Electrochemical oxidation of catechols in the presence of enaminone: exclusive α- arylation
Ni-Tao
Zhang
a,
Xiao-Guang
Gao
a,
Cheng-Chu
Zeng
*a,
Li-Ming
Hu
a,
Hong-Yu
Tian
b and
Yuan-Bin
She
c
aCollege of Life Science & Bioengineering, Beijing University of Technology, Beijing, 100124, China. E-mail: zengcc@bjut.edu.cn; Fax: 0086-10-67392001
bSchool of Food Chemistry, Beijing Technology & Business University, Beijing, 100048, China
cCollege of Environmental & Energy Engineering, Beijing University of Technology, Beijing, 100124, China
First published on 4th November 2011
Abstract
Anodic oxidation of catechols in the presence of enaminones serving as potential doubly nucleophiles is examined using cyclic voltammetry and preparative electrolysis methods. Selective α-arylation is observed, which is consistent with that from the chemical oxidation approach. The results demonstrate that formation of either indoles or α-arylated products is depended on the nature of the polarized enaminone.
1. Introduction
Electroorganic synthesis in aqueous medium combines the advantages of electroorganic synthesis1 and organic synthesis in water.2 Thus, using electroorganic synthetic techniques in aqueous medium to synthesize organic compounds possessing various biological activities or to achieve the transformation of one of the key steps in the total synthesis of a natural product will become more and more attractive.3The anodic oxidation of a catechol generates a reactive o-benzoquinone that can be used to trigger a number of interesting reactions.4–9 Tabakovic and collaborators first reported the electrochemical synthesis of benzofurans from catechol and 1,3-dicarbonyl compounds.5 Later, Nematollahi et al. carried out an extensive investigation of the anodic oxidation of catechols in different mono-nucleophiles or doubly nucleophiles and proposed an ECEC (E = electrochemical and C = chemical step) mechanism for the electrochemical formation of benzofuran cycle.6 On the other hand, we have recently shown that the electrochemical oxidation of catechols in the presence of α-oxoheterocyclic keteneN,O-acetals could generate in one-pot polyhydroxylated fused indole derivatives in reasonable yields,8 whereas the anodic oxidation reaction stopped at the intermolecular Michael addition step which resulted in exclusive α-arylated products when α-oxoheterocyclic keteneN,N-acetals were used as the nucleophilic species.9 Obviously, the nature of the starting ketene acetals influences the formation of either indole or α-arylation (Scheme 1).

In order to probe how the nature of the starting dinucleophiles govern the pathways of α-arylation or indole formation, it is necessary to know what would occur if a simple enaminone is used as a potential dinucleophile, where the alkyloxy group of the keteneN,O-acetals is replaced by an alkyl group (Fig. 1). In this work, we reported our outcome on the anodic oxidation of catechol 1 in the presence of enaminones 2 serving as nucleophiles using cyclic voltammetry and preparative electrolysis methods (Fig. 1).
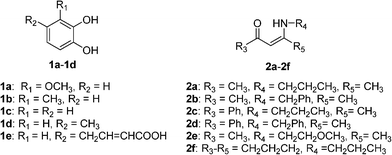 | ||
| Fig. 1 The structures of starting catechols 1 and enaminones 2. | ||
2. Results and discussion
2.1 Electrochemical investigation of catechols in the absence and presence of enaminones by cyclic voltammetry
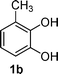
The electrochemical properties of the starting catechols were first studied. Taking 3-methylcatechol (1b) as an example, we investigated the electrochemical behaviour of catechol 1 by cyclic voltammetry (CV) in the absence and presence of enaminones 2 at room temperature in water containing 0.2 M acetate buffer (pH = 7.0) as the supporting electrolyte.As shown in Fig. 2 (curve a), upon scanning anodically, catechol 1b exhibits a well-defined quasi-reversible oxidation wave (peak A) at +0.57 V vs.Ag/AgCl (0.1 M KCl) and its corresponding cathodic peak (C) at +0.30 V. Peak A is attributed to the oxidation of the 3-methylcatechol to a cation radical that ultimately leads to the formation of an o-benzoquinone derivative and peak C to the reduction of the o-benzoquinone. The ratio of the current amplitudes between the oxidation and reduction processes is equal to unity (Ipox/Ipred), indicating that the o-benzoquinone produced at the surface of the electrode is stable in pH 7 acetate buffer solution. The side-reactions such as hydroxylation and dimerization reactions are too slow to be observed on the time scale of the cyclic voltammetry.6 Curve b of Fig. 2 is the CV of enaminone 2a; it shows one well-defined irreversible anodic wave at +1.27 V.
 | ||
Fig. 2
Cyclic voltammogram of a) 2 mM of 3-methylcatechol (1b), b) 2 mM of enaminone 2a, c) a mixture of 2 mM of 1b and 2 mM of 2a at a glassy carbon working electrode, platinum wire counter electrode and Ag/AgCl (0.1 M KCl) reference electrode, in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) acetate buffer solution/ acetonitrile (0.2 M, pH 7.0); scan rate: 100 mV s−1. :1 (v/v) acetate buffer solution/ acetonitrile (0.2 M, pH 7.0); scan rate: 100 mV s−1. | ||
When one equivalent amount of enaminone 2a was added to the solution of catechol 1b, the voltammogram of the mixture exhibits two anodic peaks A1 (+0.73 V vs.Ag/AgCl) and A2 (+1.05 V vs.Ag/AgCl), whereas the cathodic current disappears (curve c, Fig. 2). A1 is a catalytic current of the oxidation of 3-methylcatechol. Its shifted anodic peak has been attributed6 to a formation of a thin film of the product at the surface of the electrode, inhibiting (to a certain extent) the performance of the electrode process. The A2 is more like the oxidation peak of corresponding product. The observation that the cathodic current of the corresponding o-benzoquinone disappears indicates that the electrogenerated o-benzoquinone intermediate undergoes follow-up chemical reactions under these conditions.
2.2 Electrochemical oxidation of catechols in the presence of enaminones
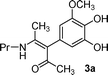
After examining the electrochemical properties of substituted catechols in the absence and presence of enaminones, we can conclude that selective oxidation of catechols, instead of enaminones, will occur if the potential is controlled at 0.5 V vs.Ag/AgCl (0.1 M KCl). In addition, due to a quite different potential gap between enaminones and catechols (1.27 V vs. 0.57 V), selective oxidation of catechols may also achieve by using constant current technique. In this work, constant current electrolysis technique was used to perform preparative scale of electrolyses.At the outset, 3-methoxycatechol (1a) was subjected to anodic oxidation in the presence of enaminones 2 (Scheme 2 and Table 1) under the optimized conditions developed earlier.9a Thus, in a pilot experiment, a solution of an equimolar quantity of 1a and 2a was electrolyzed at constant current of 15 mA (∼5 mA cm−2) at pH 7.0 acetate buffer solution. In the course of the reaction, the solution turned from yellow to brown. After 3-methoxycatechol was consumed (the charge passed was approximately 2.2 F/mol), the reaction mixture was worked up and product 3a was obtained in 39% yield (Table 1, Entry 1). The structure of compound 3a was assigned to be α-arylated product on the basis of its spectral and analytical data.
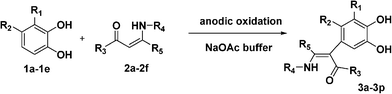 | ||
| Scheme 2 Electrochemical oxidation of catechols in the presence of enaminones 2. | ||
| Entry | Catechol 1 | Enaminone 2 | Product | Workup methodabc | Yield |
|---|---|---|---|---|---|
| a Method A: Column chromatograph was done to afford products 3a–3j. b Method B: After removal of partial supporting electrolyte solvent, a precipitate formed, which was recrystallized using mixed solvents of chloroform and acetone to get pure products 3p–3r. c Method C: A precipitate formed and high quality of products 3k–3n were obtained after recrystallization from methanol. | |||||
| 1 |

|

|

|
C | 39 |
| 2 | 1a |

|

|
C | 41 |
| 3 | 1a |

|

|
C | 44 |
| 4 | 1a |
|

|
C | 22 |
| 5 |

|
|
|
C | 27 |
| 6 | 1a |
|

|
C | 29 |
| 7 |
|
2a |

|
C | 37 |
| 8 | 1b | 2b |

|
C | 46 |
| 9 | 1b | 2c |

|
C | 33 |
| 10 |

|
2c |

|
C | 22 |
| 11 |

|
2a |

|
A | 67 |
| 12 | 1d | 2b |
|
A | 59 |
| 13 | 1d | 2c |
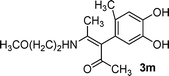
|
A | 69 |
| 14 | 1d | 2d |

|
A | 72 |
| 15 | 1d | 2e |

|
A | 63 |
| 16 |
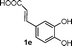
|
2a |

|
B | 52 |
| 17 | 1e | 2b |
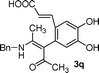
|
B | 39 |
| 18 | 1e | 2d |

|
B | 34 |
To elucidate how the enaminones influence the α-arylation, enaminones 2b and 2c, wherein the propyl group of 2a was replaced by a benzyl or a methoxyethyl, were employed to react with the electrochemically generated o-benzoquinone. Consistently, substrates 2b and 2c also afforded the corresponding α-arylated products 3b and 3c under the same reaction conditions, in yields of 41% and 44%, respectively (Table 1, Entries 2 and 3).
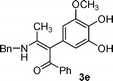
For enaminones 2a, 2b and 2c, the electro-withdrawing group is an acetyl group. To further investigate the effect of the electron withdrawing group on the reaction, benzoyl-substituted enamines 2d and 2e were synthesized and tested. As shown in Table 1, again, α-arylated products were afforded and adducts 3d and 3e were isolated in 22% and 27%, respectively (Entries 4 and 5).
The initially generated adducts 3 possess Z-geometry due to an intramolecular hydrogen bond between the NH group and the carbonyl group. In order to form indole cycle, enamino-to-imino isomerisation and imino-to-enamino isomerisation are required. Consequently, Z-configuration of the initially formed intermediate 3 is geometrically non-favored for the sequent annulation to form indole (Fig. 3).
 | ||
| Fig. 3 Preferable geometry for the formation of indole. | ||
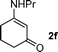
Clearly, either the presence of the Z-configuration of the intermediate 3 or the use of the compounds 2 types of enaminones stops the further intramolecular Michael addition. In order to separate the effect of the Z-configuration of the intermediate 3 on the formation of indole from that of the compounds 2 types of enaminones, E-configuration of 3-(propylamino)cyclohex-2-enone 2f was then synthesized.
The electrolysis of 1a in the presence of 2f was conducted using nearly identical conditions to those employed for the above oxidation of substrates 2. Once again, only α-arylated product 3f was isolated in 29% yield and the expected indole product was not detected.
The exclusive formation of α-arylated products indicated that this type of enaminone as dinucleophiles stopped at the α-arylated reaction step. Further electrochemical oxidation and intramolecular Michael addition could not proceed. To confirm this observation, the obtained 3a itself was used as starting material to undergo electrolysis. Consistently no corresponding indole was detected; only polymerization and decomposition were observed (Scheme 3).
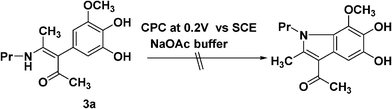 | ||
| Scheme 3 The anodic oxidation of 3a | ||
To evaluate the generality of the α-arylated reaction between catechols and enaminones under electrochemical conditions, other 3-substituted catechol or catechol itself were also investigated. For example, the anodic oxidation of the mixture of 3-methylcatechol (1b) and enaminone 2a, 2b or 2c gave corresponding 3g, 3h and 3i in respective 37%, 46% and 33% yields (Table 1, Entries 7, 8 and 9). In a similar way, the electrochemical oxidation of catechol 1c in the presence of 2c gave 3j in 22% yield (Table 1, Entry 10). Here, the major side reaction is polymerization of catechol itself.
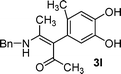
In addition to 3-substituted catechols, o-benzoquinones in situ generated from 4-substituted catechols, such as 4-methylcatechol were also treated with enaminones and the results are summarized in Table 1. For example, when 4-methylcatechol was electrolyzed in the presence of 2a, in the course of electrolysis, precipitate formed and the starting 4-methylcatechol was consumed when 2.2 F/mol current was passed. After simple filtration and washing, corresponding α-arylated products 3k was obtained in 67% yield (Table 1, Entry 11). Similar outcomes were also observed, where corresponding 3l–3o were obtained in 59–72% yields (Table 1, Entries 12–15). The yields from 4-methylcatechol are higher than that from other catechols. The actual reasons are not sure but the simple workup process, wherein the column chromatograph is not required, may be one of the reasons. Finally, caffeic acid 1e, bearing catechol subunit, was also investigated. As shown in Table 1, 34%–52% yields of corresponding α-arylated products 3p–3r were obtained (Entries 16–18).
The above results demonstrate that the reaction between electro-generated o-benzoquinone derivatives and enaminone 2 is quite similar to the behaviour of heterocyclic keteneN,N-acetals,9 but different from keteneN,O-acetals.8 These results also imply that the formation of either indoles or α-arylated product is depended on the nature of the polarized enamines when the electro-withdrawing group is carbonyl group. When an alkyloxy group is introduced at the α-position of the enamine skeleton (such as keteneN,O-acetal), sequent Michael addition may take place, in a contrast, it stops at the α-arylation step in the cases of keteneN,N-acetals or simple enaminones 2.
To further confirm that it is the nature of the starting polarized enaminone that determines α-arylation or indole formation, chemical oxidation procedure was used to repeat the reactions. In this work, PhI(OAc)2 was employed as an oxidant. Once again, it was observed that only α-arylated products were obtained when the mixture of 1 equiv. mole of a catechol and an enaminone was subjected to oxidation by 2.2 equiv. of PhI(OAc)2. As shown in Table 2, the reactions of catechol 1a, 1b and 1c with 2a and 2c also gave corresponding adducts in 40–52% of yields, which had a slightly higher yields compared with these from electrochemical method (Entries 1–5, Table 2). However, in the case of 1d, lower yields were obtained (Entries 6–7, Table 2).












These results further demonstrate that the oxidation of catechols in the presence of polarized enaminones stops at the α-arylation step or leads to the formation of indole depends on the nature of starting dinucleophiles. For enaminone, only α-arylated products formed under both electrochemical oxidation condition and chemical oxidant.
It is noteworthy that these polyhydroxylated compounds were quite difficult to separate by silica gel column chromatography because they adhered strongly to the chromatographic support.10 Therefore, somewhat lower yields of 3a–j were obtained when they were isolated using column chromatography, although almost quantitative of conversion was observed viaTLC in all cases.
Reaction mechanism
As the reaction mechanism is concerned, it is well documented6–7,9 that the anodic oxidation of catechol and its derivatives in aqueous medium leads to the formation of the corresponding o-benzoquinone intermediates. These intermediates are converted to other intermediates or products, following a pattern of an EC or an ECEC mechanism, depended on the nature of the nucleophiles and structures of the starting catechols. Accordingly, it is safe to speculate that the anodic oxidation of catechols in the presence of enaminones 2 to generate compounds 3 also follows the similar mechanism. As described in Scheme 4, the initial step is an electrochemical process that involves the oxidation of catechols 1 on the anodic electrode surface and generates the corresponding o-benzoquinones. Subsequently, a chemical reaction in the bulk electrolytic solution occurs, that is the active o-benzoquinone intermediate underwent a Michael addition reaction with the α-carbon of the enaminones 2 followed by the aromatization process leading to products 3. | ||
| Scheme 4 Proposed mechanism for the electrochemical synthesis of 3a–r. | ||
Conclusions
In a summary, to probe how the nature of doubly nucleophile governs the reaction pathways, enaminone 2 serving as potential dinucleophiles were synthesized and subjected to reaction with catechols under anodic oxidations. It was observed that α- arylation occurred exclusively for not only 4-substituted catechols, but also 3-substituted catechol and catechol itself. The electrochemical outcome was proved to be consistent with that from chemical oxidant, which further implies that the formation of either indoles or a-arylated product is depended on the nature of the enaminone. The present work extends the application of electrochemical synthesis of o-benzoquinone and its in situ transformation which would help to develop a general electrochemical approach for the indole synthesis. Further investigation using other doubly nucleophile (such as N,S-acetals) for the formation of indoles is in progress.Experimental section
Instruments and reagents
All melting points were measured with a XT4A Electrothermal melting point apparatus and are uncorrected. IR spectra were recorded as KBr pellets. 1H and 13C NMR spectra were recorded with an AV 400M Bruker spectrometer (400 MHz 1H frequency, 100 MHz 13C frequency). Chemical shifts are given as δ values (internal standard: TMS). The MS spectra (ESI) were recorded on a Bruker Esquire 6000 mass spectrometer.Catechols 1a–e were reagent-grade and obtained from Alfa Aesar China (Tianjin) Co. Ltd. Compounds 2a–f were synthesized by following known procedures.11 Other chemicals and solvents were from Beijing Chemicals Co. and used without further purification. All electrodes for CV experiments were from CH Instruments, Inc. USA. Doubly distilled de-ionized water was used for the preparation of the aqueous acetate buffer solution. All experiments were performed at room temperature and ambient pressure.
Cyclic voltammetry
Cyclic voltammograms were measured by a 273A Potentiostat/Galvanostat equipped with an electrochemical analysis software, using a conventional three-electrode cell. The working electrode was a glassy carbon disk electrode (ca. φ = 3 mm). The auxiliary and reference electrodes in these studies were Pt wire and saturated Ag/AgCl (in 3 M KCl), respectively. Glassy carbon was polished with polishing cloth before each measurement. Acetate buffer solution was prepared by NaOAc and HOAc monitored by a digital pH meter. Scan rate was 100 mV s−1. The concentration of 1 and 2 were 2 mmol L−1, while that of the supporting electrolyte was 0.2 mol L−1.General procedure for the synthesis of compounds 3a–3r by constant current electrolysis
A 100 mL of H–type cell was equipped with a medium glass frit as a membrane. The anode compartment contained an assembly of 7 graphite rods as the anode, whose upper rims were wrapped by a copper wire, and a platinum wire as the counter electrode was immersed in the cathode compartment. The current throughout electrolysis was controlled by a DC regulated power. During electrolysis, a magnetic stirrer stirred the mixture.To the anode compartment which is kept in water at room temperature was added 75 mL acetate buffer solution. Subsequently, catechols 1 (1 mmoL) and 2 (1 mmoL) were added to the anodic compartment and electrolyzed at constant current of 15 mA (∼5 mA cm−2). The electrolysis was terminated when the starting 1 was consumed as determined by TLC. After electrolysis, the anolyte was worked up by one of the methods shown below.
In the cases of caffeic acid 1e and enaminones 2b–d, a mixed solvent of acetate buffer solution and acetonitrile (2:1 ratio of acetate buffer to acetonitrile) was used as supporting electrolyte due to the low solubility of 1e and 2b–d.
Method A: The analyte was acidified to pH = 1 with 1 mol L−1 aqueous HCl and a precipitate was generated, pure compounds were finally obtained by filtration, wash with water and recrystallization from methanol.
Method B: Partial analyte solution was removed under reduced pressure and precipiate formed. After removing partial acetonitrile, pure compounds were finally obtained by filtration, washing with water and recrystallization from mixture of chloroform and acetone.
Method C: The analyte was acidified to pH = 1 with 1 mol L−1 aqueous HCl, Ethyl acetate was used to extract (3 × 30 mL) and the combined organic layer was dried over MgSO4. High quality of product could obtained after column chromatograph eluted by a solvent mixture of petroleum ether and acetone.
General procedure for the synthesis of compounds 3 by PhI(OAc)2 oxidation
To a solution of catechol 1 (1 mmol) and enaminone 2 (1 mmol) in CH2Cl2 (15 mL) was added dropwise a solution of PhI(OAc)2 (1.1 mmol or 2.2 mmol) in CH2Cl2 (15 mL) within 30 min with stirring at room temperature. TLC was used to monitor the reaction process. After the consumption of 1, the reaction solution was washed with saturated NaCl solution (2 × 30 mL) and then water. The organic layer was separated and dried using anhydrous MgSO4. The solvent was removed under vacuum and the residue was purified by silica gel chromatography, using a mixture of petroleum ether and acetone as eluent, to give the desired products.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 9.40 (br, 2H, OH), 11.98 (t, 1H, J = 5.6 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.7, 16.7, 23.3, 28.8, 47.8, 105.1, 113.3, 116.0, 120.2, 126.2, 134.5, 142.7, 145.3, 148.6, 163.1, 168.4, 192.7; ESI-MS: m/z 317.6 (M−-1).
CH), 6.54 (s, 1H, Ar–H), 7.14 (s, 1H, Ar–H), 7.26–7.41 (m, 5H, Ar–H), 7.47 (d, 1H, J = 16.0 Hz, CHCH), 12.16 (t, 1H, J = 6.0 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 16.8, 29.0, 46.7, 105.9, 113.4, 116.7, 120.1, 126.3, 127.2, 127.6, 129.2, 134.1, 139.1, 142.2, 145.4, 148.7, 163.0, 168.8, 193.6; ESI-MS: m/z 365.6 (M−-1), 732.8 (2M−-1).
CH), 6.41 (s, 1H, Ar–H), 6.97 (s, 1H, Ar–H), 6.98–7.11 (m, 5H, Ar–H), 7.52 (d, 1H, J = 15.6 Hz, CHCH), 9.22 (br, 1.5H, OH), 11.98 (br, 0.5H, OH), 12.72 (t, 1H, J = 5.6 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.8, 17.0, 23.2, 45.2, 104.7, 113.1, 115.8, 121.3, 126.8, 127.5, 128.3, 133.8, 143.1, 143.6, 145.0, 148.1, 166.3, 168.4, 189.6; ESI-MS: m/z 379.7 (M−-1).
CH), 9.40 (br, 2H, OH), 11.98 (t, 1H, J = 5.6 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.7, 16.7, 23.3, 28.8, 47.8, 105.1, 113.3, 116.0, 120.2, 126.2, 134.5, 142.7, 145.3, 148.6, 163.1, 168.4, 192.7; ESI-MS: m/z 317.6 (M−-1).
CH), 6.54 (s, 1H, Ar–H), 7.14 (s, 1H, Ar–H), 7.26–7.41 (m, 5H, Ar–H), 7.47 (d, 1H, J = 16.0 Hz, CHCH), 12.16 (t, 1H, J = 6.0 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 16.8, 29.0, 46.7, 105.9, 113.4, 116.7, 120.1, 126.3, 127.2, 127.6, 129.2, 134.1, 139.1, 142.2, 145.4, 148.7, 163.0, 168.8, 193.6; ESI-MS: m/z 365.6 (M−-1), 732.8 (2M−-1).
CH), 6.41 (s, 1H, Ar–H), 6.97 (s, 1H, Ar–H), 6.98–7.11 (m, 5H, Ar–H), 7.52 (d, 1H, J = 15.6 Hz, CHCH), 9.22 (br, 1.5H, OH), 11.98 (br, 0.5H, OH), 12.72 (t, 1H, J = 5.6 Hz, NH); 13C NMR (100 MHz, DMSO-d6): δ 11.8, 17.0, 23.2, 45.2, 104.7, 113.1, 115.8, 121.3, 126.8, 127.5, 128.3, 133.8, 143.1, 143.6, 145.0, 148.1, 166.3, 168.4, 189.6; ESI-MS: m/z 379.7 (M−-1).
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (No. 2009CB930200, 2011CB933101), The National Key Technology R&D Program (2011BAD23B01), Beijing Natural Science Foundation (No. 7112008) and Beijing City Education Committee (KM201010005009).References
- Organic synthesis in water (Ed.: P. A. Grieco), Blacky Academic and Professional: London, 1998 Search PubMed.
- E. Steckhan, T. Arns, W. R. Heineman, G. Hilt, D. Hoormann, J. Jörissen, L. Kröner, B. Lewall and H. Pütter., Chemosphere, 2001, 43, 63 CrossRef CAS.
- Z.-G. Zha, A.-L. Hui, Y.-Q. Zhou, Q. Miao, Z.-Y. Wang and H.-C. Zhang, Org. Lett., 2005, 7, 1903 CrossRef CAS.
- M. Sainsbury, J. Chem. Soc. C, 1971, 2888–2889 RSC.
- (a) Z. Grujie, I. Tabakovic and M. Trkovnik, Tetrahedron lett., 1976, 4832 Search PubMed; (b) I. Tabakovic, Z. Grujie and Z. Bejtovic, J. Heterocycl. Chem., 1983, 20, 635 CrossRef CAS.
- (a) S. M. Golabi and D. Nematollahi, J. Electroanal. Chem., 1997, 420, 127 CrossRef CAS; (b) S. M. Golabi, D. Nematollahi and D. J. Electroanal, Chem., 1997, 430, 141 CAS; (c) D. Nematollahi and H. Goodarzi, J. Org. Chem., 2002, 67, 5036 CrossRef CAS and cited wherein; (d) A. Maleki and D. Nematollahi, Org. Lett., 2011, 13, 1928–1931 CrossRef CAS.
- (a) C.-C. Zeng, D.-W. Ping, S.-C. Zhang, R.-G. Zhong and J. Y. Becker, J. Electroanal. Chem., 2008, 622, 90–96 CrossRef CAS; (b) C.-C. Zeng, F.-J. Liu, D.-W. Ping, Y.-L. Cai, R.-G. Zhong and J. Y. Becker, J. Electroanal. Chem., 2009, 625, 131–137 CrossRef CAS.
- C.-C. Zeng, F.-J. Liu, D.-W. Ping, L.-M. Hu, Y.-L. Cai and R.-G. Zhong, J. Org. Chem., 2009, 74, 6386–6389 CrossRef CAS.
- (a) C.-C. Zeng, D.-W. Ping, L.-M. Hu, X.-Q. Song and R. G. Zhong, Org. Biomol. Chem., 2010, 8, 2465–2472 RSC; (b) C.-C. Zeng, D.-W. Ping, Y.-S. Xu, L.-M. Hu and R.-G. Zhong, Eur. J. Org. Chem., 2009, 5832–5840 CrossRef CAS.
- M. E. Jung and F. Perez, Org. Lett., 2009, 11, 2165–2167 CrossRef CAS.
- V. Sridharan, C. Avendaño and J. C. Menéndez, Synlett, 2007, 6, 881–884 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |