Alkoxides of group 4 metals containing the bis(imino)phenoxide ligand: synthesis, structural characterization and polymerization studies†‡
Tanmoy
Kumar Saha
,
Bijja
Rajashekhar
and
Debashis
Chakraborty
*
Department of Chemistry, Indian Institute of Technology Madras, Chennai-600 036, Tamil Nadu, India. E-mail: dchakraborty@iitm.ac.in; Fax: +914422574202; Tel: +914422574223
First published on 7th November 2011
Abstract
A variety of group 4 metal compounds containing the bis(imino)phenoxide backbone were synthesized from the reaction of suitable ligands and group 4 metal alkoxides. They were completely characterized with different spectroscopic techniques and single-crystal X-ray diffraction studies on a few of them. These compounds were found extremely active towards the bulk polymerization of ε-caprolactone (CL), δ-valerolactone (VL), rac-butyrolactone (rac-BL), L-lactide (L-LA) and rac-lactide (rac-LA) yielding polymers with high number average molecular weight (Mn) and controlled molecular weight distribution (MWD). The kinetics and mechanistic studies associated with these polymerizations have been performed. In addition, the catalytic activity of these compounds towards the polymerization of ethylene was investigated.
Introduction
In the recent years, studies on ring-opening polymerization of cyclic esters and lactides yielding biodegradable polymers have become a topical area of research.1 This has occurred as a result of depletion of petroleum resources, ultimately initiating research that uses annually renewable feed stock as raw materials leading to the formation of green polymers.1,2 Aliphatic polyesters have become important for the biomedical industry like delivery medium for the controlled release of medicines and in the manufacturing of biodegradable surgical sutures.3 These polymers have practical applications as a result of the permeability, biocompatibility and biodegradability.4 One of the important methods in producing them is the ring-opening polymerization of cyclic esters and lactide using metal catalysts. Among the different mechanistic pathways available, the coordination-insertion mechanism is the most popular because it yields polymer with good molecular weights and narrow molecular weight distributions.5Group 4 metals have been popular towards the synthesis of new catalysts for the polymerization of cyclic esters and lactide.6,7 Our continued interest in pursuing new chemistry with group 4 metals8 prompted us to undertake this investigation for synthesizing new catalysts and evaluate their catalytic activity in ring-opening polymerization of cyclic esters and lactide.The pioneering work contributed by Ziegler and Natta towards the synthesis of high density polyethylene and the discovery of the capability of MAO (methylalumoxane) in activating group 4 metallocene complexes created a very high global impetus in polyolefin research.9Metallocenes are active in controlling the molecular weight and molecular weight distribution of the resultant polymers. A huge span of literature is available regarding the synthesis and polymerization activity of homogeneous single-site catalysts.10 Recent endeavors in this area involves the synthesis of nonmetallocene complexes, creating opportunities to synthesize polymers with novel properties and usage.11–15
Here, we report the synthesis of a family of catalysts containing the bis(imino)phenoxide ligating backbone and group 4 metals and their activity in the polymerization of cyclic esters, lactide and ethylene. A preliminary communication describing the catalytic activity of bis(imino)phenoxide compounds of zirconium towards the polymerization of cyclic esters and lactide has already appeared from our group.8b Our previous communication describing the capability of zirconium complexes of the bis(imino)phenoxide ligand shows that the variation of substituents of the aryl ring containing the N center does not have significant effect on the polymerizations of cyclic esters and lactide.8b Hence, it was required to study the effect of substituents on the central aryl ring contained in the bis(imino)phenoxide backbone.
Results and discussion
Syntheses and structural characterization
Synthesis of group 4 metal complexes was carried out using ligands L1–L4 as shown in Scheme 1. A 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric reaction between ligands L1–L4 with group 4 metal alcoholates in toluene resulted in the quantitative formation of compounds 1–10 respectively (Scheme 1). Nevertheless, a 1:1 stoichiometric reaction between the reactants resulted in the formation of 1–10 along with the presence of unreacted metal alcoholate, as understood by the spectroscopic analysis of the crude reaction mixture. The resulting products were purified by crystallization from toluene and isolated in high yields and purity as orange to yellow solids. Compounds 1–10 were unambiguously characterized using various spectroscopic techniques and single-crystal X-ray diffraction studies on a few of them. Compound 10 was synthesized with the intention of the possibility of growing single crystals suitable for X-ray diffraction studies. Repeated attempts in growing suitable single crystals with 7–9 did not give fruitful results.
:1 stoichiometric reaction between ligands L1–L4 with group 4 metal alcoholates in toluene resulted in the quantitative formation of compounds 1–10 respectively (Scheme 1). Nevertheless, a 1:1 stoichiometric reaction between the reactants resulted in the formation of 1–10 along with the presence of unreacted metal alcoholate, as understood by the spectroscopic analysis of the crude reaction mixture. The resulting products were purified by crystallization from toluene and isolated in high yields and purity as orange to yellow solids. Compounds 1–10 were unambiguously characterized using various spectroscopic techniques and single-crystal X-ray diffraction studies on a few of them. Compound 10 was synthesized with the intention of the possibility of growing single crystals suitable for X-ray diffraction studies. Repeated attempts in growing suitable single crystals with 7–9 did not give fruitful results.
 | ||
| Scheme 1 Bis(imino)phenoxide compounds of group 4 metals. | ||
The 1H NMR of 1–10 reveal all the signals in the correct integration ratio. All these compounds contain a molecule of coordinated toluene. In case of compounds 1–6, the protons from the CH moiety present in the OiPr group appears much downfield shifted as compared to the signals of the CH unit from OiPr group of the ligand. The interesting observation is that the CH protons from the CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N fragments do not appear as a single peak uniformly for all the compounds. In the case of 1, 2 and 9, the CHN fragments are present as a single broad peak with the correct integration. For 8, it appears as a set of two signals in 1:1 integration ratio. This may be rationalized by considering the structure of these molecules wherein there are two N centers coordinated and two free. In all the other compounds, the imine protons appear as a set of three signals in 2:1:1 integration ratio.
N fragments do not appear as a single peak uniformly for all the compounds. In the case of 1, 2 and 9, the CHN fragments are present as a single broad peak with the correct integration. For 8, it appears as a set of two signals in 1:1 integration ratio. This may be rationalized by considering the structure of these molecules wherein there are two N centers coordinated and two free. In all the other compounds, the imine protons appear as a set of three signals in 2:1:1 integration ratio.
The 13C NMR of 1–9 shows the presence of moieties corresponding to the different carbon environments in these complexes. For 1, 2, 8 and 9, the CHN appears as a single signal whereas for the others, two signals, one for the coordinated CHN and other for the non coordinated moiety are observed. The overall conclusions drawn from 13C NMR studies are in agreement with the conclusions drawn from the 1H NMR spectra of 1–10.
Electrospray ionization mass spectrometric (ESI–MS) studies on 1–10 reveals clearly the presence of the molecular ion peak and suggests that these compounds appear in the monomeric state. Elemental analyses of 1–10 shows that the experimentally determined values are in good agreement with the calculated ones, suggesting acceptable purity of these isolated compounds.
Zirconium and hafnium alkoxides retain their dimeric structure even in solution.16 However, in this work it is observed that the dimeric core of these alkoxides is disturbed. This is contrary to our observation with salens where the cavity provided by the ligand is sufficient to accommodate the dimeric core of the metal alkoxide.8e
Variable temperature 1H NMR studies were performed using compound 1 and 7 (Fig. 1) (See ESI for 1).† It was observed that as the temperature is lowered gradually, the signals for CHN group become gradually broad. This signifies that there is scope for ligand scrambling due to the presence of coordinatively unsaturated metal center. In 1, the signal corresponding to the OiPr group remains as a single broad signal at all temperatures. This proves that the preferred structure is monomeric and the equilibrium with oligomeric species is completely absent.

Single-crystal X-ray diffraction studies
Crystals of 3, 5 and 10 suitable for X-ray diffraction studies were grown from saturated toluene solution of the respective compounds at −24 °C, over a period of two weeks. These compounds are monomeric in the solid state containing a single metal center that adopts a distorted octahedral geometry (Fig. 2, Fig. 3 and Fig. 4). Out of the four nitrogen centers, two are coordinated to the metal center where as the other two remain free. Such a product configuration is thermodynamically preferred as compared to the situation where both the nitrogen centers remain simultaneously coordinated to the metal. All the bond lengths and angles match well with the literature data.6e,6k,6w The crystal data and structure refinement details for 3, 5 and 10 are enumerated in Table 1. | ||
| Fig. 2 Molecular structure of 3: thermal ellipsoids drawn at 30% probability level, hydrogen atoms have been omitted for clarity. Selected bond lengths (A°) and angles (°): Ti(1)–O(1) 1.7887(17), Ti(1)–O(2) 1.8062(18), Ti(1)–O(3) 1.9946(17), Ti(1)–O(4) 1.9773(16), Ti(1)–N(1) 2.2252(19), Ti(1)–N(2) 2.2285(19), N(1)–Ti(1)–N(2) 175.05(7), O(3)–Ti(1)–N(1) 92.71(7). | ||
 | ||
| Fig. 3 Molecular structure of 5: thermal ellipsoids drawn at 30% probability level, hydrogen atoms have been omitted for clarity. Selected bond lengths (A°) and angles (°): Zr(1)–O(1) 2.110(4), Zr(1)–O(2) 1.920(4), Zr(1)–O(3) 2.093(4), Zr(1)–O(4) 1.933(4), Zr(1)–N(1) 2.376(5), Zr(1)–N(2) 2.382(5), N(1)–Zr(1)–N(2) 175.01(16), O(3)–Zr(1)–N(1) 97.78(16). | ||
 | ||
| Fig. 4 Molecular structure of 10: thermal ellipsoids drawn at 30% probability level, hydrogen atoms have been omitted for clarity. Selected bond lengths (A°) and angles (°): Hf(1)–O(1) 2.0284(17), Hf(1)–O(2) 1.9227(16), Hf(1)–N(1) 2.4206(19), N(1)–Hf(1)–N(1) 78.38(9), O(2)–Hf(1)–N(1) 165.99(7). | ||
| Compounds | 3 | 5 | 10 |
|---|---|---|---|
| Molecular formula | C79H104N4O6Ti | C70H90Cl2N4O4Zr | C54H60N4O8Hf |
| Formula weight | 1253.54 | 1213.58 | 1071.55 |
| T/K | 173(2) | 173(2) | 173(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71703 |
| Crystal system, | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | C2/c |
| a/Å | 12.0283(3) | 12.070(2) | 19.017(3) |
| b/Å | 28.2315(9) | 22.740(5) | 19.507(3) |
| c/Å | 21.6400(8) | 27.720(6) | 15.973(2) |
| α (°) | 90.00 | 90.00 | 90.00 |
| β (°) | 98.2510(10) | 100.89(3) | 117.708(6) |
| γ (°) | 90.00 | 90.00 | 90.00 |
| V/Å3 | 7272.4(4) | 7471(3) | 5245.9(13) |
| Z, Calculated density (g cm−3) | 4, 1.145 | 4, 1.079 | 4, 1.357 |
| Absorption coefficient (mm−1) | 0.171 | 0.262 | 2.043 |
| F(000) | 2704 | 2576 | 2192 |
| Crystal size/mm | 0.35 × 0.28 × 0.25 | 0.38 × 0.35 × 0.22 | 0.23 × 0.20 × 0.15 |
| Limiting indices | −12 ≤ h ≤ 16 | −10 ≤ h ≤ 15 | −31 ≤ h ≤ 29 |
| −38 ≤ k ≤ 38 | −30 ≤ k ≤ 29 | −31 ≤ k ≤ 31 | |
| −22 ≤ l ≤ 27 | −32 ≤ l ≤ 36 | −24 ≤ l ≤ 25 | |
| Reflections collected/unique | 56295 | 52626 | 36623 |
| Independent reflections | 17605 | 17045 | 11509 |
| Data/restraints/parameters | 17605/0/834 | 17045/0/750 | 11509/0/309 |
| Goodness-of-fit on F2 | 0.968 | 0.988 | 1.009 |
| Final R indices [I >2σ(I)] | R = 0.0608 | R = 0.0988 | R = 0.0411 |
| wR = 0.1471 | wR = 0.2589 | wR = 0.0807 | |
| R indices (all data) | R = 0.1316 | R = 0.2163 | R = 0.0719 |
| wR = 0.1847 | wR = 0.3318 | wR = 0.0917 |
Ring-opening polymerization
These compounds have proved to be powerful catalysts for the bulk ring-opening polymerization of different cyclic esters and lactide. We did not perform any polymerization studies with 10 since it was synthesized for structural characterization alone. The results are depicted in Table 2.:1 ratio
| Entry | Catalyst | Monomer | T/°C | Yield/% | Timea/min | M n b/kg mol−1 | M w / M n | P r c |
|---|---|---|---|---|---|---|---|---|
| a Time of polymerization measured by quenching the polymerization reaction when all monomer was found consumed. b Measured by GPC at 27 °C in THF relative to polystyrene standards. c Calculated using homonuclear-decoupled 1H NMR spectrum.17 | ||||||||
| 1 | 1 | CL | 80 | 99 | 5 | 70.8 | 1.32 | |
| 2 | 1 | VL | 80 | 97 | 3 | 61.3 | 1.22 | |
| 3 | 1 | rac-BL | 80 | 96 | 9 | 40.1 | 1.33 | |
| 4 | 1 | L-LA | 130 | 98 | 9 | 52.9 | 1.11 | |
| 5 | 1 | rac-LA | 130 | 99 | 6 | 46.6 | 1.13 | 0.72 |
| 6 | 2 | CL | 80 | 97 | 7 | 66.9 | 1.35 | |
| 7 | 2 | VL | 80 | 95 | 5 | 58.7 | 1.25 | |
| 8 | 2 | rac-BL | 80 | 97 | 12 | 37.5 | 1.34 | 0.73 |
| 9 | 2 | L-LA | 130 | 99 | 10.5 | 50.2 | 1.12 | |
| 10 | 2 | rac-LA | 130 | 98 | 8 | 45.2 | 1.14 | |
| 11 | 3 | CL | 80 | 98 | 10 | 64.8 | 1.38 | |
| 12 | 3 | VL | 80 | 96 | 8 | 57.7 | 1.29 | |
| 13 | 3 | rac-BL | 80 | 98 | 17 | 35.2 | 1.30 | |
| 14 | 3 | L-LA | 130 | 99 | 13 | 48.4 | 1.14 | |
| 15 | 3 | rac-LA | 130 | 99 | 11 | 44.6 | 1.15 | 0.75 |
| 16 | 5 | CL | 80 | 99 | 9 | 96.6 | 1.32 | |
| 17 | 5 | VL | 80 | 96 | 6 | 65.1 | 1.21 | |
| 18 | 5 | rac-BL | 80 | 97 | 15 | 38.2 | 1.34 | |
| 19 | 5 | L-LA | 130 | 99 | 18 | 92.6 | 1.13 | |
| 20 | 5 | rac-LA | 130 | 98 | 8 | 88.0 | 1.15 | 0.74 |
| 21 | 6 | CL | 80 | 97 | 12 | 95.0 | 1.31 | |
| 22 | 6 | VL | 80 | 98 | 8 | 63.2 | 1.23 | |
| 23 | 6 | rac-BL | 80 | 97 | 18 | 36.9 | 1.34 | |
| 24 | 6 | L-LA | 130 | 99 | 21 | 90.2 | 1.16 | |
| 25 | 6 | rac-LA | 130 | 99 | 11 | 87.4 | 1.14 | 0.73 |
| 26 | 7 | CL | 80 | 97 | 12 | 52.4 | 1.34 | |
| 27 | 7 | VL | 80 | 98 | 11 | 49.7 | 1.27 | |
| 28 | 7 | rac-BL | 80 | 97 | 20 | 36.8 | 1.31 | |
| 29 | 7 | L-LA | 130 | 99 | 19 | 47.2 | 1.19 | |
| 30 | 7 | rac-LA | 130 | 99 | 10 | 45.6 | 1.20 | 0.75 |
| 31 | 8 | CL | 80 | 97 | 15 | 50.3 | 1.38 | |
| 32 | 8 | VL | 80 | 96 | 14 | 47.4 | 1.29 | |
| 33 | 8 | rac-BL | 80 | 98 | 23 | 34.4 | 1.34 | |
| 34 | 8 | L-LA | 130 | 99 | 25 | 46.5 | 1.11 | |
| 35 | 8 | rac-LA | 130 | 98 | 13 | 44.7 | 1.12 | 0.77 |
| 36 | 9 | CL | 80 | 99 | 19 | 49.4 | 1.39 | |
| 37 | 9 | VL | 80 | 98 | 18 | 45.6 | 1.21 | |
| 38 | 9 | rac-BL | 80 | 98 | 30 | 34.0 | 1.55 | |
| 39 | 9 | L-LA | 130 | 97 | 28 | 44.7 | 1.14 | |
| 40 | 9 | rac-LA | 130 | 99 | 17 | 43.8 | 1.13 | 0.76 |
Analysis of the data depicted in Table 2 shows that there is a reasonable degree of control in these polymerizations. In general, the reactivity decreases down the group from titanium to hafnium with the exception of rac-LA polymerization, where the reactivity of titanium and zirconium catalysts coincidentally remain almost the same. The ring-opening polymerizations are anticipated to proceed via a coordination-insertion mechanism for which Lewis acidity of metal center is important. If we compare the electron-releasing effects of three substituents in phenolic moiety of ligand, the order will be OMe > Me > Cl. As the electron donating tendency of substituent on the phenyl ring increases, the Lewis acidity of metal will decrease. Hence, it leads to loss of reactivity towards polymerization. So the reactivity order will be Cl > Me > OMe for a given monomer, although the differences in Mn are marginal. Another important observation for each monomer is that the zirconium complexes yield the respective polymers with maximum Mn, followed by titanium, with hafnium yielding the least. Hence, we conclude that the presence of different para substituents on the central aryl ring in these ligands do not influence the polymerization to a significant degree. Also, we have concluded previously that the presence of different alkyl substituents at different positions of the aniline ring does not produce notable effects during these polymerizations.8b Comparison with the literature shows that our system is far better in terms of time taken for polymerization, Mn and MWDs.6b,6d,6e,6j,6p,6q,6w We have done a closer comparison with the results reported for the imino(phenoxide) complexes.6v We find here that our results are better even for tacticity in addition to time taken for polymerization, Mn and MWDs.
We have concluded from homonuclear decoupling 1H NMR that the polymerization of L-LA using 1–9 yields heterotactic enriched polymer (Table 2). The polymerization of rac-BL is highly stereoselective leading to the formation of syndiotactic polymer. In the 13C NMR, the r-centered diad (δ = 169.38 ppm) shows a very strong contribution, indicating syndiotacticity. This conclusion is supported by the presence of a prominent signal (δ = 40.85 ppm) in the methylene region which corresponds to the rr triad.18
Since polymerization of L-LA yielded best results, we studied the variations of Mn and MWDs with [L-LA]o/[Cat]o for L-LA using 2, 5 and 8. The results are depicted in Table 3 and Fig. 5. The plot suggests that there is a continuous rise in the in the Mn with an increase in the [L-LA]o/[Cat]o for the catalysts indicated. Again the MWDs are found to vary linearly with the increase in [L-LA]o/[Cat]o ratio.
![M
n
vs.
[L-LA]o/[Cat]ovs. Mw/Mn plot for 2, 5 and 8.](/image/article/2012/RA/c1ra00543j/c1ra00543j-f5.gif) | ||
| Fig. 5 M n vs. [L-LA]o/[Cat]ovs. Mw/Mn plot for 2, 5 and 8. | ||
| Entry | Catalyst | [L-LA]o/[Cat]o | Timea/min | Yield/% | M n b/kgmol−1 | M w/Mn |
|---|---|---|---|---|---|---|
| a Time of polymerization measured by quenching the polymerization reaction when all monomer was found consumed. b Measured by GPC at 27 °C in THF relative to polystyrene standards. | ||||||
| 1 | 2 | 200 | 10.5 | 99 | 50.2 | 1.12 |
| 2 | 2 | 400 | 15 | 99 | 92.9 | 1.15 |
| 3 | 2 | 600 | 18 | 98 | 149.7 | 1.16 |
| 4 | 2 | 800 | 22 | 97 | 211.3 | 1.17 |
| 5 | 2 | 1000 | 27 | 96 | 254.1 | 1.19 |
| 6 | 2 | 1200 | 33 | 99 | 307.3 | 1.22 |
| 7 | 5 | 200 | 18 | 98 | 92.6 | 1.13 |
| 8 | 5 | 400 | 22 | 99 | 184.3 | 1.15 |
| 9 | 5 | 600 | 27 | 99 | 295.0 | 1.16 |
| 10 | 5 | 800 | 32 | 97 | 374.3 | 1.18 |
| 11 | 5 | 1000 | 36 | 96 | 451.2 | 1.21 |
| 12 | 5 | 1200 | 41 | 98 | 552.8 | 1.24 |
| 13 | 8 | 200 | 25 | 99 | 44.6 | 1.21 |
| 14 | 8 | 400 | 29 | 99 | 88.4 | 1.24 |
| 15 | 8 | 600 | 35 | 97 | 143.7 | 1.26 |
| 16 | 8 | 800 | 39 | 98 | 185.6 | 1.29 |
| 17 | 8 | 1000 | 43 | 99 | 225.2 | 1.30 |
| 18 | 8 | 1200 | 49 | 97 | 259.0 | 1.32 |
Again, the plot of Mn vs % conversion for 2, 5 and 8 (Fig. 6) was found linear, suggesting a good degree of control in these polymerizations. Fig. 5 and 6 seem to suggest that the polymerization of L-LA is highly controlled and propagates in a living manner.
 | ||
| Fig. 6 M n vs % conversion for 2, 5 and 8. | ||
Next, we proceeded with the kinetic studies for the polymerization of L-LA using 3, 6 and 9 in ratio [L-LA]o/[Cat]o = 200 at 130 °C. We decided to choose these catalysts since they yielded polymer with highest Mn values (Table 2). The results are presented in Fig. 7. There is a first-order dependence of the rate of polymerization on L-LA concentration with complete absence of induction period. The plot of [L-LA]o/[L-LA]tvs. time was found to be linear. The values of the apparent rate constant (kapp) for L-LA polymerization catalyzed by 3, 6 and 9 were evaluated from the slope of these lines and were found to be 0.61395 min−1, 0.44329 min−1 and 0.22971 min−1 respectively. This implies that the rate of polymerization follows the order Ti > Zr > Hf.
![Semi-logarithmic plots of L-LA conversion in time initiated by 3, 6 and 9: [L-LA]o/[Cat]o = 200 at 130 °C.](/image/article/2012/RA/c1ra00543j/c1ra00543j-f7.gif) | ||
| Fig. 7 Semi-logarithmic plots of L-LA conversion in time initiated by 3, 6 and 9: [L-LA]o/[Cat]o = 200 at 130 °C. | ||
In order to have a complete idea on the polymerization mechanism, we synthesized a low molecular weight oligomer of L-LA. Compound 5 was reacted with L-LA in 1:10 stoichiometric ratio at 130 °C for 30 mins. The residue was dissolved in minimum amount of CH2Cl2 and poured into cold methanol. The oligomer was isolated and was subjected to MALDI-TOF and 1H NMR studies. The results (Fig. 8 and Fig. 9) indicate that the polymerization proceeds with the coordination-insertion mechanism as shown in Scheme 2 and the phenolate ligand (L2) is incorporated as one of the end terminal groups. The possibility of activated monomer mechanism does not exist since no alcoholic initiators have been added externally to the polymerization reactions.
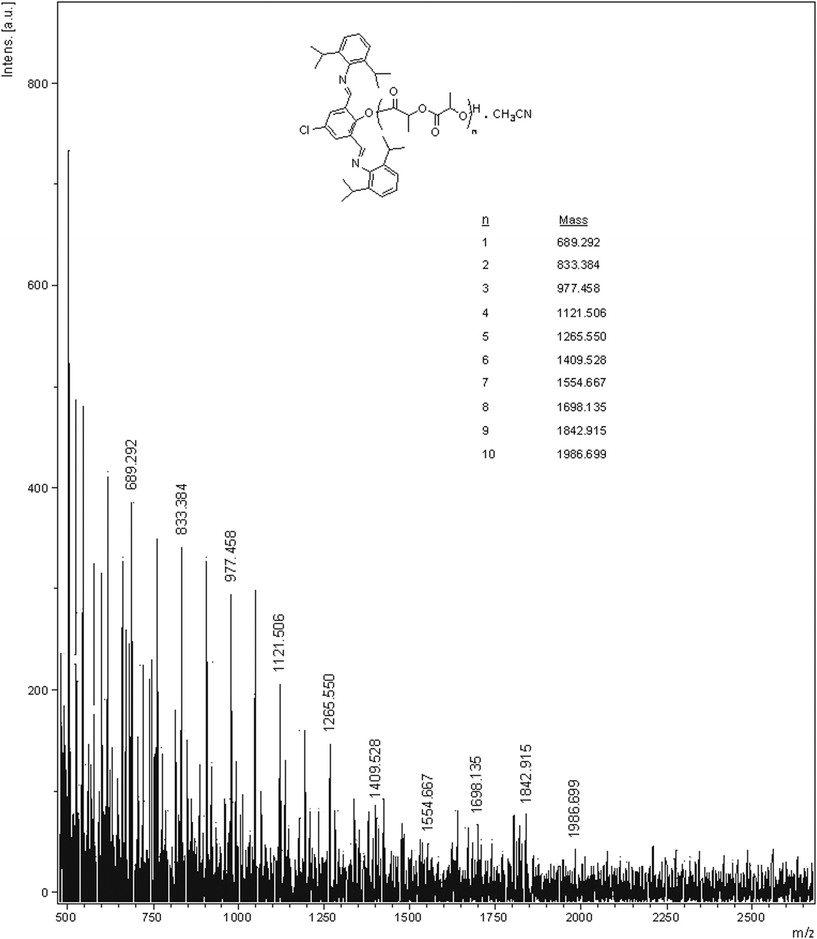 | ||
| Fig. 8 MALDI-TOF spectrum of the crude product obtained from a reaction between 5 and L-LA in 1:10 ratio at 130 °C. | ||
 | ||
| Fig. 9
1H NMR spectrum of the crude product obtained from a reaction between 5 and L-LA in 1:10 ratio at 130 °C (x = peaks corresponding to L-LA). | ||
 | ||
| Scheme 2 Mechanism of ring-opening polymerization. | ||
Ethylene polymerization
The catalytic activity of compounds 1–9 activated by MAO were tested towards the polymerization of ethylene. The results are depicted in Table 4.| Entry | Catalyst a | Activityb | Yieldc (g) | M w/ kgmol−1 | M n/ kgmol−1 | M w/Mn |
|---|---|---|---|---|---|---|
| a All experiments were performed in toluene at MAO:catalyst ratio = 1000, 80 °C for 30 min, catalyst = 8 mg, solvent = 7 mL. b Activity in (g PE/mol cat ×h) × 105. c gm of polymer obtained after 30 min. | ||||||
| 1 | 1 | 4.28 | 1.5 | 132.8 | 75.0 | 1.77 |
| 2 | 2 | 4.05 | 1.4 | 131.5 | 73.4 | 1.79 |
| 3 | 3 | 3.75 | 1.3 | 130.1 | 71.9 | 1.81 |
| 4 | 4 | 5.22 | 1.8 | 149.3 | 82.0 | 1.82 |
| 5 | 5 | 4.70 | 1.6 | 149.0 | 80.5 | 1.85 |
| 6 | 6 | 4.36 | 1.5 | 149.2 | 79.8 | 1.87 |
| 7 | 7 | 4.25 | 1.3 | 128.4 | 66.9 | 1.92 |
| 8 | 8 | 4.17 | 1.3 | 126.3 | 64.8 | 1.95 |
| 9 | 9 | 3.82 | 1.2 | 125.0 | 62.8 | 1.99 |
The trials were performed in toluene at 80 °C using MAO as the activator. Analysis of the data reveals that the zirconium compounds yield the best results in terms of activity, Mn and Mw. This is followed by the results using titanium catalysts and finally for hafnium catalysts.
Conclusions
In summary, we have synthesized a library of new alkoxides containing group 4 metals and bis(imino)phenoxide backbone as the ancillary ligand. These compounds are extremely active towards the polymerization of different cyclic esters, lactide and ethylene. The zirconium catalysts were found to yield better polymerization results in comparison to the titanium and hafnium analogues. Presence of different substituents either on the central aryl ring or on different anilines present in the various ligands do not affect the polymerizations in a significant manner.Experimental
General experimental details
All reactions were performed under a dry argon atmosphere employing standard Schlenk techniques or in a glove box where the oxygen and moisture levels were maintained normally below 1 ppm. The reactions to synthesize 1–10 were done in toluene which was rigorously dried over sodium benzophenone ketyl and distilled fresh prior to use. The deuterated solvents used for NMR studies were dried using standard procedures, distilled and stored in the glove box throughout the program of study. A Bruker Avance 400 instrument was used to record all 1H and 13C NMR with the chemical shifts referenced to residual solvent resonances and are reported as parts per million relative to SiMe4. A Waters Q-Tof micro mass spectrometer was utilized to record the ESI-MS spectra and MALDI-TOF measurements were performed on a Bruker Daltonics instrument in dihydroxy benzoic acid matrix. Elemental analyses reports were obtained using a Perkin Elmer Series 11 analyzer. The starting materials namely Ti(OiPr)4, Zr(OiPr)4·iPrOH and Hf(OtBu)4 were purchased from Aldrich and used as received. The various monomers used for this study were purchased from aldrich, dried/purified and stored in the glove box. The ligands L1, L2 and L4,19 were synthesized using reported literature methods. MAO was purchased from Aldrich.Synthesis of L3
To a stirred methanolic solution of 2,6-diformyl-4-methoxy phenol (1g, 5.5 mmol), 2,6-diisopropyl aniline (1.97 g, 11.1 mmol) was added drop wise and after completion of addition the orange solution was set to reflux for 12 h. The reaction mixture was cooled to ambient temperature during which the orange product precipitated. The orange color solid was filtered off and washed with cold methanol and dried. Yield = 76%. Anal. Calc. for C33H42N2O2: C, 79.48; H, 8.49; N, 5.62. Found: C, 79.81; H, 8.52; N, 5.58. 1H NMR (400 MHz, CDCl3): δ = 1.24–1.31 (d, CH(CH3)3, 24H), 3.03–3.09 (m, CH(CH3)3, 4H), 3.96 (s, Ar–O–CH3, 3H), 7.22–7.30 (m, Ar–H, 8H), 8.62 (s, CHN, 2H), 13.15 (s, O–H, 1H). 13C NMR (100 MHz, CDCl3): δ = 23.64 (CH(CH3)3), 27.92 (CH(CH3)3), 55.84 (Ar–O–CH3), 118.85 (Ar–C), 118.92 (Ar–C), 122.86 (Ar–C), 123.39 (Ar–C), 137.65 (Ar–C), 149.32 (Ar–C), 155.21 (Ar–O–CH3), 157.15 (Ar–O), 162.72 (CHN). m/z calculated for [M]+ calculated 498.698 found 498.
Synthesis of catalysts
A general procedure describing the synthesis of 1–10 is outlined:In an argon filled glove box, to a stirred solution of metal alkoxide (0.13 mmol) in 5 mL toluene at −24 °C was added a solution of L1–L4 (0.26 mmol) in 5 mL toluene. The reaction mixture was allowed to warm up to room temperature and stirred additionally for 24 h. The solvent was removed under reduced pressure and the residue obtained was crystallized from concentrated toluene solution at −24 °C.
N, 4H). 13C NMR (100 MHz, CDCl3): δ = 21.29 (Ar–CH3), 23.38 (O–CH(CH3)2), 23.69 (CH(CH3)2), 25.36 (CH(CH3)2), 25.53 (CH(CH3)2), 26.27 (CH(CH3)2), 27.36 (CH(CH3)2), 28.06 (CH(CH3)2), 28.22 (CH(CH3)2), 28.42 (CH(CH3)2), 76.78 (O–CH(CH3)2), 121.71 (Ar–C), 123.16 (Ar–CH3), 123.48 (Ar–C), 123.93 (Ar–C), 124.10 (Ar–C), 125.32 (Ar–C), 125.40 (Ar–C), 126.59 (Ar–C), 138.53 (Ar–CH3), 139.45 (Ar–C), 141.11 (Ar–C), 150.45 (Ar–C), 158.44 (Ar–O), 164.39 (CHN). ESI m/z calculated for [M]+. C72H96N4O4Ti: 1129.424 found 1129.520.
N, 4H). 13C NMR (100 MHz, CDCl3): δ = 23.51 (O–CH(CH3)2), 24.68 (CH(CH3)2), 24.87 (CH(CH3)2), 25.27 (CH(CH3)2), 25.44 (CH(CH3)2), 25.84 (CH(CH3)2), 27.85 (CH(CH3)2), 28.07 (CH(CH3)2), 28.16 (CH(CH3)2), 76.96 (O–CH(CH3)2), 119.90 (Ar–C), 123.19 (Ar–C), 124.64 (Ar–C), 127.63 (Ar–Cl), 128.27 (Ar–C), 138.18 (Ar–C), 138.76 (Ar–C), 139.58 (Ar–C), 149.62 (Ar–C), 158.01 (Ar–O), 162.29 (CHN). ESI m/z calculated for [M + Na]+. C70H90Cl2N4O4Ti: 1170.261 found 1193.055.
N, 2H), 8.62 (s, CHN, 2H). 13C NMR (100 MHz, CDCl3): δ = 23.55 (O–CH(CH3)2), 24.60 (CH(CH3)2), 24.86 (CH(CH3)2), 25.27 (CH(CH3)2), 25.46 (CH(CH3)2), 25.74 (CH(CH3)2), 27.81 (CH(CH3)2), 28.00 (CH(CH3)2), 28.13 (CH(CH3)2), 56.16 (Ar–OCH3), 76.64 (O–CH(CH3)2), 119.75 (Ar–C), 123.09 (Ar–C), 124.68 (Ar–C), 128.17 (Ar–C), 138.18 (Ar–CH3), 138.86 (Ar–C), 139.48 (Ar–C), 149.97 (Ar–C), 150.67 (Ar–OCH3), 150.81 (Ar–OCH3), 157.94 (Ar–O), 161.69 (CHN), 164.38 (CHN). ESI m/z calculated for [M]+. C72H96N4O6Ti: 1161.423 found 1161.464.
N, 2H), 8.45 (s, CHN, 1H), 8.60 (s, CHN, 1H). 13C NMR (100 MHz, CDCl3): δ = 22.14 (Ar–CH3), 22.75 (O–CH(CH3)2), 23.30 (CH(CH3)2), 24.02 (CH(CH3)2), 25.32 (CH(CH3)2), 25.50 (CH(CH3)2), 26.23 (CH(CH3)2), 27.00 (CH(CH3)2), 27.42 (CH(CH3)2), 28.27 (CH(CH3)2), 70.63 (O–CH(CH3)2), 121.33 (Ar–C), 122.73 (Ar–C), 123.33 (Ar–CH3), 123.61 (Ar–C), 124.36 (Ar–C), 127.02 (Ar–Cl), 128.37 (Ar–C), 129.18 (Ar–CH3), 133.81 (Ar–C), 137.56 (Ar–C), 138.26 (Ar–CH3), 140.68 (Ar–C), 140.94 (Ar–C), 149.89 (Ar–C), 150.40 (Ar–C), 157.84 (Ar–O), 164.32 (CHN), 171.09 (CHN). ESI m/z calculated for [M]+. C70H90Cl2N4O4Zr: 1213.618 found 1213.622.
N, 2H), 8.08 (s, CHN, 1H), 8.59 (s, CHN, 1H). 13C NMR (100 MHz, CDCl3): δ = 22.82 (O–CH(CH3)2), 23.74 (CH(CH3)2), 25.32 (CH(CH3)2), 25.48 (CH(CH3)2), 25.65 (CH(CH3)2), 26.31 (CH(CH3)2), 27.29 (CH(CH3)2), 27.35 (CH(CH3)2), 28.26 (CH(CH3)2), 56.42 (Ar–OCH3), 70.16 (O–CH(CH3)2), 121.78 (Ar–C), 123.26 (Ar–CH3), 123.78 (Ar–C), 124.06 (Ar–C), 125.45 (Ar–C), 126.67 (Ar–C), 128.38 (Ar–C), 129.18 (Ar–CH3), 133.81 (Ar–C), 137.56 (Ar–C), 138.42 (Ar–CH3), 140.82 (Ar–C), 140.94 (Ar–C), 149.70 (Ar–C), 150.02 (Ar–OCH3), 150.28 (Ar–O), 158.98 (Ar–O), 161.34 (CHN), 171.64 (CHN). ESI m/z calculated for [M]+. C72H96N4O6Zr: 1204.780 found 1204.790.
N, 2H), 8.27 (s, CHN, 1H). 8.58 (s, CHN, 1H). 13C NMR (100 MHz, CDCl3): δ = 22.05(O–Ar–CH3), 22.75 (CH(CH3)2), 23.53 (CH(CH3)2), 24.89 (CH(CH3)2), 27.82 (CH(CH3)2), 28.07 (CH(CH3)2),32.10 (CH(CH3)2), 32.38 (CH(CH3)2), 33.09 (CH(CH3)2), 33.37 (O–C(CH3)3), 33.94 (O–C(CH3)3), 76.32 (O–C(CH3)3), 118.74 (Ar–C), 122.05 (Ar–C), 122.65 (Ar–C), 123.12 (Ar–CH3), 124.38 (Ar–C), 125.30 (Ar–C), 128.19 (Ar–C), 128.95 (Ar–C), 137.52 (Ar–C), 149.45 (Ar–C), 158.02 (Ar–O), 162.19 (CHN), 163.42 (CHN). ESI m/z calculated for [M]+. C74H100N4O4Hf: 1288.100 found 1288.353.
N, 2H), 8.27 (s, CHN, 2H). 13C NMR (100 MHz, CDCl3): δ = 21.40 (Ar–CH3), 22.38 (CH(CH3)2), 22.78 (CH(CH3)2), 24.99 (CH(CH3)2), 27.73 (CH(CH3)2), 27.86 (CH(CH3)2), 28.01 (CH(CH3)2), 31.64 (CH(CH3)2), 32.16 (CH(CH3)2), 32.33 (O–C(CH3)3), 33.68 (O–C(CH3)3), 76.31 (O–C(CH3)3), 118.65 (Ar–C), 122.03 (Ar–C), 122.69 (Ar–C), 123.15 (Ar–CH3), 124.36 (Ar–C), 125.24 (Ar–C), 127.70 (Ar–Cl), 128.16 (Ar–C), 128.97 (Ar–C), 137.67 (Ar–C), 149.65 (Ar–C), 157.92 (Ar–O), 162.27 (CHN). ESI m/z calculated for [M+H]+. C72H94Cl2N4O4Hf: 1328.937 found 1329.931.
N, 4H). 13C NMR (100 MHz, CDCl3): δ = 21.38 (Ar–CH3), 22.37 (CH(CH3)2), 22.89 (CH(CH3)2), 23.53 (CH(CH3)2), 27.68 (CH(CH3)2), 27.84 (CH(CH3)2), 27.96 (CH(CH3)2), 31.17 (CH(CH3)2), 31.17 (CH(CH3)2), 32.22 (O–C(CH3)3), 33.08 (O–C(CH3)3), 56.11 (Ar–OCH3), 69.85 (O–C(CH3)3), 118.43 (Ar–C), 122.49 (Ar–C), 122.68 (Ar–C), 123.06 (Ar–CH3), 125.22 (Ar–C), 128.15 (Ar–C), 128.95 (Ar–C), 137.77 (Ar–C), 138.08 (Ar–CH3), 139.48 (Ar–C), 148.52 (Ar–C), 152.15 (Ar–OCH3), 155.87 (Ar–O), 162.68 (CHN). ESI m/z calculated for [M]+. C74H100N4O6Hf: 1320.099 found 1320.368.
N, 2H), 8.96 (s, CHN, 2H). 13C NMR (100 MHz, CDCl3): δ = 20.06 (O–Ar–CH3), 32.95 (Ar–O–CH3), 55.36 (Ar–O–CH3), 55.44 (Ar–O–CH3), 75.52 (O–C(CH3)3), 113.22 (Ar–C), 114.28 (Ar–C), 122.27 (Ar–C), 123.17 (Ar–C), 123.61 (Ar–C), 125.85 (Ar–C), 126.03 (Ar–C), 132.62 (Ar–C), 137.52 (Ar–C), 145.14 (Ar–C), 145.84 (Ar–C), 155.84 (Ar–O), 157.63 (Ar–O–CH3), 157.85 (Ar–O–CH3), 161.02 (CHN), 166.44 (CHN).
Crystallographic data
Crystals of appropriate dimensions of 3, 5 and 10, suitable for X-ray diffraction studies were grown from toluene at −24 °C. A Bruker AXS (Kappa Apex 2) CCD diffractometer equipped with graphite monochromated Mo (Kα) (λ = 0.7107 Å) radiation source was employed for the collection of crystal data. ω and ϕ scans was employed to collect the data. The frame width for ω was set to 0.5° for all data collection. The data were collected with 100% completeness for θ up to 25°. The frames were integrated and the data collected were reduced for Lorentz and polarization corrections using SAINT-NT. The data set was subjected to the multi-scan absorption correction. All structures were solved using SIR-92 and refined using SHELXL-97.20 The non hydrogen atoms were refined with anisotropic displacement parameter. All the hydrogen atoms could be located in the difference Fourier map. The hydrogen atoms bonded to carbon atoms were fixed at chemically meaningful positions and were allowed to ride with the parent atom during refinement. These data were deposited with CCDC.Typical procedure for the bulk polymerization of CL, VL, rac-BL, L-LA and rac-LA
The procedure for the bulk polymerizations in 200:1 ratio between respective monomers and 1–9 are outlined below:
For CL polymerization, in an ampoule sealed under argon atmosphere, 11.28 μmol of 1–9 was added to 0.25 mL of monomer. The contents were stirred and immersed in a bath preheated to 80 °C. A rise in viscosity of the polymerization reaction was observed and finally the stirring ceased. The contents were dissolved into minimum quantity of CH2Cl2 and poured into cold methanol. The polymer precipitated immediately and was isolated by filtration. The filtered product was dried in vacuum in a vacuum oven until a constant weight was attained. Similarly for, VL polymerization, 13.47 μmol of 1–9 was used for 0.25 mL of monomer. The polymerization was performed at 80 °C and same procedure for work up was followed.
Again, for rac-BL polymerization, 15.33 μmol of 1–9 was used for 0.25 mL of monomer. The polymerization was performed at 80 °C and same procedure for work up was followed.
For L-LA or rac-LA, polymerization, 8.67 μmol of 1–9 and 0.25 g L-LA or rac-LA were taken in an ampoule under an argon atmosphere. The contents were rapidly stirred at 130 °C. Once the monomer melted completely, rise in viscosity of the polymerization reaction was observed and finally the stirring ceased. The contents were dissolved into minimum quantity of CH2Cl2 and poured into cold methanol. The polymer precipitated immediately and was isolated by filtration. The filtered product was dried in vacuum until a constant weight was attained.
Typical procedure for ethylene polymerization
The polymerizations were done in a 50 mL flask equipped with a magnetic stirrer. The flask was charged in an argon filled glove box with 8 mg of 1–9, 7 mL toluene along with the required amount of MAO. The flask was connected to a Schlenk line and the contents were subjected to repeated freeze thaw cycles. Finally, the flask was immersed in an oil bath preheated to 80 °C and ethylene gas was bubbled. Polymerization was noticed immediately. The ethylene feed was passed for 30 mins and subsequently the polymerization was quenched with acidic methanol. The polymer produced was collected by filtration and dried till constant weight was achieved.Characterization of polymers
Data concerning molecular weights and the polydispersity indices of the polymer samples obtained by the ring-opening polymerization of various cyclic ester monomers and lactide were determined by using a GPC instrument with Waters 510 pump and Waters 410 differential refractometer as the detector. Three columns namely WATERS STRYGEL-HR5, STRYGEL-HR4 and STRYGEL-HR3 each of dimensions (7.8 × 300 mm) were connected in series. Measurements were done in THF at 27 °C. Number average molecular weights (Mn) and polydispersity (Mw/Mn) (MWDs) of polymers were measured relative to polystyrene standards.Molecular weights (Mn and Mw) and the polydispersity indices (Mw/Mn) of ethylene samples were determined by GPC instrument with Waters 510 pump and Waters 2414 differential refractometer as the detector. The column namely WATERS STRYGEL-HR4 of dimensions (4.6 × 300 mm) was connected during the experiment. Measurements were done in tri-chloro benzene (TCB). Number average molecular weights (Mn) and molecular weight distributions (MWDs) of polymers were measured relative to polystyrene standards.
Polymerization kinetics
Bulk polymerization of different cyclic ester monomers and lactide using 3, 6 and 9 were carried out at 130 °C under an argon atmosphere. At different appropriate intervals of time, 0.2 mL aliquots were removed from the reaction mixture and poured directly into CDCl3 containing 250 ppm BHT. The contents of the quenched aliquots obtained at various time intervals were analyzed by 1H NMR for the determination of conversion. The [L-LA]o/[L-LA]t ratio was calculated by integration of the peak corresponding to the methine proton for the polymer and monomer. Apparent rate constant were obtained from the slopes of the best fit lines.Acknowledgements
This work was supported by the research grant received from Department of Science and Technology, New Delhi, Government of India. TKS and BR thank the Council of Scientific and Industrial Research and the University Grants Commission, New Delhi for a research fellowship. The authors thank Mr. V. Ramkumar, Department of Chemistry, Indian Institute of Technology Madras, for X-ray diffraction studies. The authors express their gratitude to the referees for their comments and suggestions.References
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- (a) J. Lunt, Polym. Degrad. Stab., 1998, 59, 145–152 CrossRef CAS; (b) S. K. Ritter, Chem. Eng. News, 2002, 80, 26–27; (c) E. T. H. Vink, K. R. Rábago, D. A. Glassner and P. R. Gruber, Polym. Degrad. Stab., 2003, 80, 403–419 CrossRef CAS.
- (a) A. –C. Albertsson and I. K. Varma, Adv. Polym. Sci., 2000, 157, 1–40 CrossRef; (b) R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS; (c) M. Okada, Prog. Polym. Sci., 2002, 27, 87–133 CrossRef CAS.
- (a) H. R. Kricheldorf, Chemosphere, 2000, 43, 49–54; (b) Y. Ikada and H. Tsuji, Macromol. Rapid Commun., 2000, 212, 117–132 CrossRef; (c) O. Coulembier, P. Degée, J. L. Hedrick and P. Dubois, Prog. Polym. Sci., 2006, 31, 723–747 CrossRef CAS.
- (a) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (b) G. W. Coates, J. Chem. Soc., Dalton Trans., 2002, 467–475 RSC; (c) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef; (d) J. Wu, T. –L. Yu, C. –T. Chen and C. –C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS; (e) S. Penczek, M. Cypryk, A. Duda, P. Kubisa and S. Slomkowski, Prog. Polym. Sci., 2007, 32, 247–282 CrossRef CAS; (f) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 Search PubMed; (g) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC; (h) M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484–3504 RSC; (i) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (j) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- (a) H. R. Kricheldorf, M. Berl and N. Scharnagl, Macromolecules, 1988, 21, 286–293 CrossRef CAS; (b) D. Takeuchi, T. Nakamura and T. Aida, Macromolecules, 2000, 33, 725–729 CrossRef CAS; (c) Y. Kim and J. G. Verkade, Macromol. Rapid Commun., 2002, 23, 917–921 CrossRef CAS; (d) Y. Kim and J. G. Verkade, Organometallics, 2002, 21, 2395–2399 CrossRef; (e) Y. Kim, G. K. Jnaneshwara and J. G. Verkade, Inorg. Chem., 2003, 42, 1437–1447 CrossRef CAS; (f) Y. Takashima, Y. Nakayama, T. Hirao, H. Yasuda and A. Harada, J. Organomet. Chem., 2004, 689, 612–619 CrossRef CAS; (g) Y. Kim and J. G. Verkade, Macromol. Symp., 2005, 224, 105–118 CrossRef CAS; (h) S. K. Russell, C. L. Gamble, K. J. Gibbins, K. C. S. Juhl, W. S. Mitchell, A. J. Tumas and G. E. Hofmeister, Macromolecules, 2005, 38, 10336–10340 CrossRef CAS; (i) C. K. A. Gregson, I. J. Blackmore, V. C. Gibson, N. J. Long, E. L. Marshall and A. J. P. White, Dalton Trans., 2006, 3134–3140 RSC; (j) D. Patel, S. T. Liddle, S. A. Mungur, M. Rodden, A. Blake and P. L. Arnold, Chem. Commun., 2006, 1124–1126 RSC; (k) S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS; (l) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS; (m) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2006, 887–889 RSC; (n) K. –C. Hsieh, W. –Y. Lee, L. –F. Hsueh, H. M. Lee and J. –H. Huang, Eur. J. Inorg. Chem., 2006, 2306–2312 CrossRef CAS; (o) J. Cayuela, V. Bounor-Legaré, P. Cassagnau and A. Michel, Macromolecules, 2006, 39, 1338–1346 CrossRef CAS; (p) C. J. Chuck, M. G. Davidson, M. D. Jones, G. Kociok-Köhn, M. D. Lunn and S. Wu, Inorg. Chem., 2006, 45, 6595–6597 CrossRef CAS; (q) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC; (r) F. Gornshtein, M. Kapon, M. Botoshansky and M. Eisen, Organometallics, 2007, 26, 497–507 CrossRef CAS; (s) R. C. J. Atkinson, K. Gerry, V. C. Gibson, N. J. Long, E. L. Marshall and L. J. West, Organometallics, 2007, 26, 316–320 CrossRef CAS; (t) J. Lee, Y. Kim and Y. Do, Inorg. Chem., 2007, 46, 7701–7703 CrossRef CAS; (u) S. Mun, J. Lee, S. H. Kim, Y. Hong, Y. –H. Ko, Y. K. Shin, J. H. Lim, C. S. Hong, Y. Do and Y. Kim, J. Organomet. Chem., 2007, 692, 3519–3525 CrossRef CAS; (v) A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437–1443 RSC; (w) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC; (x) Y. Ning, Y. Zhang, A. R. Delgado and E. Y. –X. Chen, Organometallics, 2008, 27, 5632–5640 CrossRef CAS; (y) R. F. Munha, L. G. Alves, N. Maulide, M. T. Duarte, I. E. Marko, M. D. Fryzuk and A. M. Martins, Inorg. Chem. Commun., 2008, 11, 1174–1176 CrossRef CAS; (z) K. Krauzy-Dziedzic, J. Ejfler, S. Szafert and P. Sobota, Dalton Trans., 2008, 2620–2626 RSC.
- (a) P. Dobrzynski and J. Polym, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 1886–1900 CrossRef CAS; (b) J. Cheng, J. Sun, K. Wu and X. Yan, Huaxue Gongye Yu Gongcheng Jishu, 2006, 27, 5–7 Search PubMed; (c) A. J. Chmura, D. M. Cousins, M. G. Davidson, M. D. Jones, M. D. Lunn and M. F. Mahon, Dalton Trans., 2008, 1437–1443 RSC; (d) A. Grafov, S. Vuorinen, T. Repo, M. Kemell, M. Nieger and M. Leskelä, Eur. Polym. J., 2008, 44, 3797–3805 CrossRef CAS; (e) G. Zi, Q. Wang, L. Xiang and H. Song, Dalton Trans., 2008, 5930–5944 RSC; (f) A. L. Zelikoff, J. Kopilov, I. Goldberg, G. W. Coates and M. Kol, Chem. Commun., 2009, 6804–6806 RSC; (g) A. D. Schwarz, A. L. Thompson and P. Mountford, Inorg. Chem., 2009, 48, 10442–10454 CrossRef CAS; (h) R. H. Howard, C. Alonso-Moreno, L. M. Broomfield, D. L. Hughes, J. A. Wright and M. Bochmann, Dalton Trans., 2009, 8667–8682 RSC; (i) E. Kim, E. W. Shin, I. –K. Yoo and J. S. Chung, J. Mol. Catal. A: Chem., 2009, 298, 36–39 CrossRef CAS; (j) E. L. Whitelaw, M. D. Jones, M. F. Mahon and G. Kociok-Kohn, Dalton Trans., 2009, 9020–9025 RSC; (k) M. D. Jones, M. G. Davidson and G. Kociok-Kohn, Polyhedron, 2010, 29, 697–700 CrossRef CAS; (l) F. Zhang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1993–1999 CrossRef CAS; (m) E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Inorg. Chem., 2010, 49, 3977–3979 CrossRef CAS; (n) A. D. Schwarz, K. R. Herbert, C. Paniagua and P. Mountford, Organometallics, 2010, 29, 4171–4188 CrossRef CAS; (o) M. Hu, M. Wang, H. Zhu, L. Zhang, H. Zhang and L. Sun, Dalton Trans., 2010, 39, 4440–4446 RSC; (p) M. G. Davidson, S. D. Bull, T. R. Forder, C. J. Frankis and M. D. Jones, Polymer Preprints, 2010, 51, 390–391 CAS; (q) A. Stopper, I. Goldberg and M. Kol, Inorg. Chem. Commun., 2011, 14, 715–718 CrossRef CAS; (r) S. L. Hancock, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 2033–2037 RSC.
- (a) R. R. Gowda, D. Chakraborty and V. Ramkumar, Eur. J. Inorg. Chem., 2009, 2981–2993 CrossRef CAS; (b) T. K. Saha, B. Rajashekhar, R. R. Gowda, V. Ramkumar and D. Chakraborty, Dalton Trans., 2010, 39, 5091–5093 RSC; (c) R. R. Gowda, D. Chakraborty and V. Ramkumar, Polymer, 2010, 51, 4750–4759 CrossRef CAS; (d) R. R. Gowda, D. Chakraborty and V. Ramkumar, J. Organomet. Chem., 2011, 696, 572–580 CrossRef CAS; (e) T. K. Saha, V. Ramkumar and D. Chakraborty, Inorg. Chem., 2011, 50, 2720–2722 CrossRef CAS.
- (a) K. Ziegler, H. Kolzkamp, H. Breil and H. Martin, Angew. Chem., 1955, 67, 541–544 CrossRef; (b) G. Natta, P. Pino, P. Corradini, F. Danusso, E. Mantica, G. Mazzanti and G. Moraglio, J. Am. Chem. Soc., 1955, 77, 1708–1710 CrossRef CAS; (c) G. Natta, Angew. Chem., 1956, 68, 393–396 CrossRef CAS; (d) H. Sinn and W. Kaminsky, Adv. Organomet. Chem., 1980, 18, 99–149 CAS; (e) H. Sinn, W. Kaminsky, H. J. Vollmer and R. Woldt, Angew. Chem., Int. Ed. Engl., 1980, 19, 390–392 CrossRef.
- (a) G. W. Coates and R. M. Waymouth, Science, 1995, 267, 217–219 CrossRef CAS; (b) E. Hauptman, R. M. Waymouth and J. W. Ziller, J. Am. Chem. Soc., 1995, 117, 11586–11587 CrossRef CAS; (c) R. Kravchenko, A. Masood and R. M. Waymouth, Organometallics, 1997, 16, 3635–3639 CrossRef CAS; (d) J. L. Maciejewski-Petoff, M. D. Bruce, R. M. Waymouth, A. Masood, T. K. Lal, R. W. Quan and S. J. Behrend, Organometallics, 1997, 16, 5909–5916 CrossRef; (e) M. D. Bruce, G. W. Coates, E. Hauptman, R. M. Waymouth and J. W. Ziller, J. Am. Chem. Soc., 1997, 119, 11174–11182 CrossRef CAS; (f) R. Kravchenko and R. M. Waymouth, Macromolecules, 1998, 31, 1–6 CrossRef CAS; (g) R. Kravchenko, A. Masood, R. M. Waymouth and C. L. Myers, J. Am. Chem. Soc., 1998, 120, 2039–2046 CrossRef CAS; (h) Y. Hu, M. T. Krejchi, C. D. Shah, C. L. Myers and R. M. Waymouth, Macromolecules, 1998, 31, 6908–6916 CrossRef CAS; (i) E. D. Carlson, M. T. Krejchi, C. D. Shah, T. Terakawa, R. M. Waymouth and G. G. Fuller, Macromolecules, 1998, 31, 5343–5351 CrossRef CAS; (j) S. Lin and R. M. Waymouth, Acc. Chem. Res., 2002, 35, 765–773 CrossRef CAS.
- (a) A. O. Patil and G. G. Haltky, ACS Symp. Ser., 2003, 857, 1–11 CrossRef CAS; (b) V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283–316 CrossRef CAS.
- (a) J. D. Scollard and D. H. McConville, J. Am. Chem. Soc., 1996, 118, 10008–10009 CrossRef CAS; (b) F. G. N. Cloke, T. J. Geldbach, P. B. Hitchcock and J. B. Love, J. Organomet. Chem., 1996, 506, 343–345 CrossRef CAS; (c) Z. Ziniuk, I. Goldberg and M. Kol, Inorg. Chem. Commun., 1999, 2, 549–551 CrossRef CAS; (d) K. Nomura, K. Oya and Y. Imanishi, Polymer, 2000, 41, 2755–2764 CrossRef CAS; (e) C. H. Lee, Y. –H. La and J. W. Park, Organometallics, 2000, 19, 344–351 CrossRef CAS; (f) E. Y. Tshuva, I. Goldberg and M. Kol, J. Am. Chem. Soc., 2000, 122, 10706–10707 CrossRef CAS; (g) E. Y. Tshuva, I. Goldberg, M. Kol and Z. Goldschmidt, Organometallics, 2001, 20, 3017–3028 CrossRef CAS; (h) S. J. Kim, I. N. Jung, B. R.Yoo, S. H. Kim, J. J. Ko, D. J. Byun and S. O. Kang, Organometallics, 2001, 20, 2136–2144 CrossRef CAS; (i) S. Danie`le, P. B. Hitchcock, M. F. Lappert and P. G. Merle, J. Chem. Soc., Dalton Trans., 2001, 13–19 RSC; (j) H. Hagimoto, T. Shiono and T. Ikeda, Macromol. Rapid Commun., 2002, 23, 73–76 CrossRef CAS.
- (a) H. Mack and M. S. Eisen, J. Organomet. Chem., 1996, 525, 81–87 CrossRef CAS; (b) R. R. Schrock, R. Baumann, S. M. Reid, J. T. Goodman, R. Stumpf and W. M. Davis, Organometallics, 1999, 18, 3649–3670 CrossRef CAS; (c) D. D. Graf, R. R. Schrock, W. M. Davis and R. Stumpf, Organometallics, 1999, 18, 843–852 CrossRef CAS; (d) L. –C. Liang, R. R. Schrock, W. M. Davis and D. H. McConville, J. Am. Chem. Soc., 1999, 121, 5797–5798 CrossRef CAS; (e) P. Mehrkhodavandi, P. J. Bonitatebus Jr. and R. R. Schrock, J. Am. Chem. Soc., 2000, 122, 7841–7842 CrossRef CAS; (f) L. H. Gade, Chem. Commun., 2000, 173–181 RSC; (g) R. R. Schrock, P. J. Bonitatebus Jr. and Y. Schrodi, Organometallics, 2001, 20, 1056–1058 CrossRef CAS; (h) R. R. Schrock, P. J. Bonitatebus Jr. and Y. Schrodi, Organometallics, 2001, 20, 3560–3573 CrossRef CAS; (i) J. T. Goodman and R. R. Schrock, Organometallics, 2001, 20, 5205–5211 CrossRef CAS; (j) E. Shaviv, M. Botoshansky and M. S. Eisen, J. Organomet. Chem., 2003, 683, 165–180 CrossRef CAS.
- (a) V. Busico, R. Cipullo, L. Caporaso, G. Angelini and A. L. Segre, J. Mol. Catal. A: Chem., 1998, 128, 53–64 CrossRef CAS; (b) J. Richter, F. T. Edelmann, M. Noltemeyer, H.-G. Schmidt, M. Shmulinson and M. S. Eisen, J. Mol. Catal. A: Chem., 1998, 130, 149–162 CrossRef CAS; (c) V. Volkis, M. Shmulinson, C. Averbuj, A. Lisovskii, F. T. Edelmann and M. S. Eisen, Organometallics, 1998, 17, 3155–3157 CrossRef CAS; (d) K. C. Jayaratne and L. R. Sita, J. Am. Chem. Soc., 2000, 122, 958–959 CrossRef CAS; (e) R. J. Keaton, K. C. Jayaratne, D. A. Henningsen, L. A. Koterwas and L. R. Sita, J. Am. Chem. Soc., 2001, 123, 6197–6198 CrossRef CAS; (f) K. C. Jayaratne and L. R. Sita, J. Am. Chem. Soc., 2001, 123, 10754–10755 CrossRef CAS; (g) V. Volkis, E. Nelkenbaum, A. Lisovskii, G. Hasson, R. Semiat, M. Kapon, M. Botoshansky, Y. Eishen and M. S. Eisen, J. Am. Chem. Soc., 2003, 125, 2179–2194 CrossRef CAS.
- (a) Y. Nakayama, K. Watanabe, N. Ueyama, A. Nakamura, A. Harada and J. Okuda, Organometallics, 2000, 19, 2498–2503 CrossRef CAS; (b) R. M. Gauvin, J. A. Osborn and J. Kress, Organometallics, 2000, 19, 2944–2946 CrossRef CAS; (c) P. Sobota, K. Przybylak, J. Utko, L. B. Jerzykiewicz, A. J. L. Pombeiro, M. da Silva and K. Szczegot, Chem.–Eur. J., 2001, 7, 951–958 CrossRef CAS; (d) J. Saito, M. Mitani, J. Mohri, Y. Yoshida, S. Matsui, S. Ishii, S. Kojoh, N. Kashiwa and T. Fujita, Angew. Chem., Int. Ed., 2001, 40, 2918–2920 CrossRef CAS; (e) N. Matsukawa, S. Matsui, M. Mitani, J. Saito, K. Tsuru and T. Fujita, J. Mol. Catal. A: Chem., 2001, 169, 99–104 CrossRef CAS; (f) S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123, 6847–6856 CrossRef CAS.
- M. Veith, S. Mathur, C. Mathur and V. Huch, J. Chem. Soc., Dalton Trans., 1997, 2101–2108 RSC.
- L. F. Sánchez–Barba, A. Garcés, J. Fernández-Baeza, A. Otero, C. Alonso–Moreno, A. Lara–Sánchez and A. M. Rodríguez, Organometallics, 2011, 30, 2775–2789 CrossRef CAS.
- A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J. –F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS.
- (a) L. Han, J. Du, H. Yang, H. Wang, X. Leng, A. Galstyan, S. Zariæ and W. –H. Sun, Inorg. Chem. Commun., 2003, 6, 5–9 CrossRef CAS; (b) J. Du, L. Han, Y. Cui, J. Li, Y. Li and W. –H. Sun, Aust. J. Chem., 2003, 56, 703–706 CrossRef CAS; (c) Al–Jeboori, M. Jaber, Al–Shihri and S. Ayed, J. Saudi Chem. Soc., 2001, 5, 341–352 Search PubMed.
- G. M. Sheldrick, SHELXL97. Program for crystal structure refinement, Göttingen, Germany Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC reference numbers 836424–836426. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00543j |
| ‡ This paper is dedicated to Prof. Herbert W. Roesky on the occasion of his 75th birthday. |
| This journal is © The Royal Society of Chemistry 2012 |