Palladium-catalyzed sequential reaction viaSonogashira coupling, isomerization, Claisen rearrangement and [4 + 2] cycloaddition sequence for the rapid synthesis of tricyclo[3.2.1.02,7]oct-3-ene derivatives†
Shugao
Zhu
a,
Luling
Wu
*a and
Xian
Huang‡
ab
aDepartment of Chemistry, Zhejiang University, Hangzhou, 310028, P. R. China
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, P. R. China. E-mail: Wululing@zju.edu.cn
First published on 3rd November 2011
Abstract
An interesting sequential reaction consisting of Sonogashira coupling, propargyl–allenyl isomerization, allenyl–benzyl Claisen rearrangement and [4 + 2] cycloaddition was developed. This reaction provides an efficient synthesis of fused tricyclo[3.2.1.02,7]oct-3-enes.
A sequential reaction, which incorporates two or more different transformations forming multiple bonds, stereocenters, and rings in a single stroke, holds great promise for the rapid buildup of molecular complexity and diversity.1 Recently much attention has been focused on the development of such reactions.2 Among them, the palladium-catalyzed sequential reaction is one of the most appealing strategies to construct complex molecular skeletons with high efficiencies in organic synthesis.
Müller et al. pioneered the Sonogashira coupling/propargyl–allenyl isomerization reactions for the synthesis of a variety of useful compounds including chalcones, pyrazolines, pyrroles, fluorescent spirocycles, and some other pharmaceutically interesting heterocycles.3 They reported a new coupling–isomerization–Claisen domino reaction starting from organic halides with an electron-withdrawing group and 1-(hetero)aryl propargyl trityl ethers dichotomizes in the concluding steps of the sequence.4 Our group previously established a series of sequential reactions wherein an allene intermediate, generated in situ, would undergo [4 + 2] cycloaddition reaction under mild conditions, providing an efficient synthesis of structurally complex polycycles with 2,3-dihydrofuran units,5 structurally diverse fused dihydroisobenzofuran derivatives,6 and polysubstituted pyridines and isoquinolines.7
As a continuing exploration, we envisioned that the coupling product A first undergoes a base-catalyzed isomerization to generate the vinylalleneB, followed by an intramolecular [3,3]-sigmatropic rearrangement (allenyl–benzyl Claisen rearrangement) to form intermediate C, which may subsequently undergo intramolecular [4 + 2] cycloaddition with several possibilities (C1–C5, Scheme 1). We anticipated that the reaction may proceed via possible transition states C1 or C2 to form compound 3a or 4a because the transition states C3–C5 may be unfavorable due to the electronic effects.8 Herein we wish to report our recent observation for such a transformation.
![Possible sequential reaction involving Pd-catalyzed Sonogashira coupling, propargyl–allenyl isomerization, allenyl–benzyl Claisen rearrangement and [4 + 2] cycloaddition with various possibilities](/image/article/2012/RA/c1ra00452b/c1ra00452b-s1.gif) | ||
| Scheme 1 Possible sequential reaction involving Pd-catalyzed Sonogashira coupling, propargyl–allenyl isomerization, allenyl–benzyl Claisen rearrangement and [4 + 2] cycloaddition with various possibilities | ||
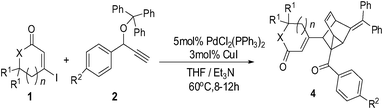
We began our investigations on this reaction using 3-iodocyclohex-2-enone 1a, triphenyl(1-phenylprop-2-ynyloxy)methane 2a in the presence of a catalytic amount of Pd(PPh3)2Cl2 and CuI in toluene and Et3N at 100 °C. The reaction successfully gave polycyclic product 4a in a 20% yield (Table 1, entry 1). Although it is difficult to determine the structure just based on 1H NMR, 13C NMR, MS and IR analysis at first, we were pleased to determine its structure by an X-ray single crystal diffraction study of its analogue 4b (Fig. 1). Obviously, the exclusive formation of 4a clearly indicates that this reaction adopts a C2-type transition state rather than the C1-type (as depicted in Scheme 1) to undergo an intramolecular [4 + 2] cycloaddition to conclude the sequence, which may be attributed to the repulsive interaction between the 6-membered ring and the approaching diene. Although the exact reasons why the reaction favors a C2-type transition state rather than the C1-type are not known at present and will require further investigation, this finding undoubtedly shows the potential of a concise and direct approach to a class of interesting compounds with the not-readily-available tricyclo[3.2.1.02,7]oct-3-ene skeleton. Tricyclo[3.2.1.02,7]oct-3-enes, which consist of fused three-, five-, and six-membered rings, has been found as the core of a few molecules of biological interest9 and in intermediate compounds in the synthesis of cafestol.10 Although some methods have been developed for their synthesis,11 lengthy or complicated procedures as well as harsh reaction conditions are usually applied. Thus, we next focus our effort on optimizing the conditions to improve the yields of 4a. As shown in Table 1, decreasing the reaction temperature significantly slowed down the reaction in toluene, and did not improve the yields of 4a (entries 2 and 3). Comparatively, the reaction proceeded much more efficiently when MeCN was used as solvent, whereas the yields were still unsatisfying (entries 4–6). Gratifying, when THF was employed as the solvent, the reaction conducted at 60 °C for 8 hours gave the best results, furnishing 4a in 73% yield (entry 8). The reaction conducted in 1,4-dioxane furnished 4a in inferior yields (entry 9). Under the optimal conditions, the scope of this reaction was explored. Some typical results are summarized in Table 2. The reaction of aryl propargyl trityl ethers 2 with 3-iodocyclohex-2-enones 1 proceeded smoothly to afford the fused 3 in 73–82% yields at 60 °C (Table 2, entries 1,3,4,5,6), the aryl propargyl trityl ethers 2 that bear aromatic groups including p-Me, p-MeO, p-F, and p-Br substituted phenyl groups were applicable. The reaction was also successfully extended to other electron-deficient vinyl iodides such as 3-iodo-5,5-dimethylcyclohex-2-enone 1b and 3-iodocyclopent-2-enone 1c (entries 2,7–11). When 3-iodobutenolide 1d was employed under the established conditions, the expected product was obtained smoothly and in good yield as well (entry 12). All of the products were characterized by spectroscopic methods, and some special 1H NMR signals were assigned in 4b (Fig. 1).
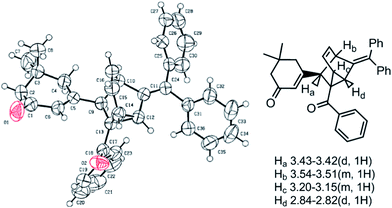 | ||
| Fig. 1 ORTEP representation of 4b. | ||
|
|
||||
|---|---|---|---|---|
| entry | solvent | temp (°C) | time | yield of 4a (%)b |
| a The reaction was carried out using 1 (0. 5 mmol) and 2 (0.6 mmol) in the presence of 5 mol% [Pd(PPh3)2Cl2] and 3 mol% of CuI in 6 mL of solvent and 2 mL of Et3N. b Isolated yields. | ||||
| 1 | Toluene | 100 | 1 h | 20 |
| 2 | Toluene | 60 | 1 d | 30 |
| 3 | Toluene | rt | 3 d | 26 |
| 4 | CH3CN | rt | 2 d | 43 |
| 5 | CH3CN | 60 | 4 h | 55 |
| 6 | CH3CN | reflux | 2 h | 48 |
| 7 | THF | rt | 3 d | 40 |
| 8 | THF | 60 | 8 h | 73 |
| 9 | 1,4-dioxane | 60 | 10 h | 37 |
|
|
|||||
|---|---|---|---|---|---|
| entry | 1 | 2 (R2) | yield of 4 (%)b | ||
| X | n | (R1) | |||
| a The reaction was carried out using 1 (0. 5 mmol) and 2 (0.6 mmol) in the presence of 5 mol% [Pd(PPh3)2Cl2] and 3 mol% of CuI in 6 mL of THF and 2 mL of Et3N at 60 °C for 8–12 h. b Isolated yields. | |||||
| 1 | C | 1 | H(1a) | H(2a) | 73(4a) |
| 2 | CH3(1b) | 2a | 70(4b) | ||
| 3 | 1a | CH3(2b) | 79(4c) | ||
| 4 | 1a | OCH3(2c) | 82(4d) | ||
| 5 | 1a | F(2d) | 80(4e) | ||
| 6 | 1a | Br(2e) | 75(4f) | ||
| 7 | 1b | 2c | 82(4g) | ||
| 8 | 1b | 2e | 77(4h) | ||
| 9 | C | 0 | H(1c) | 2b | 75(4i) |
| 10 | 1c | 2d | 78(4j) | ||
| 11 | 1c | 2e | 72(4k) | ||
| 12 | O | 0 | CH3(1d) | 2d | 75(4l) |
In conclusion, we have developed a convenient sequential palladium-catalyzed coupling, propargyl–allenyl isomerization, allenyl–benzyl Claisen rearrangement and [4 + 2] cycloaddition, leading to a facile and efficient synthesis of tricyclo[3.2.1.02,7]oct-3-ene derivatives. In respect to the easy availability of the starting materials, simple manipulation, mild conditions and high efficiency, this reaction will be synthetically useful in organic chemistry. Further efforts to explore the reaction scope as well as the mechanism study are currently under way.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (Project Nos. 20732005 and 20872127) and National Basic Research Program of China (973 Program, 2009CB825300) for financial support.References
- (a) For recent examples, see: A. Ajamian and J. L. Gleason, Angew. Chem., Int. Ed., 2004, 43, 3754 CrossRef CAS; (b) Y. Yamamoto, Y. Nakagai and K. Itoh, Chem.–Eur. J., 2004, 10, 231 CrossRef CAS; (c) J. Cossy, F. Bargiggia and S. Bouz, Org. Lett., 2003, 5, 459 CrossRef CAS; (d) A. N. Thadani and V. H. Rawal, Org. Lett., 2002, 4, 4317 CrossRef CAS; (e) S. U. Son, K. H. Park and Y. K. Chung, J. Am. Chem. Soc., 2002, 124, 6838 CrossRef CAS; (f) J. Louie, C. W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312 CrossRef CAS; (g) P. A. Evans and J. E. Robinson, J. Am. Chem. Soc., 2001, 123, 4609 CrossRef CAS.
- (a) L. F. Tietze and N. Rackelmann, Pure Appl. Chem., 2004, 76, 1967 CrossRef CAS; (b) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC; (c) P. J. Parsons, C. S. Penkett and A. Shell, Chem. Rev., 1996, 96, 195 CrossRef CAS; (d) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS; (e) A. Heumann and M. Reglier, Tetrahedron, 1996, 52, 9289 CrossRef CAS; (f) M. Malacria, Chem. Rev., 1996, 96, 289 CrossRef CAS; (g) G. Poli, G. Giambastiani and A. Heumann, Tetrahedron, 2000, 56, 5959 CrossRef CAS.
- (a) R. U. Braun, M. Ansorge and T. J. J. Müller, Chem.–Eur. J., 2006, 12, 9081 CrossRef CAS; (b) D. M. D'Souza, F. Rominger and T. J. J. Müller, Angew. Chem., Int. Ed., 2005, 44, 153 CrossRef CAS; (c) D. M. D'Souza, A. Kiel, D.-P. Herten and T. J. J. Müller, Chem.–Eur. J., 2008, 14, 529 CrossRef CAS; (d) D. M. D'Souza, F. Rominger and T. J. J. Müller, Chem. Commun., 2006, 4096 RSC; (e) D. M. D'Souza, W.-W. Liao and T. J. J. Müller, Org. Biomol. Chem., 2008, 6, 532 RSC; (f) O. G. Schramm, née. Dediu and T. J. J. Müller, Adv. Synth. Catal., 2006, 348, 2565 CrossRef CAS; (g) B. U. Braun, K. Zeitler and T. J. J. Müller, Org. Lett., 2001, 3, 3297 CrossRef; (h) B. U. Braun, K. Zeitler and T. J. J. Müller, Org. Lett., 2000, 2, 4181 CrossRef; (i) O. G. Schramm, née. Dediu, T. Oeser and T. J. J. Müller, J. Org. Chem., 2006, 71, 3494 CrossRef CAS.
- D. M. D'Souza, F. Rominger and T. J. J. Müller, Chem. Commun., 2006, 4096 RSC.
- (a) R. Shen and X. Huang, Org. Lett., 2008, 10, 3283 CrossRef CAS; (b) R. Shen, S. Zhu and X. Huang, J. Org. Chem., 2009, 74, 4118 CrossRef CAS.
- R. Shen, X. Huang and L. Chen, Adv. Synth. Catal., 2008, 350, 2865 CrossRef CAS.
- F. Sha and X. Huang, Angew. Chem., Int. Ed., 2009, 48, 3458 CrossRef CAS.
- (a) N. Chopin, H. Gérard, I. Chataigner and S. R. Piettre, J. Org. Chem., 2009, 74, 1237 CrossRef CAS; (b) A. J. H. Klunder, J. Zhu and B. Zwanenburg, Chem. Rev., 1999, 99, 1163 CrossRef CAS; (c) J. G. Martin and R. K. Hill, Chem. Rev., 1961, 61, 537 CrossRef CAS; (d) H. Kwart and K. King, Chem. Rev., 1967, 415 Search PubMed.
- (a) S. W. Pelletier, N. V. Mody, Z. Djarmati and S. D. Lajsic, J. Org. Chem., 1976, 41, 3042 CrossRef CAS; (b) F. Bohlmann, J. Jakupovic, M. Ahmed, M. Grenz, H. Suding, H. Robinson and R. M. King, Phytochemistry, 1981, 20, 113 CrossRef CAS.
- E. J. Corey, G. Wess, Y. B. Xiang and A. K. Singh, J. Am. Chem. Soc., 1987, 109, 4717 CrossRef CAS.
- (a) D. M. Hodgson and R. E. Marriott, Tetrahedron Lett., 1997, 38, 887 CrossRef CAS; (b) G. Mehta and C. Ravikrishna, Tetrahedron Lett., 1996, 37, 2655 CrossRef CAS; (c) R. Kiattansakul and J. K. Snyder, Tetrahedron Lett., 1999, 40, 1079 CrossRef CAS; (d) A. Abad, C. Agullo, A. C. Cunat, I. De Alfonso, I. Navarro and N. Vera, Molecules, 2004, 9, 287 CrossRef CAS; (e) J. E. Moses, J. E. Baldwin, R. M. Adlington, A. R. Cowley and R. Marquez, Tetrahedron Lett., 2003, 44, 6625 CrossRef CAS; (f) S. M. Ng, C. M. Beaudry and D. Trauner, Org. Lett., 2003, 5, 1701 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Crystal data for 4b: C36H32O2, MW = 496.6, monoclinic, space group P2(1)/n, final R indices [I >2σ (I)], R1 = 0.0408, wR2 = 0.0865; R indices (all data), R1 = 0.0715, wR2 = 0.0985; a = 16.5695(6) Å, b = 15.0759(6) Å, c =11.2960(6) Å, α = 90°, β =90°, γ = 90°, V = 2821.7(2) Å3, T = 293(2) K, Z = 4, reflections collected/unique 11515/4234 (Rint = 0.0531), parameters 67.03. Supplementary crystallographic data has been deposited at the Cambridge Crystallographic Data Centre, CCDC 821989. See DOI: 10.1039/c1ra00452b/ |
| ‡ Professor Huang passed away on March 6, 2010. |
| This journal is © The Royal Society of Chemistry 2012 |