4-dimethylaniline | 25 °C | >98 (100:0) |
| 6 | 4-tertbutylaniline | 25 °C | 88 (100:0) |
| 7 | 3-methoxyaniline | 25 °C | 62 (100:0) |
| 8 | 4-methoxyaniline | 25 °C | >98b (100:0) |
| 9 | 4-(methylthio)aniline | 25 °C | 56 (100:0) |
| 10 | 4-dimethylaminoaniline | 25 °C | >98b (100:0) |
| 11 | 3-chloroaniline | 70 °C | 36 (72:28) |
| 12 | 4-chloroaniline | 70 °C | 57 (75:25) |
| 13 | 3-fluoroaniline | 70 °C | 50 (100:0) |
| 14 | 4-fluoroaniline | 70 °C | 65 (100:0) |
| 15 | 4-bromo-3-methylaniline | 70 °C | 17 (100:0) |
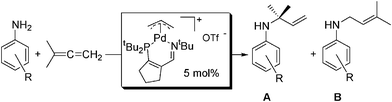
Although the catalytic hydroamination of 1,1-dimethylallene at room temperature yielded excellent results, reaction at elevated temperatures caused an oligomerization of 1,1-dimethylallene that was clearly visible in the 1H and 13C{1H} NMR spectra. Control experiments showed that 5 effectively catalyzed this oligomerization in the absence of amine, indicating that this process is independent of the hydroamination reaction. In contrast, at room temperature no oligomerization was detectible on the normal catalytic timescale. To help circumvent this side reaction, 2 equiv. of allene were used during the catalytic studies. For each catalytic hydroamination (Table 1), reaction progress was readily monitored by 1H NMR spectroscopy. As the allene and aryl amine resonances disappeared, a distinct set of product allylic amine resonances developed, consistent with known examples in literature.6,7,18
It was found that the hydroamination of 1,1-dimethylallene could be coupled to an aryl amino Claisen rearrangement reaction to produce substituted 2-allyl-anilines in one-pot, starting from 1,1-dimethylallene and an aniline (Table 2). This two-step, one-pot reaction sequence worked very well for a wide variety of the substrates originally screened for catalytic hydroamination. One-pot reactions of this type reduce the total amount of solvent needed by elimination of the intermediate purification step between the hydroamination reaction and Claisen rearrangement.
|
|
|||
|---|---|---|---|
| Entrya | Amine | t (h) | Conversionb (B:C) or (B:C:D) |
| a Entry numbers correspond to amines from Table 1. b Catalytic procedure: amine (0.5 mmol) was added to a mixture of [(3IPtBu)Pd(allyl)]OTf (5 mol%) and benzene-d6 (0.8 ml). Subsequently allene (1.0 mmol) was added and the reaction was sealed. After 20 h, triflic acid (7.5 mg, 10 mol%) was added and the reaction was heated to 70 °C for the time indicated. Conversion and product ratios were determined viaNMR spectroscopy. | |||
| 1 | (R, R' = H) | 72 | 51 (0:100) |
| 2 | (R = H; R' = Me) | 20 | >98 (0:50:50) |
| 3 | (R = H, R' = Et) | 20 | 70 (0:50:50) |
| 4 | (R = Me, R' = H) | 72 | >98 (12:88) |
| 6 | (R = tBu, R' = H) | 20 | 90 (0:100) |
| 9 | (R = SMe, R' = H) | 20 | 53 (0:100) |
| 13 | (R = H, R' = F) | 72 | 61(26:74:0) |
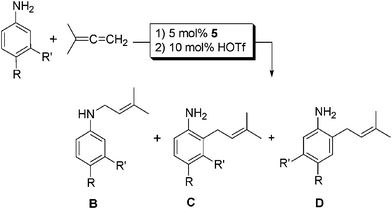
One notable characteristic of this reaction is that the amino Claisen rearrangement of N-allylic-3-fluoroaniline proceeded with regiospecificity to yield exclusively 2-allyl-3-fluoroaniline. When methyl or ethyl groups were located at the 3 position 1:1 mixtures of 2- and 6-allyl-anilines were observed (Table 2, entries 2 and 3). In order to effect these transformations, after hydroamination for 20 h with 5 (5 mol%), the reaction was uncapped and triflic acid (10 mol%) was added. The reaction was then heated to 70 °C and the conversion to the 2-(3-methyl-2-butenyl)-anilines was monitored via1H NMR spectroscopy. Products were identified in the 1H NMR spectra by characteristic triplets of multiplets at approximately 5 ppm corresponding to the allylic proton and doublets around 3 ppm for the methylene protons of the 3-methyl-2-butenyl group, an evident change in the splitting pattern compared to the intermediate N-allylic species.
Overall, these experiments demonstrate that alicyclic 3-iminophosphines are superb ligands for the palladium catalyzed, room-temperature hydroamination of 3-methyl-1,2-butadiene with anilines. This catalytic process is an effective means for the synthesis of N-(1,1-dimethyl-2-propenyl)-anilines in good to near quantitative conversion with 100% atom economy. These N-allylic anilines are the kinetic products of this hydroamination reaction and possess a terminal vinylic moiety, making them especially useful product amines. Although conversions were lower and heating of the reaction was required for the halogenated substrates, the hydroamination of fluorinated anilines proceeded in moderate conversion. To our knowledge, this is the first example of the hydroamination of 3-methyl-1,2-butadiene with fluorinated anilines. A subsequent acid-catalyzed aryl amino Claisen rearrangement of the product N-(1,1-dimethyl-2-propenyl)-anilines allowed for a one-pot synthesis of 2-(3-methyl-2-butenyl)-anilines from 1,1-dimethylallene and substituted anilines. This avoids intermediate purification steps, reducing the amount of solvent required. The use of a catalyst and the 100% atom economy of both the hydroamination reaction and the aryl amino Claisen rearrangement make this method for the synthesis of N-allylic and 2-allylic-anilines more environmentally friendly then previous methods. Both N-(1,1-dimethyl-2-propenyl)-anilines and 2-(3-methyl-2-butenyl)-anilines are desirable compounds because of their applicability in the synthesis of heterocycles that are common in many natural products and pharmaceuticals.28,40,41 Our ongoing investigations with this catalyst system are aimed at expansion of the substrate scope, with specific interest in utilization of different allenes and other unsaturated carbon frameworks. Additionally, we are pursuing modifications to the catalyst structure in an effort to make this catalysis more amenable to greener solvents, such as alcohols.
Acknowledgements
This material is based upon work supported by the National Science Foundation under CHE-0841611. We would like to acknowledge Dr Glenn Kuchenbeiser and Dr Andrew R. Shaffer for early contributions to this project related to catalytic screening and ligand synthesis, respectively. We would also like to thank Dr Allen Oliver (University of Notre Dame) for his assistance with X-ray crystallography.References
- G. Kuchenbeiser, A. R. Shaffer, N. C. Zingales, J. F. Beck and J. A. R. Schmidt, J. Organomet. Chem., 2011, 696, 179–187 CrossRef CAS.
- A. R. Shaffer and J. A. R. Schmidt, Organometallics, 2008, 27, 1259–1266 CrossRef CAS.
- Y. Yamashita, A. Gopalarathnam and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7508–7509 CrossRef CAS.
- A. Padwa, A. C. Flick, C. A. Leverett and T. Stengel, J. Org. Chem., 2004, 69, 6377–6386 CrossRef CAS.
- P. A. Evans, J. E. Robinson and K. K. Moffett, Org. Lett., 2001, 3, 3269–3271 CrossRef CAS.
- I. Dubovyk, I. D. G. Watson and A. K. Yudin, J. Am. Chem. Soc., 2007, 129, 14172–14173 CrossRef CAS.
- B. Plietker, Angew. Chem., Int. Ed., 2006, 45, 6053–6056 CrossRef CAS.
- A. M. Johns, Z. J. Liu and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7259–7261 CrossRef CAS.
- S. L. You, X. Z. Zhu, Y. M. Luo, X. L. Hou and L. X. Dai, J. Am. Chem. Soc., 2001, 123, 7471–7472 CrossRef CAS.
- J. W. Faller and J. C. Wilt, Org. Lett., 2005, 7, 633–636 CrossRef CAS.
- J. W. Faller and J. C. Wilt, Organometallics, 2005, 24, 5076–5083 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224–5228 CrossRef CAS.
- X. M. Zeng, G. D. Frey, R. Kinjo, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2009, 131, 8690–8696 CrossRef CAS.
- X. M. Zeng, M. Soleilhavoup and G. Bertrand, Org. Lett., 2009, 11, 3166–3169 CrossRef CAS.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2011, 50, 5560–5563 CrossRef CAS.
- R. A. Widenhoefer, Chem.–Eur. J., 2008, 14, 5382–5391 CrossRef CAS.
- A. N. Duncan and R. A. Widenhoefer, Synlett, 2010, 419–422 CAS.
- N. Nishina and Y. Yamamoto, Tetrahedron, 2009, 65, 1799–1808 CrossRef CAS.
- N. Nishina and Y. Yamamoto, Tetrahedron Lett., 2008, 49, 4908–4911 CrossRef CAS.
- N. Nishina and Y. Yamamoto, Synlett, 2007, 1767–1770 CAS.
- R. E. Kinder, Z. Zhang and R. A. Widenhoefer, Org. Lett., 2008, 10, 3157–3159 CrossRef CAS.
- V. Lillo, E. Fernández and A. M. Segarra, Tetrahedron: Asymmetry, 2007, 18, 911–914 CrossRef CAS.
- J. Q. Zhou and H. Alper, J. Org. Chem., 1992, 57, 3328–3331 CrossRef CAS.
- M. A. Cooper, M. A. Lucas, J. M. Taylor, A. D. Ward and N. M. Williamson, Synthesis, 2001, 621–625 CrossRef CAS.
- Q. Yang, H. Alper and W. J. Xiao, Org. Lett., 2007, 9, 769–771 CrossRef CAS.
- M. Johannsen and K. A. Jørgensen, Chem. Rev., 1998, 98, 1689–1708 CrossRef CAS.
- M. A. Cooper, C. L. Francis, J. W. Holman, B. Kasum, T. Taverner, E. R. T. Tiekink and A. D. Ward, Aust. J. Chem., 2000, 53, 123–129 CrossRef CAS.
- C. L. Francis and A. D. Ward, Aust. J. Chem., 1994, 47, 2109–2117 CrossRef CAS.
- K. D. Raner and A. D. Ward, Aust. J. Chem., 1991, 44, 1749–1760 CrossRef CAS.
- K. C. Nicolaou, A. J. Roecker, R. Hughes, R. van Summeren, J. A. Pfefferkorn and N. Winssinger, Bioorg. Med. Chem., 2003, 11, 465–476 CrossRef CAS.
- A. F. Yépez, A. Palma, E. Stashenko, A. Bahsas and J. M. Amaro-Luis, Tetrahedron Lett., 2006, 47, 5825–5828 CrossRef.
- S. L. G. Ayala, E. Stashenko, A. Palma, A. Bahsas and J. M. Amaro-Luis, Synlett, 2006, 2275–2277 Search PubMed.
- K. C. Majumdar, T. Bhattacharyya, B. Chattopadhyay and B. Sinha, Synthesis, 2009, 2117–2142 CrossRef CAS.
- S. Jain, N. Pandey and D. Kishore, Indian J. Chem., Sect B, 2007, 46, 529–531 Search PubMed.
- W. K. Anderson and G. F. Lai, Synthesis, 1995, 1287–1290 CrossRef CAS.
- S. Jolidon and H. J. Hansen, Helv. Chim. Acta, 1977, 60, 978–1032 CrossRef.
- C. S. Yi and S. Y. Yun, Org. Lett., 2005, 7, 2181–2183 CrossRef CAS.
- E. A. Petrushkina, N. E. Mysova and A. V. Orlinkov, Russ. J. Gen. Chem., 2005, 75, 910–914 CrossRef CAS.
- E. A. Petrushkina, V. N. Kalinin, G. B. Ivanova and V. A. Kheinman, Russ. J. Gen. Chem., 2006, 76, 1953–1957 CrossRef CAS.
- J. Barluenga, F. Rodríguez and F. J. Fañanás, Chem.–Asian J., 2009, 4, 1036–1048 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 823823. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00795e |
| This journal is © The Royal Society of Chemistry 2012 |