Exciton migration and charge transfer in chemically linked P3HT–TiO2 nanorod composite†
Tingting
Xu
a,
Ming
Yan
a,
James D.
Hoefelmeyer
b and
Qiquan
Qiao
*a
aCenter for Advanced Photovoltaics, Department of Electrical Engineering and Computer Sciences, South Dakota State University, Brookings, SD 57006, USA. E-mail: Qiquan.Qiao@sdstate.edu; Fax: +1-605-688-4401; Tel: +1-605-688-6965
bDepartment of Chemistry, University of South Dakota, Vermillion, SD 57069, USA
First published on 23rd November 2011
Abstract
Exciton migration and charge transfer in the chemically linked P3HT–TiO2 nanorod composite (P3HT–Si–nr–TiO2) solution were investigated in comparison with pristine P3HT and physically mixed P3HT/LA–nr–TiO2 solutions. The chemically linked P3HT–Si–nr–TiO2 was made by covalently linking in situ polymerized P3HT onto nr–TiO2 using triethoxy-2-thienylsilane as a linker to replace the initial linoleic acid (LA) capping agent on nr–TiO2. The physically mixed P3HT/LA–nr–TiO2 was prepared by adding ex situ synthesized P3HT into the LA–capped nr–TiO2 solution. In the chemically linked sample, charge transfer from P3HT to TiO2 nanorods was found to occur evidenced by photoluminescence (PL) quenching and ultrafast decay dynamics with a timescale of 0.75 ps. However, both the emission spectra and femtosecond dynamics in physically mixed sample overlapped very well with those from pristine P3HT solution, indicating no PL quenching or charge transfer from P3HT to nr–TiO2. In addition, blue shift in absorbance and PL spectra, larger Stokes shift, and structureless PL spectra found in the chemically linked sample indicated that P3HT formed a more coil-like conformation with more twisted torsion disorders than those in pristine P3HT and physically mixed samples. This is consistent with the femtosecond measurement result that torsional relaxation occurred with a longer decay time and higher amplitude. Moreover, intersystem crossings (ISC) from singlet state (S1) to triplet state (T1) in P3HT of the three samples were all found to occur in a comparable timescale of ∼1 ns and showed no dependence on conformational disorders such as torsional defects.
1. Introduction
Hybrid nanocomposites of conjugated polymers and inorganic nanocrystals have received intense investigation for applications in optoelectronic devices such as light-emitting diodes (LEDs),1,2 biosensors3 and photovoltaic cells.4 The hybrid system has advantages derived from use of organic and inorganic materials. There is great potential in these systems due to the ability to precisely synthesize inorganic nanocrystals and organic polymers with high degree of structural control, to optimize properties of the components, and to build functional materials. The organic component offers low-cost solution-based processing while the inorganic component provides unique characteristics including relatively high electron mobility and electron affinity, and thermal stability.5 However, the interface between conjugated polymers and nanocrystals is a challenge due to their dramatically different properties. Nanocrystals require surface passivation to prevent aggregation and enhance their dispersion in solutions. Commonly used surfactants such as alkyl carboxylic acid, trioctylphosphine oxide (TOPO) and alkyl thiols are non-conjugated saturated chains that do not effectively facilitate electron transfer between the organic and inorganic materials.6–8 Therefore, it is difficult to achieve simultaneously high miscibility and efficient electronic communication.Greenham et al. found that neither Förster energy transfer nor charge transfer occurs in the MEH-PPV/TOPO-capped CdS composite film because spectral overlap between MEH-PPV emission and CdS absorption that is needed for Förster energy transfer to occur does not exist, while charge transfer is blocked by a layer of saturated TOPO.7 When the saturated TOPO was exchanged with the pyridine group, charge transfer from MEH-PPV to CdS was observed. Förster energy transfer did occur when TOPO-capped CdS was replaced by TOPO-capped CdSe in which the MEH-PPV emission spectrum showed overlap with CdSe absorption spectrum.7 In addition, Beek et al. reported that significant electron transfer occurs in an oligothiophene/TiO2 nanoparticle dispersion in which the oligomer was chemically anchored onto TiO2 nanoparticlesvia a carboxylic acid functionalized group.9 However, when the carboxylic acid group was replaced with a non-anchoring aldehyde group on the oligothiophene, no photoluminescence (PL) quenching (charge transfer) was observed in the solution. Förster energy transfer was not found because TiO2 nanoparticle has a larger bandgap than the oligothiophene, in other words the oligothiophene emission spectrum does not overlap with nr–TiO2 absorption. The dependence of alkyl linker length on electron transfer was also studied in the oligothiophene/TiO2 nanoparticle assembly.9 The electron transfer rate was found to decrease as the alkyl spacer length increased.9 Zhang et al. grew regiorandom P3HT onto TiO2 nanotubesvia a covalent silane linker, which exhibited more efficient charge transfer from P3HT to TiO2 than physisorbed P3HT.10 However, the alkyl group used in the linker might cause electron transfer to take place in a weakly-coupled nonadiabatic regime. In such a regime, the electron transfer rate was found to attenuate with the spacer distance as lnkct = lnk0 − βσ, where kct is the electron transfer rate, β is the attenuation coefficient, and σ is number of sigma bonds.9 The β value was found to be 0.3∼0.6 per bond in theoligothiophene–TiO2 assembly.9 Various approaches have been reported to chemically attach or in situ synthesize conjugated polymers onto inorganic nanocrystals, quantum dots, and nanotubes.6,9,11–13 However, little work has been reported on energy transfer or charge transfer from P3HT to nr–TiO2, and their dependence on the polymer chain conformation in the chemically linked organic/inorganic hybrid systems.
In this work, triethoxy-2-thienylsilane was used to covalently link poly(3-hexylthiophene) (P3HT) onto TiO2 nanorods (nr–TiO2). The energy migration in P3HT and charge transfer from P3HT to nr–TiO2 in the chemically linked P3HT–Si–nr–TiO2 solution were investigated in comparison with pristine P3HT and physically mixed P3HT/LA–nr–TiO2. The charge transfer from P3HT to nr–TiO2 was found to occur evidenced by photoluminescence (PL) quenching and a decay time constant of 0.75 ps in the chemically linked samples, but was not observed in the physically mixed P3HT/LA–nr–TiO2 solution with a rise process that is almost the same as the pristine P3HT. In addition, P3HT was found to have a more coil-like conformation in the chemically linked samples with more twisted defects that led to torsional relaxation with a larger amplitude and a longer decay time than those in pristine P3HT and physically mixed samples. Moreover, intersystem crossings (ISC) from singlet state (S1) to triplet state (T1) in P3HT of the three samples were all found to occur in a comparable timescale of ∼1 ns and showed no dependence on conformational disorders such as torsional defects.
2. Experimental section
Reagents titanium tetrabutoxide, trimethylamine N-oxide dihydrate, thiophene, iron(III) chloride, and anhydrous toluene were purchased from Acros. Triethoxy-2-thienylsilane was ordered from Sigma Aldrich. They were used as received. Chloroform was dried over CaH2 and freshly distilled. All other solvents were purchased from commercial sources and used without further purification.2.1 Synthesis of linoleic acid-capped nr-TiO2
The nr–TiO2 with a diameter of 3∼4 nm and a length of 30∼40 nm was prepared following the method reported by Liet al.14 Briefly, titanium tetrabutoxide was added to a solution of hexane, linoleic acid (LA), and triethylamine, and then the mixture was placed in a sealed acid digestion bomb. The system was heated in a 150 °C oven for 24 h. The TiO2 nanorods were recovered through fractional precipitation upon addition of aliquots of ethanol, followed by centrifugation. Purification was achieved with a total of three cycles of precipitation/redispersion using hexane/ethanol solvent/non-solvent pair. The LA-capped nr–TiO2 was dispersed in organic solvent such as hexane or toluene to give a transparent pale-yellow dispersion that can be kept sealed in the dark indefinitely. We note that the dispersion is not stable in visible light, and that after 24 h a white precipitate appears.2.2. Synthesis of triethoxy-2-thienylsilanefunctionalized nr–TiO2 (Si–nr–TiO2)
After washed by ethanol for more than three times, the majority of LA groups can be removed evidenced by the precipitation of nr–TiO2 due to its lost solubility in hexane. The lost solubility can be recovered by adding several drops of LA, indicating that LA can be removed by washing with ethanol. This agrees well with previous reports where other groups have removed surface-capping agents such as oleic acid.15–18 The triethoxy-2-thienylsilane group was then covalently linked to TiO2 surface following a method reported by Zhang et al.10 A 50 mL flask was charged with 20 mg nr–TiO2 whose majority of LA ligand has been removed, 0.1 g triethoxy-2-thienylsilane and 20 mL anhydrous toluene. The solution was then heated at reflux for 12 h under a nitrogen atmosphere. The successful exchange of LA by triethoxy-2-thienylsilane can be supported by the FTIR measurements discussed later. The mixture was washed in fresh toluene and methanol and then centrifuged to obtain wet slurry.2.3 Synthesis of P3HT grafted onto triethoxy-2-thienylsilane functionalized nr–TiO2 (P3HT–Si–nr–TiO2)
3-Hexylthiophene was synthesized after modification of the reported method with 75% yield.191H–NMR (400MHz, CDCl3): δ = 0.88 (3H, m), δ = 1.30 (6H, m), δ = 1.61 (2H, m), δ = 2.61 (2H, t), δ = 6.91 (2H, 2d), δ = 7.21 (1H, 2d). 20 mg Si–nr–TiO2 wet slurry was stirred in 10 mL anhydrous chloroform solution under N2 at room temperature, and then 80 mg FeCl3 and 20 mg 3-hexylthiophene were added subsequently.20 The mixture was then stirred for 24 h to complete the polymerization. The product was precipitated with methanol to remove residual FeCl3. The dark-red solid was washed by Soxhlet extraction overnight in methanol. The red solid was finally dried under vacuum at 40 °C for 24 h for further analysis. The red solid is the chemically linked P3HT–Si–nr–TiO2 sample, which will be dissolved in CHCl3 for UV-Vis absorbance, fluorescence and ultrafast spectroscopic studies. When the chemically linked P3HT–Si–nr–TiO2 was re-dissolved in CHCl3, some nr–TiO2 aggregates were found. The aggregates were most possibly formed during the precipitation process when residual FeCl3 was removed by methanol from the chemically linked P3HT–Si–nr–TiO2. These aggregates can cause light scattering in the UV-Vis absorbance, PL and ultrafast spectroscopic measurement, affecting the comparison between these samples. Therefore, the chemically linked P3HT–Si–nr–TiO2 solutions were filtrated using a 0.45 μm filter to remove any nr–TiO2 aggregates and get a clear light yellow solution. For comparison, physically blended P3HT/LA-capped nr–TiO2 solutions were also prepared. No filtration was conducted in the physically blended solutions because no aggregates were observed.2.4 Synthesis of pristine P3HT
Pristine P3HT was prepared using FeCl3 as a chemical oxidant, and then purified by Soxhlet extraction and drying in vacuum. The molecular weight of regiorandom P3HT is Mn = 55156, PDI = 3.22. P3HT was characterized by 1H NMR (figure S1†) and the results showed that P3HT has regiorandom conformation with head-to-tail (HT) and head-to-head (HH) ratio of around 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.
:1.
2.5 Materials characterizations
1H NMR spectra were recorded on a Bruker 400 NMR spectrometer. Transmission electron microscopy (TEM) images were acquired using a Hitachi TEM. The samples were prepared by dropping dilute dispersion of nr–TiO2 in chloroform onto carbon-coated copper grids whereby the solvent was allowed to evaporate in air.Powder X-ray diffraction (XRD) patterns were collected on a PANalytical X'Pert Pro from 5–90° (2θ) in the Center for Nanophase Materials Sciences at Oak Ridge National Laboratory. The X-ray generator was operated at 45 kV and 40 mA, and the copper anode produced Cu-Kα radiation (λ = 1.54 Å). Dry nr–TiO2 powders were deposited on a single crystal silicon wafer cut off-axis as low-background holder.
FTIR spectra were obtained using a Bruker Tensor-27 FTIR spectrometer. Measurements were obtained from KBr pellets with embedded nr–TiO2 powder. The FTIR spectra were acquired by scanning the specimens from 600–4000 cm−1 at a resolution of 2 cm−1.
The UV-visible absorbance spectra were obtained from an HP Agilent 8352 spectrophotometer. The fluorescence emission spectra were measured with a FS920 fluorescence spectrometer (Edinburgh Instruments, Ltd.) with a Xenon arc lamp as the light source.
A femtosecond up-conversion fluorescence system (FOG100) was employed to investigate energy migration and chargetransfer dynamics. A Ti:sapphire laser (Tsunami) was used to produce 57 fs pulses centered at 800 nm with a repetition rate of 86 MHz. The excitation pulses at 400 nm, produced by the second harmonic generation of the Ti:sapphire laser through a 2 mm beta-barium-borate (BBO) crystal, were focused into a 0.4 mm thick sample cell. To avoid high-intensity laser induced bleaching effects, the sample cell was rotated. The excited sample fluorescence was crossed with the gate pulses at another BBO crystal to generate the up-converted signal which was spectrally filtered out by a grating pair and then detected by a photomultiplier tube (PMT) with quite low dark counts. Usually, the instrument response of this fluorescence dynamics system was measured to be a Gaussian function with a full width at half maximum (FWHM) of ∼260 fs.
3. Results and discussion
3.1 Characterization of LA-capped nr–TiO2
Fig. 1a is a TEM image of nr–TiO2. No aggregation of nanorods was observed because they were capped by LA. The nr–TiO2 has a high aspect ratio with uniform length of 30∼40 nm and diameter of 3∼4 nm. The XRD pattern of nr–TiO2 in Fig. 1 shows the presence of anatase phase TiO2. The width of the (004) peak is narrower than the others, consistent with a preferred growth orientation parallel to the c-axis of anatase lattice, which implies the formation of rod-like nanocrystals.15,21 | ||
| Fig. 1 (a) Transmission electron microscopy (TEM) image and (b) X-ray Diffraction (XRD) of LA-capped nr–TiO2. | ||
3.2 Fourier transform infrared spectroscopy (FTIR) measurement
Scheme 1 shows the process of surface functionalization of nr–TiO2viatriethoxy-2-thienylsilane followed by in situpolymerization of P3HT onto the nr–TiO2 surface. | ||
| Scheme 1 Grafting P3HT onto the triethoxy-2-thienylsilane functionalized nr–TiO2. | ||
The nature of organic coating on nr–TiO2 surface was investigated by FTIR. The spectra from four samples are shown in Fig. 2: (a) LA-capped nr–TiO2, (b) triethoxy-2-thienylsilane surface modified nr–TiO2, (c) chemically linked P3HT–Si–nr–TiO2 composite by in situpolymerization, and (d) pristine P3HT. The LA capped nr–TiO2 (Fig. 2a) exhibits an intense antisymmetric and symmetric C–H stretching vibration of –CH2– group in the hydrocarbon group at 2923 and 2852 cm−1, respectively, while the shoulder at 2950 cm−1 is assigned to the asymmetric stretch of the terminal –CH3group of LA ligand.22 A weak but distinct peak at 3008 cm−1 results from the C–H stretching in –CH3 of LA.22 The intense bands at 1519 cm−1 and 1431 cm−1 are assigned to the COO− antisymmetric and symmetric stretching vibrations of the carboxylate complexes anchored to the surface Ti centers.23 The broad band at about 720 cm−1 is ascribed to the characteristic vibrations of Ti–O–Ti network.17 After being washed with ethanol for more than three times, nr–TiO2 became insoluble in hexane and could be redispersed when several drops of LA were added again. The loss of solubility by washing with ethanol and the redispersion by adding several drops of LA indicated that the LA ligands can be easily removed from and re-attached onto nr–TiO2.15 This agrees well with previous reports where other groups have removed oleic acid surfactant by washing with ethanol.15–18 After that, triethoxy-2-thienylsilane was attached onto nr–TiO2 as a new capping agent to replace LA. As shown in Scheme 1, the triethoxy-2-thienylsilane capping agent has much fewer –CH2– groups than the LA capping group. By keeping the intensity of Ti–O–Ti peak as constant at about 720 cm−1, we can compare the intensity at 2923 and 2852 cm−1 to determine whether the majority of LA groups have been exchanged by triethoxy-2-thienylsilane. Compared to Fig. 2a, the much lower intensity at 2923 and 2852 cm−1 for the same amount of nr–TiO2 indicates that the majority of LA groups have been replaced by triethoxy-2-thienylsilane. This can be further supported by the absence of the peaks at 1519 cm−1 and 1431 cm−1 for COO−group in the triethoxy-2-thienylsilane capped nr–TiO2. The peak at 3102 cm−1 is related to the C–H stretching mode in the thiophene rings,24 while those at 1408 cm−1 and 1498 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching mode in triethoxy-2-thienylsilane capped nr–TiO2.13 The bands at 837 and 1064 cm−1 are assigned to the out-of-plane and in-plane bending vibration of CH in thiophene ring,13,24 while the broad and strong peaks at 1124 cm−1 result from the Si–O group in triethoxy-2-thienylsilane capped nr–TiO2.25 This supports that the triethoxy-2-thienylsilane linker has been bound to the nr–TiO2 surface. Fig. 2c shows the FTIR spectrum of in situ polymerized P3HT onto nr–TiO2 surface linked by triethoxy-2-thienylsilane. Similar to that found in triethoxy-2-thienylsilane modified nr–TiO2, the Si–O signal at 1124 cm−1 in Fig. 2c further supports that triethoxy-2-thienylsilane has been bound to the nr–TiO2 surface.26 The band around 2800∼3000 cm−1 is ascribed to the C–H stretch of P3HT side chain, including C–H stretching vibration mode of the –CH2– and –CH3 end group, which is identical to pristine P3HT (Fig. 2d) and suggests that P3HT has been polymerized and anchored onto nr–TiO2.
C stretching mode in triethoxy-2-thienylsilane capped nr–TiO2.13 The bands at 837 and 1064 cm−1 are assigned to the out-of-plane and in-plane bending vibration of CH in thiophene ring,13,24 while the broad and strong peaks at 1124 cm−1 result from the Si–O group in triethoxy-2-thienylsilane capped nr–TiO2.25 This supports that the triethoxy-2-thienylsilane linker has been bound to the nr–TiO2 surface. Fig. 2c shows the FTIR spectrum of in situ polymerized P3HT onto nr–TiO2 surface linked by triethoxy-2-thienylsilane. Similar to that found in triethoxy-2-thienylsilane modified nr–TiO2, the Si–O signal at 1124 cm−1 in Fig. 2c further supports that triethoxy-2-thienylsilane has been bound to the nr–TiO2 surface.26 The band around 2800∼3000 cm−1 is ascribed to the C–H stretch of P3HT side chain, including C–H stretching vibration mode of the –CH2– and –CH3 end group, which is identical to pristine P3HT (Fig. 2d) and suggests that P3HT has been polymerized and anchored onto nr–TiO2.
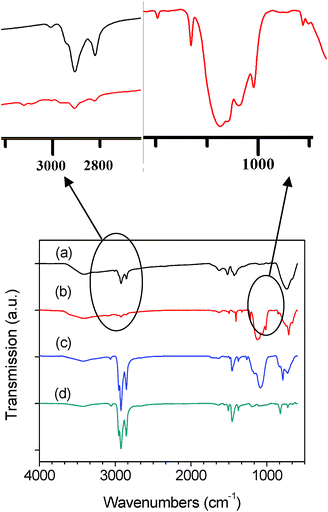 | ||
| Fig. 2 The FTIR spectra of (a) LA-capped nr–TiO2 (LA–nr–TiO2), (b) triethoxy-2-thienylsilane functionalized nr–TiO2 (Si–nr–TiO2), (c) chemically linked P3HT–Si–nr–TiO2 composite by in situpolymerization, and (d) pristine P3HT. | ||
3.3 Steady-state absorption and emission
Fig. 3 shows the normalized absorbance and PL spectra of pristine P3HT, physically blended P3HT/LA–nr–TiO2 mixture and chemically linked P3HT–Si–nr–TiO2 in chloroform solutions. The absorbance spectrum of physically mixed P3HT/LA–nr–TiO2 presents the absorbance contributed from the P3HT and TiO2 components. However, the chemically linked P3HT–Si–nr–TiO2 sample exhibits a blue-shift absorbance (438 nm for physically mixed solution → 404 nm for chemically linked solution) and PL (573 nm for physically mixed solution → 563 nm for chemically linked solution). It has been reported that the blue-shift indicates that the polymer chains are more coil-like or twisted in solutions or films.27–29 A possible reason for the blue-shift is that P3HT is tightly linked onto nr–TiO2 with more coil-like chains in the chemically linked system, leading to a reduced effective conjugation length of segments on the chain. In addition, the Stokes shift is larger in chemically linked solution (155 nm) than that in physically mixed sample (135 nm). The PL of the pristine P3HT and physically mixed solutions exhibited a vibronic shoulder at about 620 nm, indicating a more structured behavior in fluorescence spectra, while that of the chemically linked solution seemed more structureless without any vibronic peaks or shoulders. It is generally known that photoexcitation can induce a change of polymer conformation from a more flexible ground state (aromatic) to a more rigid planar excited state (quinoidal) geometry.30 Therefore, the Stokes shift can be attributed to the conformational change from nonplanar (e.g., torsionally disordered) ground state to a planar excited state upon photoexcitation. Previous reports also showed that PL spectra are more structured with vibronic peaks or shoulders than absorbance spectra when polymers become planar after photoexcitation.30,31 Unlike pristine P3HT and physically mixed samples exhibiting a vibronic shoulder at ∼620 nm, the PL spectrum in the chemically linked solution was less structured without any vibronic peaks or shoulders (Fig. 3), suggesting that its excited state was less planar. Moreover, the larger Stokes shift further indicated that polymer chains are more coil-like or twisted in the chemically linked solution. In the polymerization here, the TiO2 nanorods were first surface-functionalized by triethoxy-2-thienylsilane to serve as initiator sites and then P3HT grew directly from these sites. The final P3HT conformation on the surface depends on the density of the 2-thienylsilane functionalized initiator sites. Milner reported that polymers stretch away from the surface and form a polymer “brush” if the initiator sites are very dense with a high coverage on the surface for polymerization.32 However, at a low surface-anchored initiator density, polymers can collapse to form a coil-like conformation.33 Here, the observed blue shift and structureless PL, as well as the larger Stokes shift indicated that P3HT formed a more coil-like conformation on the surface of nr–TiO2 in the chemically linked samples. Such a more coil-like conformation can induce twisting and/or bending along the polymer backbone that leads to reduced effective conjugation length of polymer segments.40,41 It is also important to mention that the P3HT was regiorandom in all the three samples; however it exhibited a relatively coil-like twisted conformation in the chemically linked samples, which was evidenced by the blue shift and structureless PL, as well as the larger Stokes shift. | ||
| Fig. 3 Normalized UV-Vis absorbance spectra of (a) chemically linked P3HT–Si–nr–TiO2 (dark) (b) pristine P3HT (red), (c) physically mixed P3HT/LA–nr–TiO2 (blue); and normalized photoluminescence (PL) of (d) chemically linked P3HT–Si–nr–TiO2 (magenta) (e) pristine P3HT (violet), and (f) physically mixed P3HT/LA–nr–TiO2 (green). Note, pristine P3HT PL (violet) and physically mixed composite PL (green) overlaps so well that they look like one plot. In the PL measurement, the solution samples were excited at 420 nm. | ||
The steady-state fluorescence quenching spectra of the chemically linked and physically blended P3HT and nr–TiO2 composites are shown in Fig. 4a. As discussed above, the PL of the chemically linked solution is blue-shifted and more structureless compared to pristine P3HT (or the physical mixture). Again, this could indicate that polymer conformation is more coil-like in the chemically linked samples. In order to more appropriately compare the PL quenching in physically mixed solution with that in the chemically linked sample, the ratios of P3HT to nr–TiO2 and their concentrations in the physically mixed solutions were prepared as the same as those in the chemically linked samples. According to Beer's law, the absorbance is linear with the concentration (or number density of absorbers) following A = εlc, where A is absorbance, ε is extinction coefficient, l is light path length, and c is concentration of absorbing species in the solution. Since the same cell and materials (P3HT and nr–TiO2) were used for absorbance measurements, ε and l should be the same. Therefore, the same absorbance A indicated the same amount P3HT and nr–TiO2 in the physically mixed and chemically linked samples. Therefore, the maximum absorbance at λmax of P3HT in the physically mixed and chemically linked solutions was controlled to be equal so that the amount of P3HT is roughly comparable in the two solutions. Similarly, nr–TiO2 was also made at the same amount in both solutions using their absorption spectra. The solutions were excited at 420 nm and monitored with a spectral range from 450 to 800 nm. Since the absorption maxima of these solutions shift from one sample to the other (Fig. 4), exciting at 420 nm may lead to quantitatively different amount of PL intensity between samples, which can make the comparison between the PL quenching complex. However, as shown in Fig. 3, the excitation at 420 nm is very close to the absorbance peak and the differences can be neglected when absorbance maxima were controlled the same. Since PL and charge transfer compete with each other, the decrease of PL intensity in polymers is typically regarded as a good sign for Förster energy transfer and/or charge transfer from the polymers to acceptor materials (e.g.TiO2, CdS, and CdSe).7,16
 | ||
| Fig. 4 (a) The fluorescence spectra of pristine P3HT (green), physically mixed P3HT/LA–nr–TiO2, and chemically linked P3HT–Si–nr–TiO2 (red) with an excitation wavelength at λex = 420 nm. (b) The nr–TiO2 absorbance (dark) and pristine P3HT (red) emission spectra. | ||
Förster energy transfer requires spectral overlap between polymer emission and acceptor absorbance,34 while charge transfer typically requires a donor/acceptor interface with suitable energy level alignment between the two materials.7 However, as shown in Fig. 4b, the PL of P3HT is in a range of 450–800 nm, while nr–TiO2 has an absorbance less than 350 nm, showing no spectral overlap between them. Thus, Förster energy transfer from P3HT to nr–TiO2 cannot occur. On the other hand, the lowest unoccupied molecular orbital (LUMO) of P3HT is −3.2 eV, while the conduction band (CB) of TiO2 is −4.2 eV, supporting a photoinduced charge transfer from P3HT to nr–TiO2.35,36 Also, LA-capping agent was exchanged by triethoxy-2-thienylsilane that served as a chemical linker between P3HT and nr–TiO2 and helped to form a close contact. Therefore, the reduced PL intensity in the chemically linked samples was attributed to photoinduced charge transfer from P3HT to nr–TiO2. This is consistent with other groups' reports for P3HT and terthiophene.8,10 For example, PL quenching was observed due to electron transfer from terthiophene to TiO2 when terthiophene was chemically linked to TiO2 nanoparticles through a carboxyl group.8 Recently, Botiz et al. reported that non-radiative processes can also lead to PL quenching.37 However, the energy levels of P3HT and PCBM are well aligned and no spectral overlap between P3HT and PCBM is found, so it seems reasonable to attribute PL quenching to charge transfer from P3HT to PCBM. However, PL spectra from the physical mixture overlapped very well with pristine P3HT and showed no decreased PL intensity compared to pristine P3HT, demonstrating that neither Förster energy transfer nor charge transfer occurred from P3HT to LA-capped nr–TiO2. The reason that Förster energy transfer did not occur was again attributed to no spectral overlap between P3HT emission and nr–TiO2 absorbance (Fig. 4b), while no charge transfer was caused by the blocking effect of LA-capping agent. It is important to mention that the LA-capping agent was not removed from nr–TiO2 surface in order to keep good dispersion of nr–TiO2 in physically mixed samples. The LA-capping agent can block charge transfer from P3HT to nr–TiO2. Then only dynamic (collisional) interaction could take effect between P3HT and LA-capped nr–TiO2, which was found to be very insignificant.8 This is in agreement with the previous finding that neither energy transfer nor charge transfer occurred from MEH-PPV to TOPO-capped CdS nanocrystals due to both little spectral overlap and blocking effects of TOPO-capping agents when they are physically mixed.7
3.4 Time-resolved fluorescence measurement
In order to further understand exciton migration and charge transfer in chemically linked P3HT-Si–nr–TiO2 in comparison with physically mixed P3HT/LA–nr–TiO2, a femtosecond fluorescence up-conversion (FFU) technique was used to study their ultrafast processes. The measured fluorescence transient Ifl(t) is the convolution of real transient and instrument response function (IRF). The real fluorescence transient can be deconvoluted using a sum of multiple-exponential functions:38 | (1) |
Generally upon photoexcitation, excited states follow self-trapping (dynamic localization) of excitons by nuclear motion within 100 fs,40 exciton relaxation [i.e. energetically downhill resonant energy transfer (RET) or downhill excitonic energy transfer (EET)] and migration (i.e.RET or EET between segments with comparable energy) along polymer chains. Downhill EET was reported to take place from less than 1 ps to several ps after photoexcitation,41–44 much faster than EET between segments with comparable energy at the bottom of the density of states that happens at much slower timescale of ∼10–100 ps.38,39,43
Fig. 5 shows fluorescence dynamics of three different samples tested in chloroform solution, (1) pristine P3HT, (2) physically mixed P3HT/LA–nr–TiO2, and(3) chemically linked P3HT–Si–nr–TiO2. The excitation wavelength was fixed at 400 nm and fluorescence decay was detected at 580 nm near the P3HT emission peak in solutions. The obtained fluorescence curves were well-fitted by a multi-exponential decay (eqn 1) convoluted with the instrument response function. Table 1 lists the fitted data. The self-trapping of excitons that couples to initial vibrational relaxation typically occurs in a time scale less than 100 fs.38,40,45 These processes were not observed because the 100 fs time scale was shorter than the IRF (∼260 fs, fwhm) of the measurement here. In the chemically linked P3HT–Si–nr–TiO2, the initial fast timescale exhibited an ultrafast decay time constant (τ1) of 0.75 ps. Since downhill EET was previously found to occur from less than 1 ps to several ps,41–44 this τ1 process can be assigned to downhill EET from short segments with energy larger than 580 nm (2.14 eV) to relative long segments with energy at about 580 nm (2.14 eV). The downhill RET is sometimes also called downhill relaxation of self-trapped excitons or downhill excitation energy transfer (EET).39 The τ1 process is a decay dynamics, showing that downhill EET from higher energy segments to the detected 580 nm emission segments was much weaker than the loss from the 580 nm emission segments to others.46 Such loss may include several possibilities: (1) downhill EET from the 580 nm emission segments to even lower energy segments, (2) Förster energy transfer from polymers to acceptors, and (3) charge transfer from the 580 nm emission segments to acceptor materials. The first possibility (downhill EET to even lower energy segments) only occurs in P3HT without the involvement of nr–TiO2 in the chemically linked samples. Thus it should have similar dynames as that in pristine P3HT. However, the rising dynamics in pristine P3HT suggested that the downhill EET from the 580 nm emission segments to even lower energy segments is not strong enough to cause a decay dynamics in the initial fast stage (τ1). Therefore the first possibility can be excluded in explaining the decay process in the chemically linked samples. The second possibility (Förster energy transfer) can also be omitted since P3HT emission spectra do not have spectral overlap with nr–TiO2 absorbance spectra (Fig. 4b). Therefore, only the third possibility (photoinduced charge transfer) can explain initial fast decay dynamics. As discussed above, the triethoxy-2-thienylsilane linker can improve electronic communication; and the P3HT and nr–TiO2 interface is aligned with suitable energy level alignment that supports photoinduced charge transfer from P3HT to nr–TiO2. This is consistent with the finding of PL quenching in chemically linked samples (Fig. 4a). However for the physically mixed solution, the normalized femtosecond fluorescence measurement in Fig. 5b shows that the ultrafast dynamics of physically mixed sample almost overlaps with that of pristine P3HT. In the physically mixed P3HT/LA–nr–TiO2, the initial fast process (τ1) exhibited a rising dynamics with comparable timescale (0.5 ps for physical mixture and 0.6 ps for pristine P3HT) and intensity (−25.5% for both). Since no acceptor materials were used in the pristine P3HT, evidence was clear that no charge transfer occurred from P3HT to nr–TiO2. Therefore, the rising dynamics with comparable timescales and intensities in both pristine P3HT and physically mixed samples indicated that there was no charge transfer in the physically mixed samples either. Then in such a system, only collisional quenching can take place with insignificant PL quenching. This is in good agreement with the previous observation by Beek et al. that collisional quenching in the absence of Förster energy transfer and charge transfer is an insignificant process in a physical mixture of TiO2 nanoparticles and an aldehyde-functionalized terthiophene.8
 | ||
| Fig. 5 Time-resolved fluorescence dynamics of pristine P3HT (pink), P3HT/nr–TiO2 physical mixture (green), and chemically linked P3HT–nr–TiO2 nanocomposite (magenta). These curves were de-convoluted with a sum of multiple exponential functions. | ||
| Sample | τ 1(ps) | A1 (%) | τ 2(ps) | A2 (%) | τ 3 (ps) | A3 (%) |
|---|---|---|---|---|---|---|
| Pure P3HT | 0.6 | −25.5 | 9.1 | 29.2 | 950 | 45,3 |
| P3HT/LA–nr–TiO2 physical mixture | 0.5 | −25.5 | 10.1 | 29.2 | 950 | 45.3 |
| Chemically linked P3HT–Si–nr–TiO2composite | 0.75 | 16.8 | 19 | 47.3 | 980 | 35.9 |
The energetically downhill EET was reported to take place from less than 1 ps to several ps, which is much faster than EET between segments with comparable energy that happens at a much slower timescale of ∼10—100 ps.38,39,41,43 However, conjugated polymer chains typically have torsional defects at nonplanar equilibrium locations in the ground state, which can inhibit exciton migration and thus affects charge mobility.31,47 Darling reported that density functional theory (DFT) calculation can be used to isolate the effects of torsional defects on the delocalization of frontier molecule orbitals as well as bandgaps in thiopehenes oligomers.48 Conformational defects can be reduced after torsional relaxation that typically happens at timescale from several ps to a few tens of ps.38,39 Nakamura et al. reported that the torsional relaxation took place in a time scale of ∼3–10 ps for thiophene oligomers.38 Westenhoff et al. reported that torsional relaxation occurred at a time scale of 15 ps.31 The τ2 processes of pristine P3HT, physical mixture, and chemically linked sample were observed to occur in a timescale of 9.1 ps, 10.1 ps, and 19 ps, respectively. Therefore, the τ2 process can be assigned to torsional relaxation that has been reported in such timescale by others and our own previous work.31,38,46,47 Torsional relaxation in pristine P3HT solution was found to occur at a timescale of 9.1 ps with an intensity of 29.2%, which are comparable those (10.1 ps and 29.2%) from physically mixed sample as shown in Table 1. This indicated that physical addition of LA-capped nr–TiO2 into P3HT solution had little effect on torsional relaxation in P3HT. However, the chemically linked sample exhibited significantly increased decay timescale (19 ps) and amplitude (48%) compared to those (∼9–10 ps and ∼29.2%) in pristine P3HT and physical mixture. The longer torsional relaxation time and larger amplitude suggested that P3HT chains in the chemically linked P3HT are more twisted with more torsional defects. This further supported that the chemically linked P3HT has a more coil-like conformation that leads to a more difficult process for planarization in excited state. Therefore, such torsional relaxation is expected to take a longer decay time in chemically linked P3HT.
The τ3 process was observed to have a comparable time constant of 0.95 ns for pristine P3HT and physically mixed P3HT/LA–nr–TiO2 solutions and 0.98 ns for the chemically linked P3HT–Si–nr–TiO2 sample. Previous work by Kraabel et al. has reported that intersystem crossing (ISC) occurred at a timescale of ∼1 ns in solutions of regiorandom poly(3-octylthiophene) (P3OT), regioregular P3HT, and thiophene oligomers.49 Since the timescale in this work was close to 1 ns, the τ3 process can also be assigned to triplet formation via ISC from singlet state (S1) to triplet state (T1) in P3HT. It is important to mention that the τ3 process is less likely to be assigned to PL decay (relaxation) of singlet state to ground state because this process was previously reported to occur in a timescale of several hundreds of ps.38,39,49,50 Although the chemically linked P3HT had a more coil-like conformation with more twisted defects than pristine P3HT and P3HT/nr–TiO2 physical mixture, the ISC time constant did not show any dependance on polymer conformation disorders such as torsional defects. This is in good agreement with previous reports from Kraabel et al. who found that ISC depends mainly on strong spin–orbit interaction induced by sulfur in P3HT, but not on chain conformational defects, chain ends, or effects caused by regularities of side chains.49
4. Conclusion
Energy migration in P3HT that is related to the polymer chain conformation, and charge transfer from P3HT to nr–TiO2 in the chemically linked P3HT–Si–nr–TiO2 solution were investigated compared with pristine P3HT and physically mixed P3HT/LA–nr–TiO2 solution. Charge transfer from P3HT to nr–TiO2 was found to occur evidenced by PL quenching and an initial decay time constant (τ1) of 0.75 ps in the chemically linked solution, while no charge transfer was observed in physically mixed solution with a initial fast rise process (τ1) that is almost the same as pristine P3HT. This can further be confirmed by the fact that no PL quenching was observed. In addition, the blue shift phenomenon found in the absorbance and PL spectra, structureless PL spectra and larger Stokes shift indicated that P3HT formed a more coil-like conformation in the chemically linked sample. Such a more coil-like conformation can induce twisting and/or bending along polymer backbone that leads to reduced effective conjugation length of polymer segments. Therefore, torsional relaxation was found to occur at a longer decay time with a stronger amplitude. Moreover, intersystem crossing (ISC) from singlet state (S1) to triplet state (T1) in P3HT of the three samples was all found to occur in a timescale of ∼1 ns and showed no dependence on conformational disorders such as torsional defects.Acknowledgements
The project was partially supported by NSF CAREER (ECCS-0950731), ACS Petroleum Research Funds DNI (48733DNI10), NSF EPSCoR (EPS-EPSCoR-0903804), NASA EPSCoR (NNX09AP67A), US-Israel Binational Science Foundation (2008265), and US-Egypt Joint Science & Technology Funds (913), Research Corporation (Cottrell College Science Award - CC6960), and South Dakota Center for Research and Development of Light-activated Materials. Research at the Oak Ridge National Laboratory's Center for Nanophase Materials Sciences and SHARE User Facility was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U.S. Department of Energy. The authors thank Dr X. Z. Yan for helping to collect the time-resolved fluorescence data.References
- V. L. Colvin, M. C. Schlamp and A. P. Alivisatos, Nature, 1994, 370, 354–357 CrossRef CAS.
- S. Coe, W.-K. Woo, M. Bawendi and V. Bulovic, Nature, 2002, 420, 800–803 CrossRef CAS.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef CAS.
- J. Bouclé, P. Ravirajan and J. Nelson, J. Mater. Chem., 2007, 17, 3141–3153 RSC.
- D. J. Milliron, A. P. Alivisatos, C. Pitois, C. Edder and J. M. J. Frechet, Adv. Mater., 2003, 15, 58–61 CrossRef CAS.
- N. C. Greenham, X. Peng and A. P. Alivisatos, Phys. Rev. B: Condens. Matter, 1996, 54, 17628–17637 CrossRef CAS.
- W. J. E. Beek and R. A. J. Janssen, Adv. Funct. Mater., 2002, 12, 519–525 CrossRef CAS.
- W. J. E. Beek and R. A. J. Janssen, J. Mater. Chem., 2004, 14, 2795–2800 RSC.
- Y. Zhang, C. Wang, L. Rothberg and M.-K. Ng, J. Mater. Chem., 2006, 16, 3721–3725 RSC.
- M. D. Goodman, J. Xu, J. Wang and Z. Lin, Chem. Mater., 2009, 21, 934–938 CrossRef CAS.
- S. L. Lu, S. S. Sun, X. X. Jiang, J. W. Mao, T. H. Li and K. X. Wan, J. Mater. Sci.: Mater. Electron., 2010, 21, 682–686 CrossRef CAS.
- S. Tepavcevic, S. B. Darling, N. M. Dimitrijevic, T. Rajh and S. J. Sibener, Small, 2009, 5, 1776–1783 CrossRef CAS.
- X. L. Li, Q. Peng, J. X. Yi, X. Wang and Y. D. Li, Chem.–Eur. J., 2006, 12, 2383–2391 CrossRef CAS.
- P. D. Cozzoli, A. Kornowski and H. Weller, J. Am. Chem. Soc., 2003, 125, 14539–14548 CrossRef CAS.
- C.-H. Chang, T.-K. Huang, Y.-T. Lin, Y.-Y. Lin, C.-W. Chen, T.-H. Chu and W.-F. Su, J. Mater. Chem., 2008, 18, 2201–2207 RSC.
- J. Liu, W. Wang, H. Yu, Z. Wu, J. Peng and Y. Cao, Sol. Energy Mater. Sol. Cells, 2008, 92, 1403–1409 CrossRef CAS.
- M. G. Manera, P. D. Cozzoli, M. L. Curri, G. Leo, R. Rella, A. Agostiano and L. Vasanelli, Synth. Met., 2005, 148, 25–29 CrossRef CAS.
- D. Appelhans, D. Ferse, H. J. P. Adler, W. Plieth, A. Fikus, K. Grundke, F. J. Schmitt, T. Bayer and B. Adolphi, Colloids Surf., A, 2000, 161, 203–212 CrossRef CAS.
- M.-D. Lu and S.-M. Yang, J. Colloid Interface Sci., 2009, 333, 128–134 CrossRef CAS.
- Y.-Y. Lin, C.-W. Chen, T.-H. Chu, W.-F. Su, C.-C. Lin, C.-H. Ku, J.-J. Wu and C.-H. Chen, J. Mater. Chem., 2007, 17, 4571–4576 RSC.
- P. J. Thistlethwaite and M. S. Hook, Langmuir, 2000, 16, 4993–4998 CrossRef CAS.
- M. Nara, H. Torii and M. Tasumi, J. Phys. Chem., 1996, 100, 19812–19817 CrossRef CAS.
- J.-S. Ji, Y.-J. Lin, H.-P. Lu, L. Wang and S.-P. Rwei, Thin Solid Films, 2006, 511-512, 182–186 CrossRef CAS.
- L. Ye, R. Pelton and M. A. Brook, Langmuir, 2007, 23, 5630–5637 CrossRef CAS.
- Z. Shi, G. Xueping, S. Deying, Y. Zhou and D. Yan, Polymer, 2007, 48, 7516–7522 CrossRef CAS.
- Y.-M. Chang, W.-F. Su and L. Wang, Macromol. Rapid Commun., 2008, 29, 1303–1308 CrossRef CAS.
- P. J. Brown, D. S. Thomas, A. Köhler, J. S. Wilson, J.-S. Kim, C. M. Ramsdale, H. Sirringhaus and R. H. Friend, Phys. Rev. B: Condens. Matter, 2003, 67, 064203 CrossRef.
- K. M. Coakley and M. D. McGehee, Chem. Mater., 2004, 16, 4533–4542 CrossRef CAS.
- S. Tretiak, A. Saxena, R. L. Martin and A. R. Bishop, Phys. Rev. Lett., 2002, 89, 097402 CrossRef CAS.
- S. Westenhoff, W. J. D. Beenken, R. H. Friend, N. C. Greenham, A. Yartsev and V. Sundstrom, Phys. Rev. Lett., 2006, 97, 166804 CrossRef.
- S. T. MILNER, Science, 1991, 251, 905–914 CAS.
- H. J. Snaith, G. L. Whiting, B. Sun, N. C. Greenham, W. T. S. Huck and R. H. Friend, Nano Lett., 2005, 5, 1653–1657 CrossRef CAS.
- J. H. Warner, A. R. Watt, E. Thomsen, N. Heckenberg, P. Meredith and H. Rubinsztein-Dunlop, J. Phys. Chem. B, 2005, 109, 9001–9005 CrossRef CAS.
- T. Xu and Q. Qiao, Energy Environ. Sci., 2011, 4, 2700–2720 CAS.
- M. Siddiki, J. Li, D. Galipeau and Q. Qiao, Energy Environ. Sci., 2010, 3, 867–883 CAS.
- I. Botiz, R. D. Schaller, R. Verduzco and S. B. Darling, J. Phys. Chem. C, 2011, 115, 9260–9266 CAS.
- T. Nakamura, Y. Araki, O. Ito, K. Takimiya and T. Otsubo, J. Phys. Chem. A, 2008, 112, 1125–1132 CrossRef CAS.
- Y. Xie, Y. Li, L. Xiao, Q. Qiao, R. Dhakal, Z. Zhang, Q. Gong, D. Galipeau and X. Yan, J. Phys. Chem. C, 2010, 114, 14590–14600 CAS.
- W. J. D. Beenken and T. Pullerits, J. Phys. Chem. B, 2004, 108, 6164–6169 CrossRef CAS.
- N. P. Wells, B. W. Boudouris, M. A. Hillmyer and D. A. Blank, J. Phys. Chem. C, 2007, 111, 15404–15414 CAS.
- T. Kobayashi, M. Yoshizawa, U. Stamm, M. Taiji and M. Hasegawa, J. Opt. Soc. Am. B, 1990, 7, 1558–1578 CrossRef CAS.
- I. G. Scheblykin, A. Yartsev, T. Pullerits, V. Gulbinas and V. Sundström, J. Phys. Chem. B, 2007, 111, 6303–6321 CrossRef CAS.
- S. Westenhoff, C. Daniel, R. H. Friend, C. Silva, V. Sundstrom and A. Yartsev, J. Chem. Phys., 2005, 122, 094903 CrossRef.
- X. Yang, T. E. Dykstra and G. D. Scholes, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 045203 CrossRef.
- J. Li, M. Yan, Y. Xie and Q. Qiao, Energy Environ. Sci., 2011 10.1039/C1031EE02055B.
- F. C. Grozema, P. T. van Duijnen, Y. A. Berlin, M. A. Ratner and L. D. A. Siebbeles, J. Phys. Chem. B, 2002, 106, 7791–7795 CrossRef CAS.
- S. B. Darling, J. Phys. Chem. B, 2008, 112, 8891–8895 CrossRef CAS.
- B. Kraabel, D. Moses and A. J. Heeger, J. Chem. Phys., 1995, 103, 5102–5108 CrossRef CAS.
- J. Guo, H. Ohkita, H. Benten and S. Ito, J. Am. Chem. Soc., 2009, 131, 16869–16880 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/ c1ra00739d/ |
| This journal is © The Royal Society of Chemistry 2012 |