DOI:
10.1039/C1RA00544H
(Paper)
RSC Adv., 2012,
2, 863-869
Chemical reactivity of hybrid particles
Received
1st August 2011
, Accepted 2nd October 2011
First published on 22nd November 2011
Abstract
Considerable effort has been invested in the development of various methods for the synthesis of hybrid organic/inorganic particles for a variety of applications. Not enough attention has been given to the use of such particles as starting reagents for chemical reactivity studies and for further chemical modifications. The complexity of such reactivity, dictated by the architecture and composition of the hybrid particles, is demonstrated here for the addition reactions of iodine and bromine with linear polyisoprene@silica and cross-linked polyisoprene@silica sub-micron particles, in both water (hydrohalogenations) and CCl4 (halogen additions). The reactivities of these new particles were comparatively studied, and correlated to the differences between the two types of particles as revealed by electron microscopy, zeta potential measurements, thermal gravimetric analysis, surface area measurements, non-reactive intra-particle diffusion, and more.
Introduction
Much attention has been given to the development of synthetic routes to hybrid materials1–3 and to particles of such materials,4–6 composed of an organic and an inorganic component, and to their physical characterization. Quite neglected, however, has been the chemical-reactivity characterization of hybrid particles, that is, the recognition that such particles can serve as substrates for further reaction, and as chemical reagents. To gain some insight into such chemical reactivity properties, we have investigated the double-bond addition reactions of iodine and bromine with linear polyisoprene (LPI)@silica and cross-linked polyisoprene (XPI)@silica sub-micron particles, in both water (hydrohalogenations) and CCl4 (halogen additions). These new particles, synthesized by our recently developed general method for the preparation of particulate polymer@silica particles,7–13 represent both semi- and full-IPN (interpenetrating network) hybrids, thus allowing the comparison of the effects of differences in the structural parameters on the reactivity. The main message of this report is that by no means can one directly extrapolate from solution chemistry to the complex situation of a hybrid material undergoing the same chemical reaction. For instance, we found that a hydrohalogenation reaction changes the hydrophobicity/hydrophilicity balance of the material while undergoing the reaction, thus accelerating in a positive feedback-loop way the ease with which further aqueous solution of the halogen diffuses into the particle; more observations are described below.
Experimental details
Chemicals
cis-Polyisoprene (PI), Mw: 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, tetraethylorthosilicate (TEOS) 98%, solid iodine, iodine water solution 0.1 N, bromine and potassium tetrachloro-platinate(II), 98% were purchased from Sigma-Aldrich. Caesium chloride, 99+% and the surfactant, triton X-100, were from Acros Organics. The cross-linker bis(3,5,5-trimethylhexanoyl) peroxide (THP) (dissolved in isododecane, 75% by weight) was donated by M. Shuster, Carmel-Olefins, Haifa.
000, tetraethylorthosilicate (TEOS) 98%, solid iodine, iodine water solution 0.1 N, bromine and potassium tetrachloro-platinate(II), 98% were purchased from Sigma-Aldrich. Caesium chloride, 99+% and the surfactant, triton X-100, were from Acros Organics. The cross-linker bis(3,5,5-trimethylhexanoyl) peroxide (THP) (dissolved in isododecane, 75% by weight) was donated by M. Shuster, Carmel-Olefins, Haifa.
Synthesis of polyisoprene@silica (LPI@silica)
A hydrophobic solution consisting of 0.2 g PI, 5.0 ml toluene and 4.0 ml TEOS was placed in an ultrasonic bath for 5 min until full dissolution of PI was achieved. The hydrophobic solution was poured, with stirring, into a hydrophilic solution consisting of 100 ml ethanol, 20 ml ammonium hydroxide (25% w/w), and 1.2 g triton X-100. A white emulsion was formed immediately. The emulsion was stirred for 24 h. The formed particles, which were collected by centrifugation of the suspension and air dried (for 24 h), contained 23% by weight of entrapped PI (from TGA—see below).
Synthesis of cross-linked polyisoprene@silica (XPI@silica)
The compositions of the hydrophobic and the hydrophilic phases remained the same as before, with the exception that 0.9 ml of cross-linking agent, THP, was added to the polymer solution. The hydrophobic solution was poured into the hydrophilic solution and the combined solution was kept at room temperature with stirring and under an argon atmosphere for 10 min during which the emulsion formed. Heating was then turned on and the emulsion was refluxed (78 °C) for 1.5 h. Stirring at room temperature continued for 24 h, after which collection of the particles was done as above; TGA showed 21% by weight of the PI component.
Common to all eight reactions, the bromination, the iodination, the hydrobromination and the hydroiodination, all performed on either LPI@silica or XPI@silica was the following procedure. The composite powder, 75 mg, containing 2.2 × 10−4 moles of double bonds, was pressed into a pellet under a pressure of 10 atm (pellet form is needed to avoid the powder re-dispersing in solution and thus interfering with the analysis). The composite pellet (either LPI@silica or XPI@silica) was placed in a quartz UV-Vis 1.0 cm cuvette and fitted in a UV-Vis spectrophotometer. Then, 4.0 ml of the reagent solution (Table 1) was transferred into the cuvette and the absorption was measured every 5 s for 30 min. A blank procedure was carried out using a composite pellet of the same weight but with 4.0 ml of the pure solvent. See Table 1 for additional details. The amount of iodine consisted of 1% (mole/mole) of the number of double bonds of the composite in order to increase the sensitivity of differences in the reaction profiles between LPI@silica and XPI@silica.
|
|
Hydroiodination |
Hydrobromination |
Iodination |
Bromination |
| Concentration/mM |
0.5 |
5 |
0.5 |
5 |
| Solvent |
Water |
Water |
CCl4 |
CCl4 |
| Absorption wavelength/nm |
350 |
398 |
516 |
398 |
| Initial number of moles of reagent |
2 × 10−6 |
1.8 × 10−5 |
2 × 10−6 |
1.8 × 10−5 |
| Number of moles of double bonds reacted after 1800 s |
LPI@silica |
1.4 × 10−6 |
7.4 × 10−6 |
3.4 × 10−7 |
1.5 × 10−5 |
| XPI@silica |
4.0 × 10−7 |
4.7 × 10−6 |
3.5 × 10−7 |
1.4 × 10−5 |
Diffusion of heavy atom compounds into the composites without reaction
Platinum complex diffusion.
4.0 ml of potassium tetrachloro-platinate(II) aqueous solution (40 mM) were poured into UV-vis cuvette in which was 22 mg of XPI@silica and (separate vessel) LPI@silica. Diffusion was tracked by taking absorption measurements at 427 nm every 5 s for 30 min.
Particle characterization and instrumentation
Particle size analysis.
The diameter of at least 150 particles was determined using Analysis software equipped in the HR-SEM. The statistical analysis was conducted using Origin software.
Thermal gravimetric analyses (TGA).
Carried out with an SDTA 851e (Mettler Toledo) apparatus under air at a rate of 10 °C min−1 using STARe software.
Zeta-potential.
Determined using Zetasizer Nanoseries by Malvern Instruments: the precipitate obtained by centrifugation of the emulsion was re-dispersed in 10 mM NaCl and injected into a cuvette.
Results and discussion
Synthesis, and structural and physical properties of the hybrid particles
Synthesis.
The combination between silica and different types of rubber, including PI, is very common and used mainly for reinforcement of the rubber with silica fillers, especially in the tire industry.14 In particulate systems, core-corona morphology was fabricated by grafting PI onto silica particle surfaces by photo15 or anionic16 polymerizations. The XPI@silica particles synthesized for the purpose of this study are of IPN (interpenetrating network) structure, which has been unknown both in the particulate and bulk forms. The fabrication of XPI@silica composite particles and of the semi-IPN LPI@silica particles was achieved by an O/W emulsion process in which polycondensating TEOS within the micelle entraps the polymer, with or without its cross-linking; see earlier reports for a detailed discussion of this synthetic approach.8,10
SEM.
SEM observations of the LPI@silica particles reveal a homogenous, narrow size distribution of sub-micron particles (the particle average diameter is 485 ± 9 nm) with smooth surface morphology (Fig. 1, left). However, the intra-particle cross-linking of PI resulted in a unique surface grainy morphology (Fig. 1, right) of smaller particles (410 ± 12 nm). It should be noted that the grainy morphology of XPI@silica was not seen in any of the previously reported polymer@silica composite particles.7–13 In order to establish the chemical nature of the grains and to determine whether they are also present on the interior of the particles, the particles were microtomically-cut and subsequently were observed with a TEM equipped with an EELS analyzer.
 |
| | Fig. 1 SEM pictures of linear (left, bar: 1 μm) and cross-linked (right, bar: 500 nm) polyisoprene@silica particles. | |
TEM and EELS.
TEM observations of the microtomically-cut particles reveal that the grains are presented all throughout the particles and not merely on their surface (Fig. 2a). Chemical mapping using EELS indicated that the grains consist of silica, as can be easily seen from Fig. 2b–d. For the carbon mapping (Fig. 2b), the grains are dark colored while for silicon and oxygen (Fig. 2c and d, respectively) they are bright. The fact that the silica lumps are seen in XPI@silica indicates that the cross-linking has induced a change in the structure of the hybrid particles, to which we return below. The proposition, which will be corroborated with further analyses, is that in XPI@silica the organic and inorganic phases are more distinctly separated throughout the particle, namely that the cross-linked polymer is more cluttered and compact due to the inter-chain bonding. The lower average particle size of XPI@silica (410 nm) compared to LPI@silica (485 nm) is in keeping with the picture of the higher compactness of XPI.
Zeta potential measurements.
The negative zeta potential values, −66 (±7) and −39 (±5) mV for LPI@silica and XPI@silica, respectively, show that the surfaces of these particles are very rich in silanols. In fact it is practically silica17 in the case of LPI@silica, but the less negative value for XPI@silica indicates partial coverage of the surface with polymer chains.
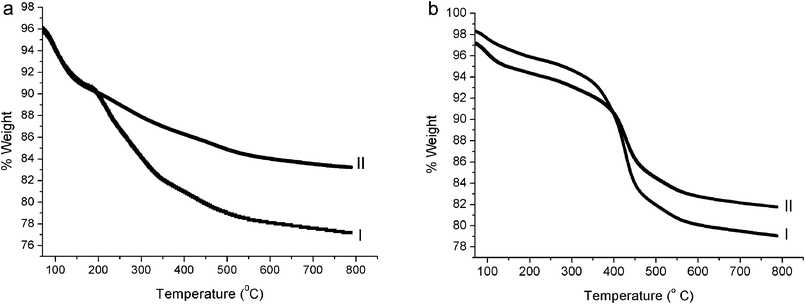
Extraction experiments and TGA.
TGA analyses before and after extraction of the polymers indicate similar behavior for the two types of hybrids: lines I in Fig. 3a and 3b show that the initial amounts of PI are similar: 23% for LPI@silica and 21% for XPI@silica. Extraction of the polymer leaves 17% organic content in LPI@silica and 18% in XPI@silica (lines II in Fig. 3a and b). This observation indicates that the entanglement of the organic component is quite pronounced for both LPI and XPI. The difference between the two types of particles is revealed by examining the particle size after a polymer extraction experiment: for LPI@silica there is a decrease in particle size to 460 (±12) nm, which is an expected result of the removal of some of the organic content from the particle. However, for XPI@silica, the extraction experiment results in an increased diameter, to 435 (±14) nm. Such solvent-induced swelling is the most typical characteristic of a cross-linked polymer, thus providing further evidence for the cross-linking of PI in the particles and for their full-IPN nature.
Surface area and porosity.
Differences between the two types of particles are also evident in surface area and porosity analyses, which are summarized in Table 2. It is seen that while LPI@silica has a surface area value which is characteristic of Stöber-type silica particles,18 XPI@silica particles have a much higher surface area. Similarly, the pore volume for XPI@silica is 20 times higher compared to LPI@silica. This is yet another indication of the compactness of the PI in the cross-linked case, which separates domains of this phase from the silica phase, increasing significantly the access to the porous silica surface and pores. Adsorption by diffusion into the pore-network of tetrachloro-platinate from water (Fig. 4) nicely demonstrated this difference: water, carrying with it the solvated complex, easily penetrates the pore network, allowing its adsorption on the internal domains of the exposed silica. The surface area and porosity values after the partial extraction are (Table 2), as expected, higher, but still keep the trend observed before this process. Finally, accessibility of the whole pore network (needed for the next section) was also ensured by an EELS follow up of the diffusion of a heavy atom, Cs, into that pore network. Fig. 5 indeed confirms that accessibility.
Table 2 Surface area and porosity for LPI@silica and XPI@silica
| |

| (1) |
 |
| | Fig. 4 Kinetics of tetrachloro-platinate diffusion into linear (L) and cross-linked (X) polyisoprene@silica particles. | |
 |
| | Fig. 5 EELS chemical mapping for caesium cation penetration into an XPI@silica particle. (b) EELS profile. | |
Reactivity
A main purpose of this work was to examine how particulate composite materials undergo chemical reactions. The cross-linking of PI takes place by a radical mechanism shown in Scheme 1. In our case, the reaction takes place inside the forming hybrid particle, in parallel to the polycondensation of TEOS and in parallel to the entrapment of PI. To ensure that the cross-linker breaks into radicals only after the initial structure of silica has formed, heating was set for 10 min after the emulsion formed. In addition, since the cross-linker was added a priori to the hydrophobic phase, the radicals formed in the vicinity of the linear PI.
Since polyisoprene has double bonds on its backbone, it can undergo addition reactions. Specifically we selected eight reactions: iodination, hydroiodination, bromination and hydrobromination, each performed on either LPI@silica or on XPI@silica (eqn (1)); the halogenations are carried out in a hydrophobic solvent, CCl4, and the hydrohalogenations in water. The progress of the reactions was followed by monitoring iodine or bromine spectroscopically, and the results are shown in Fig. 6 and 7. The overall observation is that the entrapped polymer is available for reaction with an external reagent in all cases. Iodination of both (LPI and XPI) hybrid particles in CCl4 (carried out with 1% mol/mol of iodine available for reaction with double bonds) occurs with a similar rate (Fig. 6a), and after 1800 s, 17.5% of the initial iodine has reacted, amounting to reaction with 0.16% of the double bonds. Differences between the reactivities of the two hybrid particles are seen for the hydroiodination (Fig. 6b): iodine reacts much faster with LPI@silica particles than with XPI@silica particles. For the XPI@silica particles 20% of the iodine has reacted (amounting to reaction with 0.18% of the double bonds), but the reaction with LPI@silica proceeds with 70% of the iodine (namely, with 0.63% of the double bonds). The fact that the hydroiodinations proceed faster than the iodinations, indicates that the wetting of the interior of the particle with water is easier than with CCl4, namely that the accessible interior surface area is mainly hydrophilic. A visual support for this statement is obtained from the color changes: iodine in aqueous solution is orange, while in CCl4 it is purple.
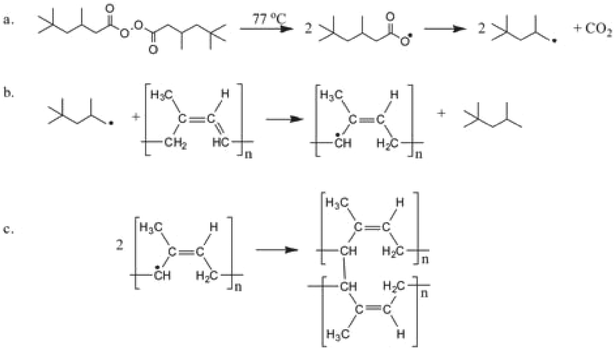 |
| | Scheme 1 Polyisoprene cross-linking reaction mechanism: a. Cleavage of 3,5,5-trimethylhexanoyl peroxide into radicals. b. Hydrogen subtraction from the polymeric chain. c. Cross linking. | |
 |
| | Fig. 6 Reaction kinetics for the reaction of iodine with linear (L) and cross-linked (X) polyisoprene@silica: (a) iodination: I2–CCl4, (b) hydroiodination: I2–H2O. | |
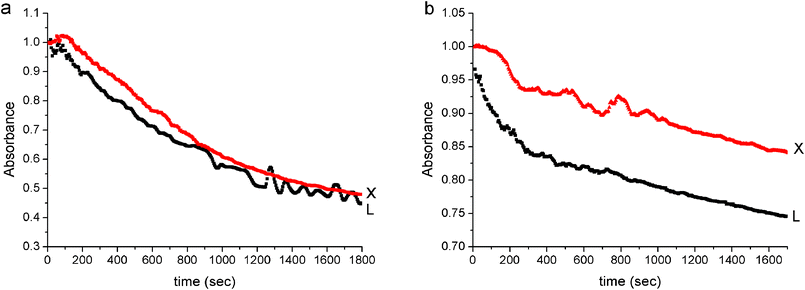 |
| | Fig. 7 Reaction kinetics for the reaction of bromine with linear (L) and cross-linked (X) polyisoprene@silica: (a) bromination: Br2–CCl4, (b) hydrobromination: Br2–H2O. | |
When reacting the composite pellet with a purple solution of iodine, the pellet is colored orange, indicating a hydrophilic nature of the composites (both LPI and XPI, Fig. 8). It is therefore a puzzle, why LPI@silica reacts faster than XPI@silica, when the surface area of the latter is larger. We attribute it to two parameters: first, in the cross-linked polymer, accessibility of the double bonds is lower because of the greater compactness of this phase compared to the linear polymer, a difference we already detected in the previous analyses described above.
 |
| | Fig. 8 Color visual support for the hydrophilic nature of the composite particles. (a) The pressed XPI@silica pellet before reaction (white). (b) Reaction in a purple iodine solution in CCl4: the pellet color changes to orange (hydrophilic environment). The diffusing purple front is clearly seen. (c) Reaction completed. Removal of iodine solution: the whole pellet turns orange. | |
The second parameter which operates on LPI@silica is interesting and unique to the hybrid situation: as the reaction progresses, the hydrophobic double-bonds are hydroxylated and become much more hydrophilic. This eases the diffusion and penetration of the aqueous solution of iodine, to perform further reaction. It is a positive feedback loop that eases the reaction as it progresses. The brominations and hydrobrominations show similar patterns of behavior (Fig. 7, Table 1), and for sake of brevity we do not repeat the description. However, it is relevant to indicate that after 9 months, full penetration of the halogen is observed (Fig. 9).
 |
| | Fig. 9 EELS mapping of Br after reaction with (a) LPI@silica, (b) XPI@silica, (microtomically cut; the stripes are due to the cut). | |
Conclusions and further discussion
LPI@silica sub-micrometre hybrid particles were fabricated by the entrapment of PI in polycondensating TEOS in surfactant-stabilized O/W micelles. Cross-linking of PI within the forming particles resulted in XPI@silica particles. It was found that cross-linking of polyisoprene increases to some extent the separation between the two phases within the particles. This phenomenon was evident from the morphology differences and from particle size and surface area measurements. Thus, the surface area obtained for XPI@silica particles was almost nine times higher than that of LPI@silica particles, indicating phase separation caused by the cross-linking, making the silica pores much more accessible; hydrophilic silica chains forming in parallel to the cross-linking of PI cannot entangle within the hydrophobic PI “islands” and so some separation forms between silica and XPI.
In order to gain some insight into the origin of the difference between the LPI and the XPI effects on the hybrid particles, we draw attention to a lot of NMR19–22 as well as IR23 evidence that the first steps of the sol–gel process involve the formation of small siloxane rings containing up to six or eight siloxane bonds. We propose here that the ring-formation is important in general to the understanding of the entrapment of organic molecules in sol–gel matrices, both small molecules and polymers, by forming rotaxane-type early stage interlocked complexes; this type of molecular association is well known.24–25Fig. 10 presents the proposition for an 8-siloxane ring and a short linear PI 5-oligomer, where the isoprene chain easily inserts into the siloxane ring (Fig. 10c). Furthermore, a single LPI chain might be held along its length by several such rings, thus contributing to the homogeneous dispersion of these chains. On the other hand, the model suggests that cross-linking renders PI too bulky for that process, so that ring-entrapment exists to a lesser degree for this case. This suggests that the experimentally observed difference between the homogeneously distributed LPI and the phase separated XPI, may also be due in part to the TEOS ring-polymerization component of its polycondensation, which helps in separating LPI chains from each other.
 |
| | Fig. 10 Suggested model for a possible contribution to the first stages of PI entrapment within the silica matrix. (a) Siloxane ring; (b) a linear isoprene oligomer; (c) a rotaxane of PI-oligomer–siloxane ring, leading to LPI@silica. Color legend: red – oxygen, yellow – silicon, and grey – carbon. Modeled with GaussView® software. | |
The chemical reactivity of the composite particles was studied using addition reactions of external reagents, iodine or bromine. It was found that in both LPI@silica and XPI@silica particles, the organic polymer is accessible for reaction with the external reagent. Moreover, in a hydrophilic solvent, water, LPI@silica particles reacted much faster and with larger amounts of external reagent than XPI@silica particles. This outcome demonstrates the special case of reactivity in hybrid particles: although PI is a hydrophobic polymer, the hybrid particle as a whole has a hydrophilic nature. In addition, although XPI@silica particles have a higher surface area, the entrapped polymer is less accessible for reaction. These observations indicate that the path of the reaction is dictated, first, by the wettability of the intrapore surface of the particle and by the unique phenomenon that as the hydrohalogenation reaction progresses, the entrapped polymer becomes more hydrophilic—it is converted into a halohydrin polymer—that further eases the penetration of the external reagent.
Acknowledgements
Supported by the NOFAR program for applied research of the Israel Ministry of Trade and Industry. We thank Hadassah Elgavi for assistance in the modeling work.
References
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893 CrossRef CAS.
- L. Nicole, L. Rozes and C. Sanchez, Adv. Mater., 2010, 22, 3208 CrossRef CAS.
- A. Brethon, C. Bied, J. J. E. Moreau and M. Wong Chi Man, J. Sol-Gel Sci. Technol., 2009, 50, 141 CrossRef CAS.
-
E. Bourgeat-Lami, Hybrid Organic/Inorganic Particles, Hybrid Materials: Synthesis, Characterization, and Applications, ed. G. Kickelbick, Weinheim, Germany, 2007 Search PubMed.
- M. Yamada, Bull. Chem. Soc. Jpn., 2009, 82, 152 CrossRef CAS.
- G. Wegner, M. M. Demir, M. Faatz, K. Gorna, R. Munoz-Espi, B. Guillemet and F. Groehn, Macromol. Res., 2007, 15, 95 CrossRef CAS.
- H. Sertchook, H. Elimelech, C. Makarov, R. Khalfin, Y. Cohen, M. Shuster, F. Babonneau and D. Avnir, J. Am. Chem. Soc., 2007, 129, 98 CrossRef CAS.
- H. Elimelech, J.-M. Nedelec, A. Hardy-Dessources, F. Babonneau and D. Avnir, J. Mater. Chem., 2010, 20, 9515 RSC.
- H. Naor, M. Shuster and D. Avnir, J. Sol-Gel Sci. Technol., 2011, 59, 194 CrossRef CAS.
- H. Sertchook and D. Avnir, Chem. Mater., 2003, 15, 1690 CrossRef CAS.
- H. Sertchook, H. Elimelech and D. Avnir, Chem. Mater., 2005, 17, 4711 CrossRef CAS.
- H. Elimelech and D. Avnir, Chem. Mater., 2008, 20, 2224 CrossRef CAS.
- L. Zalzberg and D. Avnir, J. Sol-Gel Sci. Technol., 2008, 48, 47 CrossRef CAS.
-
M. S. Evans, Tyre Compounding for Improved Performance, 12, Rapra Technology Limited, UK, 2001 Search PubMed.
- D. Derouet and C. N. Ha Thuc, J. Rubber Res., 2008, 11, 78 CAS.
- M. L. C. M. Oosterling, A. Sein and J. Schouten, Polymer, 1992, 33, 4394 CrossRef CAS.
- P. Wilhelm and D. Stephan, J. Colloid Interface Sci., 2006, 293, 88 CrossRef CAS.
- A. Labrosse and A. Burneau, J. Non-Cryst. Solids, 1997, 221, 107 CrossRef CAS.
- F. Brunet and C. Cabane, J. Non-Cryst. Solids, 1993, 163, 211 CrossRef CAS.
- J. J. van Beek, D. Seykens, J. B. H. Jansen and R. D. Schuiling, J. Non-Cryst. Solids, 1991, 134, 14 CrossRef CAS.
-
(a) L. Voon Ng, P. Thompson, J. Sanchez, C. W. Macosko and A. V. McCormick, Macromolecules, 1995, 28, 6471 CrossRef;
(b) L. Voon and A. V. McCormick, J. Phys. Chem., 1996, 100, 12517 CrossRef;
(c) S. E. Rankin, C. W. Macosko and A. V. McCormick, Chem. Mater., 1998, 10, 2037 CrossRef CAS.
-
R. K. Iler, The Chemistry Of Silica, Wiley, New York, 1979 Search PubMed.
- A. Fidalgo and L. M. Ilharco, Chem.–Eur. J., 2004, 10, 392 CrossRef CAS.
- I. T. Harrison and S. Harrison, J. Am. Chem. Soc., 1967, 89, 5723 CrossRef CAS.
- F. Aricó, J. D. Badjic, S. J. Cantrill, A. H. Flood, K. C.-F. Leung, Y. Liu and J. F. Stoddart, Top. Curr. Chem., 2005, 249, 203 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.