Electrospun carbon nanofibers decorated with various amounts of electrochemically-inert nickel nanoparticles for use as high-performance energy storage materials†
Liwen
Ji‡
,
Zhan
Lin‡
,
Mataz
Alcoutlabi‡
,
Ozan
Toprakci‡
,
Yingfang
Yao
,
Guanjie
Xu
,
Shuli
Li
and
Xiangwu
Zhang
*
Fiber and Polymer Science Program, Department of Textile Engineering, Chemistry and Science, North Carolina State University, Raleigh, NC 27695-8301, USA. E-mail: xiangwu_zhang@ncsu.edu; Fax: 919-515-6532; Tel: 919-515-6547
First published on 31st October 2011
Abstract
Carbon nanofibers decorated with various amounts of electrochemically-inert metallic nickel nanoparticles are synthesized through electrospinning and carbonization processes. The morphology and composition of Ni nanoparticles in carbon nanofibers are controlled by preparing different nanofiber precursors. The lithium-ion battery performance evaluations indicated that the content of electrochemically-inert Ni nanoparticles in carbon nanofibers has a great influence on the final electrochemical performance. For example, at certain Ni contents, these composite nanofibers display excellent electrochemical performance, such as high reversible capacities, good capacity retention, and excellent rate performance, when directly used as binder-free anodes for rechargeable lithium-ion batteries. However, when the Ni content is too low or too high, the corresponding electrodes show low reversible capacities although they still have good reversibility and rate performance.
Introduction
Rechargeable lithium-ion batteries (LIBs) have been widely used for energy storage in electronic devices such as laptops, cell phones, and digital cameras, and they are currently being studied for advanced electric and hybrid vehicles.1–7 It is very important to develop LIBs with high capacity and good rate capability to meet the energy demands of our society.1–7 Since the performance of a battery depends decisively on the properties of the electrode materials, it is necessary to seek and design novel nanostructured electrodes with large reversible storage capacity, high charge/discharge efficiency, and good rate capability. Nanostructured electrodes exhibit high surface areas and good transport properties, and they can offer an exciting solution to achieve increased storage capacity and enhanced energy/powder densities through fast interfacial kinetics.1–7 However, single-phase nanoscale electrodes are certainly not a panacea and they may not resolve all of the limitations faced by their bulk counterparts when used as electrodes for rechargeable LIBs because of their intrinsic material properties in both thermodynamics and kinetics aspects.2,4–10 In addition, single-phase nanostructure electrodes often have undesirable side reactions with the electrolyte that can lead to low reversibility and poor capacity retention.2,4–10 Therefore, the integration of different components into heterogeneous nanostructures is of particular importance because, in these well-designed multi-phase nanostructures, each component can address different problems and their combinative effects can eventually help realize the full potential of the different components in terms of energy/power density, cycle life, rate performance, etc.Nanostructured carbon-based electrodes, such as nanotubes, nanofibers and graphene, exhibit good chemical and thermal stability, excellent mechanical stress tolerance (ductility), extraordinary electronic and ionic conductivities, and also considerable Li-storage ability.2,11–21 Often, a carbon matrix is used as the host to accommodate Li-active nanoparticles, such as Si, Ge, Sn and some transition metal oxides, and to buffer the large volume change resulting from repeated lithium insertion and extraction.1,2,4,12,14,22 The resultant carbon-based multiphase nanocomposite electrodes show improved electrochemical performance compared to individual carbon and Li-active nanoparticles. Among all nanostructured carbon materials, carbon nanofibers (CNFs) are a promising one-dimensional structure with high aspect ratio and high surface-to-volume ratio, which can offer a direct pathway for efficient charge transport along the fiber axis when used as anodes for rechargeable LIBs. CNFs can also form interconnected network structures that can facilitate the percolation of electric charges throughout the electrodes and provide large surface areas that facilitate the fast diffusion of Li ions.2,23
Electrochemically-inert transition metal nanoparticles, such as Ni, Cu, Fe, and Nb, are inactive with Li.3,22 However, these transition metal nanoparticles can create conducting networks in electrodes to promote the electrical contact of the electrodes and to provide better endurance of high current densities upon charge/discharge cycling. These nanoscale transition metal nanoparticles also show the potential to stabilize the solid electrolyte interface (SEI) film during the cycle process, reduce the initial irreversible capacity, and improve the Coulombic efficiency and cycling performance of electrodes at high current densities.3,22,24–28 Hence, incorporating these transition metal particles into nanoscale carbon for use as LIB electrodes has a great beneficial impact on the overall battery performance.
In this work, CNFs decorated with different amounts of electrochemically-inert Ni nanoparticles are prepared via the electrospinning of polyacrylonitrile (PAN) solutions containing various concentrations of nickel(II) acetate tetrahydrate (Ni(OAc)2·4H2O) and sequential thermal treatments in both air and inert gas environments at high temperatures. These Ni/C composite nanofibers, in the form of freely-standing films, are directly used as binder-free anodes (negative electrodes) for LIBs. Results indicate that the Ni content in the CNF matrix has a large influence on the final electrochemical performance. At certain Ni contents, Ni/C composite nanofibers exhibit high reversible capacity, enhanced cyclability, and excellent rate capability, which have been problematic for the pure CNF electrodes. These results demonstrated that decorating CNFs with electrochemically-inert transition metal nanoparticles could provide a relatively simple, scalable, and economical route to prepare nanomaterials for use in LIB electrodes. In addition, this novel strategy may also open a new avenue to design one-dimensional transitional metal nanoparticle-loaded carbon nanofibers for lithium-air batteries, fuel cells, and other energy storage and conversation systems.
Experimental
Materials preparation and characterization
PAN (Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Fisher Scientific, and Ni(OAc)2·4H2O salt (99.99%) and solvent N,N-dimethylformamide (DMF) were purchased from Aldrich. All these reagents were used without further purification. DMF solutions of PAN (8 wt%) containing different amounts of Ni(OAc)2·4H2O (15, 30, and 50 wt%) were prepared at 60 °C. Mechanical stirring was applied for at least 72 h in order to obtain homogeneously-distributed solutions.
000) was purchased from Fisher Scientific, and Ni(OAc)2·4H2O salt (99.99%) and solvent N,N-dimethylformamide (DMF) were purchased from Aldrich. All these reagents were used without further purification. DMF solutions of PAN (8 wt%) containing different amounts of Ni(OAc)2·4H2O (15, 30, and 50 wt%) were prepared at 60 °C. Mechanical stirring was applied for at least 72 h in order to obtain homogeneously-distributed solutions.
A variable high-voltage power supply (Gamma ES40P) was used to provide a high voltage of about 15 kV for electrospinning with 0.5 ml h−1 flow rate and 15 cm needle-to-collector distance. The electrospun Ni(OAc)2·4H2O/PAN precursor nanofibers were first stabilized in an air environment at 280 °C for 6 h (heating rate was 5 °C min−1) and then carbonized at 600 °C for 8 h in an argon atmosphere (heating rate was 2 °C min−1) to form Ni/C composite nanofibers. Elemental analysis indicated that the Ni contents of Ni/C nanofibers prepared from Ni(OAc)2·4H2O/PAN precursors with 15, 30, 50 wt% Ni salt were about 5, 12, and 16 wt%, respectively.
The morphology and microstructures of Ni(OAc)2·4H2O/PAN precursor nanofibers and the carbonized nanofibers were evaluated using scanning electron microscopy (JEOL 6400F Field Emission SEM at 5 kV) and transmission electron microscopy (Hitachi HF-2000 TEM at 200 kV) coupled with energy-dispersive X-ray spectroscopy (EDX). Thermal properties of the electrospun nanofibers were evaluated using differential scanning calorimetry (DSC) from 25 to 400 °C at a heating rate of 10 °C min−1 in a nitrogen environment (Perkin Elmer Diamond Series DSC with Intracooler). Thermo-gravimetric analysis (TGA) was also used to determine the weight loss of nanofibers at 10 °C min−1 from 25 to 800 °C in air environment (TA Instruments Hi-Res TGA 2950). The structural variations of carbonized nanofibers were identified by wide angle X-ray diffraction (WAXD, Philips X’Pert PRO MRD HR X-Ray Diffraction System, Cu-Kα, λ = 1.5405 Å.). X-Ray photoelectron spectroscopy (XPS) was also used to measure elemental compositions and chemical states.
Electrochemical evaluation
Electrochemical performance was evaluated using 2032 button coin cells (Hohsen Corp.). Ni/C nanofibers were directly attached onto copper foils to be used as the binder-free working electrode. Lithium foil (0.38 mm thick, Aldrich) and Separion S240 P25 (Degussa AG, 25 μm) were used as the counter electrode and separator, respectively. The electrolyte used was 1 M lithium hexafluorophosphate (LiPF6), dissolved in 1/1 (v/v) ethylene carbonate (EC)/ethyl methyl carbonate (EMC) (Ferro Corp.). Coin cells were assembled in a high-purity argon-filled glove box. Cyclic voltammetry (CV) measurements were performed on an AQ4 Gamry Reference 600 electrochemical workstation with a voltage ranging from 0.001 to 3.0 V at a scan rate of 0.05 mV s−1. Charge (lithium insertion) and discharge (lithium extraction) were conducted using an Arbin automatic battery cycler at different current densities between cut-off potentials of 0.01 and 2.80 V.Results and discussion
Structure characterization
Fig. 1 and S1 (see ESI†) show SEM and TEM images of electrospun Ni(OAc)2·4H2O/PAN precursor nanofibers with different Ni(OAc)2·4H2O concentrations, respectively. Electrospun Ni(OAc)2·4H2O/PAN nanofibers are straight and exhibit homogeneously-distributed diameters in the range of 500 to 800 nm. However, the surface morphology of these electrospun nanofibers is irregular and uneven. In addition, with increase in Ni(OAc)2·4H2O concentration, the surface becomes rougher and some undulating structures and even pores are clearly shown. Moreover, with the increase of Ni(OAc)2·4H2O concentration, the diameter of the Ni(OAc)2·4H2O/PAN nanofibers also increases, and Ni(OAc)2·4H2O/PAN nanofibers with different Ni(OAc)2·4H2O concentrations of 15, 30, and 50 wt% have average diameters of about 500, 700, and 900 nm, respectively. With increase in Ni(OAc)2·4H2O concentration, the solution viscosity increases and results in larger diameters for Ni(OAc)2·4H2O/PAN nanofibers.29–32 | ||
| Fig. 1 SEM images of Ni(OAc)2·4H2O/PAN composite nanofibers with different Ni(OAc)2·4H2O concentrations: (A, B) 15, (C, D) 30, and (E, F) 50 wt%. | ||
The DSC analysis of pure PAN and Ni(OAc)2·4H2O/PAN nanofiber precursors was conducted at a heating rate of 10 °C min−1 in nitrogen environment, as shown in Fig. 2A. Pure PAN nanofibers exhibit a relatively large and sharp exothermic peak at around 290 °C and this might be due to multiple complex chemical reactions, such as dehydrogenation, instantaneous cyclization, and crosslinking reactions associated with the oxidative stabilization of PAN.3,11,23,29–31 In the presence of Ni(OAc)2·4H2O salt, the exothermic peak shifts to lower temperatures. At the same time, the peak intensity, which indicates the total heat of reaction, decreases as Ni(OAc)2·4H2O concentration increases. The reductions in reaction temperature and peak intensity probably resulted from the interactions between PAN and Ni(OAc)2·4H2O that decrease the formation of free radicals on the nitrile groups in PAN and subsequently lower their recombination and the heat of reaction.23,29–31,33 In addition, 50 wt% Ni(OAc)2·4H2O/PAN nanofibers display an indistinguishable exothermic peak along with a relatively wide endothermic peak. Generally, any endotherm or exotherm is an indication of structural change. PAN degrades well below the melting temperature because it undergoes certain exothermic reactions leading to the formation of ladder structures.23,29–32,34 The downshifted exothermic peak in the presence of 50 wt% Ni(OAc)2·4H2O provides the evidence that the Ni(OAc)2·4H2O compound accelerates the onset of oxidative stabilization.23,29–31,33 Furthermore, the endothermic peak only appears when the Ni(OAc)2·4H2O concentration increases to 50 wt%. The decomposition of Ni(OAc)2·4H2O was studied by Mohamed et al.,35 and their DSC results showed several endothermic events. For example, there are two overlapping endothermic peaks located at 249 and 360 °C, indicating the decomposition of Ni(OAc)2·4H2O. There is also another endothermic event above 400 °C, which can be attributed to the decomposition of NiO to Ni metal.35 Other literature results also show that the non-isothermal decomposition of Ni(OAc)2·4H2O is a complex multi-step reaction.36–38 These results agree with our DSC data for the 50 wt% Ni(OAc)2·4H2O/PAN nanofiber precursor, where the existence of the endothermic peak at around 300 °C could be the result of several complex thermal effects of Ni(OAc)2·4H2O.36–38
 | ||
| Fig. 2 DSC (A) and TGA (B) thermograms of Ni(OAc)2·4H2O/PAN composite nanofibers with different Ni(OAc)2·4H2O concentrations: (a) 0 (pure PAN), (b) 15, (c) 30, and (d) 50 wt%. | ||
The thermal studies of pure PAN and Ni(OAc)2·4H2O/PAN precursor nanofibers with Ni(OAc)2·4H2O concentrations of 15, 30, and 50 wt% were also carried out using TGA in air atmosphere, as shown in Fig. 2B. Pure PAN nanofibers degrade at about 320 °C, however, Ni(OAc)2·4H2O/PAN nanofibers begin losing weight at a lower temperature of about 250 °C due to the decomposition of Ni(OAc)2·4H2O salt. The decomposition of Ni(OAc)2·4H2O salt also destroys their interactions with PAN chains and accelerates the oxidative reactions of PAN nanofibers, which result in the lower decomposition temperature of PAN in Ni(OAc)2·4H2O/PAN nanofibers.23,29–32,34 Moreover, the final weight loss of pure PAN nanofibers is almost 100% due to its complete decomposition in air. However, the final weight percentages of Ni(OAc)2·4H2O/PAN composite nanofibers do not reach zero due to the fact that the TGA characterizations were carried out in air environment and this leads to the formation of Ni-containing compounds such as NiO after the decomposition (or oxidation) of Ni(OAc)2·4H2O salt.
Ni(OAc)2·4H2O/PAN precursor nanofibers with different Ni(OAc)2·4H2O concentrations were stabilized at 280 °C in air environment, following by carbonization at 600 °C in argon gas. The crystallinity and phase composition of heat-treated nanofibers were investigated by WAXD (Fig. 3). WAXD patterns show that the composite nanofibers exhibit diffraction peaks at 2θ of 44.4°, 51.9°, and 76.5°, which can be ascribed to the (111), (200), and (220) planes of Ni crystal, respectively.3,39,40 With increase in Ni(OAc)2·4H2O concentration, the diffraction peaks become stronger. All these peaks confirm the formation of Ni/C composite nanofibers during the heat-treatment process. Further elemental analysis indicates that the Ni contents of Ni/C composite nanofibers prepared from Ni(OAc)2·4H2O/PAN precursors with 15, 30, 50 wt% Ni salt are about 5, 12, and 16 wt%, respectively. The formation of metallic Ni may be caused by the reducing environment (e.g. some reducing products, such as hydrogen, activated carbon, charcoal, etc.) created during the carbonization process.3,41–44 In addition, a broad diffraction peak at around 25.0° (002) also appears in Fig. 3, indicating the disordered structure of typical carbon materials in the as-prepared Ni/C composite nanofibers.29,31,39
 | ||
| Fig. 3 WAXD patterns of Ni/C composite nanofibers carbonized from Ni(OAc)2·4H2O/PAN precursors with different Ni(OAc)2·4H2O concentrations: (a) 15, (b) 30, and (c) 50 wt%. | ||
Fig. 4 shows SEM images of Ni/C composite nanofibers with different Ni contents of 5, 12, and 16 wt%. It is seen that the morphology of Ni/C nanofibers is similar to that of the corresponding Ni(OAc)2·4H2O/PAN precursors shown in Fig. 1. However, spherical Ni particles with size around 20 nm are found for Ni/C nanofibers with 12 and 16 wt% Ni. Ni particles are barely seen on 5 wt% Ni/C nanofibers (Fig. 4A,B), which might be due to the relatively small amount of Ni particles in these nanofibers. For Ni/C nanofibers with 12 and 16 wt% Ni, the presence of Ni nanoparticles can be further observed from the TEM images shown in Fig. S2 and the corresponding EDX spectra in Fig. S3.† With increase in Ni content (from 5 to 12 and 16 wt%), more and more Ni particles are found in both SEM and TEM images. In addition, as shown in the EDX spectra in Fig. S3, oxygen peaks are also seen for carbonized nanofibers, which may indicate the formation of amorphous NiO impurities during the thermal treatment process. However, the XRD curves in Fig. 3 do not show any NiO peak, and this may be caused by the small content of NiO impurity or their amorphous nature.
 | ||
| Fig. 4 SEM images of Ni/C composite nanofibers with different Ni contents: (A, B) 5, (C, D) 12, and (E, F) 16 wt%. | ||
In order to determine the surface bond state of the Ni component in Ni/C nanofiber anodes, XPS analysis was conducted. Fig. 5 shows XPS spectra of Ni/C composite nanofibers. The peak at 853 eV for Ni 2p3/2 indicates the existence of Ni0.26,45 However, the Ni 2p3/2 (855 eV) peak shows that there may also exist NiO impurities (Ni2+).26,45 With the increase in Ni content, this peak becomes slightly sharper.
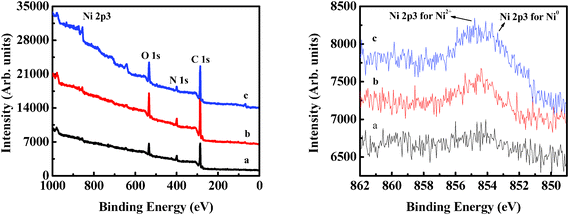 | ||
| Fig. 5 XPS results of Ni/C composite nanofibers with different Ni contents: (a) 5, (b) 12, and (c) 16 wt%. | ||
Electrochemical performance
Fig. 6 shows the CV curves of pure CNFs and Ni/C composite nanofibers. In the first cycle, pure CNF anodes exhibit a clear cathodic peak at about 0.5 V, corresponding to the formation of a SEI film.46Ni/C composite nanofiber anodes exhibit two cathodic peaks at about 0.75 and 1.70 V, respectively. These peaks were related to the formation of partially reversible SEI film and the Li insertion into NiO impurities, forming Li2O and metallic Ni.47 With increase in Ni content, the peak intensity at 0.75 V decreases, while the peak intensity at 1.70 V increases. For Ni/C nanofiber anodes, a wide peak at about 1.05 V and a very small peak at around 2.0 V are also recorded in the first anodic process. The peak at 1.05 V can be attributed to the partial decomposition of SEI, while the one at 2.0 V corresponds to the decomposition of Li2O and the conversion of Ni back to NiO. In the subsequent cycles, the reduction peak at 0.75 V disappears, and the peak at 1.70 V becomes smaller and shifts to slightly higher voltage, while all of the oxidation peaks show slightly decreased intensities. | ||
| Fig. 6 CV curves of Ni/C composite nanofibers with different Ni contents: (A) 0 (pure CNFs), (B) 5, (C) 12, and (D) 16 wt%. | ||
Fig. 7A–C shows typical charge (lithium insertion)/discharge (lithium extraction) curves of Ni/C composite nanofibers at a constant current density of 50 mA g−1 with a potential window of 0.01–2.8 V. From Fig. 7A–C, it is seen that during the first charge process, the voltages steeply decrease to about 0.9–1.0 V and then slowly decrease until total charge capacities of 901, 1245, and 895 mAh g−1 are reached, respectively, for Ni/C composite nanofibers with 5, 12, and 16 wt% Ni. The plateau at 0.9–1.0 V may be associated with the electrolyte decomposition and the formation of solid electrolyte interface (SEI) films, and it may be caused by the initial reduction of amorphous NiO impurities to Ni and Li2O.47,48 The corresponding first-cycle discharge profiles of Ni/C composite nanofibers show a slight polarization at around 0.75 V, followed by smoothly increasing curves. For Ni/C composite nanofibers with 5, 12, and 16 wt% Ni, the first-cycle discharge capacities are about 558, 728, and 475 mAh g−1, corresponding to Coulombic efficiencies of 62, 59, and 53%, respectively. These relatively small Coulombic efficiencies can be attributed to the decomposition of the electrolyte and the formation of SEI film and other inactive materials.12,49,50 However, after the first cycle, the Coulombic efficiencies of Ni/C composites remain nearly 100%. Moreover, the discharge capacities of Ni/C composite nanofiber anodes are about 488, 662, and 433 mAh g−1, respectively, at the second cycle. Compared to the charge/discharge capacities of pure CNFs reported in our previous work and others,3,11,23,29,31,51Ni/C composite nanofibers with 12 w% Ni have improved initial reversible capacity (662 mAh g−1). The reversible capacity of CNFs is about 490 mAh g−1 at 50 mA g−1.3,11,29,51 The relatively high capacities of 12 wt% Ni/C composite nanofibers might be due to the synergetic effects of the carbon matrix and Ni nanoparticles as well as the special nanostructures of the as-prepared Ni/C composites.
 | ||
| Fig. 7 Charge–discharge curves of Ni/C composite nanofibers with different Ni contents: (A) 5, (B) 12, and (C) 16 wt%. Current density: 50 mA g−1. (D) Cycling performance of Ni/C composite nanofibers with different Ni contents: (a) 5, (b) 12, and (c) 16 wt%. Current density: 50 mA g−1. (E) Rate capabilities of Ni/C composite nanofibers with different Ni contents: (a) 5, (b) 12, and (c) 16 wt%. | ||
The cycling performance of Ni/C composite nanofibers is shown in Fig. 7D. After fifty cycles, the reversible capacities remain relatively constant, i.e., 377, 549, 397 mAh g−1, respectively, for Ni/C composite nanofibers with 5, 12, and 16 wt% Ni. These capacity values correspond to 68, 75, 82% capacity retention from the first-cycle values, which indicate a relatively slow capacity fading. The reversible capacities of all three Ni/C composite nanofibers after 50 cycles are still higher than the theoretical capacity (372 mAh g−1) of graphite and other reported Ni/NiO27 or C/NiO nanocomposites.52 The relatively good cycling performance of Ni/C composite nanofibers can be ascribed to their unique composite structure, such as the increase in electrode connectivity due to the addition of inactive Ni nanoparticles, the unique one-dimensional fiber structure, and the effective lithium ion transportation at the large nanofiber surfaces.3,26
The second-cycle discharge capacities of Ni/C composite nanofibers at different current densities of 50, 100, 200, and 500 mA g−1 are shown in Fig. 7E (rate capability). At a current density of 100 mA g−1, capacities of Ni/C composite nanofibers show relatively large degradations compared to those at 50 mA g−1. However, relatively small capacity decreases are observed when the current density increases from 100 to 200 mA g−1. At a much higher current density of 500 mA g−1, Ni/C composite nanofibers with 5, 12, and 16 wt% Ni exhibit reversible capacities of approximately 280, 411, and 263 mAh g−1, respectively, indicating satisfactory rate capability, which has been a problem for the performance of many graphite anodes.23 In addition, Ni/C composite nanofibers with 12 wt% Ni have a better rate performance than pure CNFs, but Ni/CNFs with 5 and 16 wt% Ni have similar or even worse performance than pure CNFs.3,11,29,51 This indicates that the content of Ni is very important in determining the performance of Ni/C nanofiber anodes. Further explorations are still necessary to identify the potential connections between Ni content and electrochemical performance.
The Ni/C composite nanofibers with certain amounts of electrochemically-inert Ni nanoparticles show high reversible capacity, improved cyclability, and good rate capability because of the unique nanofiber structure and good structural integrity. To obtain good electrochemical performance, electrode materials should always possess the properties of high lithium storage, fast Li insertion/release kinetics, good electronic and ionic conductivities, and appropriate mechanical strength to stabilize the Li-ion transfer pathway for Li uptake/removal. A certain amount of electro-inactive Ni phase in Ni/C composite nanofibers may increase the charge transfer in electrode materials along with facilitated Li-ion diffusion, promote the electron transfer, and help avoid the breakdown of the conductive network between current collector and the electrode. The electrochemically-inert metallic Ni phase can also act as a catalyst to facilitate Li2O decomposition, and suppress or stabilize the formation of SEI layers, which in turn improves the Coulombic efficiency.3,24–26,28,47,53 At the same time, the existence of homogeneous-dispersed Ni particles can create a large amount of defects and help activate the carbon nanofibers, which in turn can enhance the Li-storage and improve the electrochemical performance of Ni/C composite nanofibers.
Conclusions
Ni/C composite nanofibers were fabricated through electrospinning and thermal treatment processes of Ni(OAc)2·4H2O/PAN precursors with different concentrations. These Ni/C composite nanofibers exhibited high reversible capacity, improved cyclic retention, and acceptable rate capability when used as binder-free anode materials for LIBs. When the Ni content was 12 wt%, Ni/C composite nanofibers delivered the highest reversible capacities of 728 and 541 mAh g−1 at the first and 50th cycles, corresponding to a capacity retention of 75%, at the current density of 50 mA g−1. When the current density increased to 500 mA g−1, Ni/C composite nanofibers showed relatively small capacity degradation compared with those at 50 mA g−1. The good electrochemical performance of Ni/C composite nanofibers is a result of the cooperative effects of Ni nanoparticles and the carbon matrix, as well as the unique one-dimensional nanofiber structure with large surface area and high length/diameter ratio.Acknowledgements
This work was supported by the U.S. Department of Energy (NO. DE-EE0001177) and the ERC Program of the National Science Foundation under Award Number EEC-08212121.References
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878 CrossRef CAS.
- L. Ji, Z. Lin, M. Alcoutlabi and X. Zhang, Energy Environ. Sci., 2011, 4, 2682 CAS.
- L. Ji, Z. Lin, A. J. Medford and X. Zhang, Chem.–Eur. J., 2009, 15, 10718 CrossRef CAS.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28 CrossRef CAS.
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- D. Liu and G. Cao, Energy Environ. Sci., 2010, 3, 1218 CAS.
- Y. S. Meng and M. E. Arroyo-de Dompablo, Energy Environ. Sci., 2009, 2, 589 CAS.
- R. Liu, J. Duay and S. B. Lee, Chem. Commun., 2011, 47, 1384 RSC.
- K. Xu, Energies, 2010, 3, 135 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS.
- L. Ji, Z. Lin, B. Guo, A. J. Medford and X. Zhang, Chem.–Eur. J., 2010, 16, 11543 CrossRef CAS.
- L. Ji, Z. Tan, T. R. Kuykendall, S. Aloni, S. Xun, E. Lin, V. Battaglia and Y. Zhang, Phys. Chem. Chem. Phys., 2011, 13, 7170 RSC.
- F.-C. Liu, W.-M. Liu, M.-H. Zhan, Z.-W. Fu and H. Li, Energy Environ. Sci., 2011, 4, 1261 CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS.
- S. W. Lee, B. M. Gallant, H. R. Byon, P. T. Hammond and Y. Shao-Horn, Energy Environ. Sci., 2011, 4, 1972 CAS.
- H. Gwon, H.-S. Kim, K. U. Lee, D.-H. Seo, Y. C. Park, Y.-S. Lee, B. T. Ahn and K. Kang, Energy Environ. Sci., 2011, 4, 1277 CAS.
- L. Hu, D. S. Hecht and G. Grüner, Chem. Rev., 2010, 110, 5790 CrossRef CAS.
- M. Pumera, Energy Environ. Sci., 2011, 4, 668 CAS.
- Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 CAS.
- G. Lota, K. Fic and E. Frackowiak, Energy Environ. Sci., 2011, 4, 1592 CAS.
- S. Chen, H. Hou, F. Harnisch, S. A. Patil, A. A. Carmona-Martinez, S. Agarwal, Y. Zhang, S. Sinha-Ray, A. L. Yarin, A. Greiner and U. Schroder, Energy Environ. Sci., 2011, 4, 1417 CAS.
- W.-J. Zhang, J. Power Sources, 2011, 196, 13 CrossRef CAS.
- C. Kim, K. S. Yang, M. Kojima, K. Yoshida, Y. J. Kim, Y. A. Kim and M. Endo, Adv. Funct. Mater., 2006, 16, 2393 CrossRef CAS.
- M.-S. Park, S. Rajendran, Y.-M. Kang, K.-S. Han, Y.-S. Han and J.-Y. Lee, J. Power Sources, 2006, 158, 650 CrossRef CAS.
- Y.-M. Kang, K.-T. Kim, J.-H. Kim, H.-S. Kim, P. S. Lee, J.-Y. Lee, H. K. Liu and S. X. Dou, J. Power Sources, 2004, 133, 252 CrossRef CAS.
- H.-R. Jung and W.-J. Lee, J. Electrochem. Soc., 2011, 158, A644 CrossRef CAS.
- X. H. Huang, J. P. Tu, B. Zhang, C. Q. Zhang, Y. Li, Y. F. Yuan and H. M. Wu, J. Power Sources, 2006, 161, 541 CrossRef CAS.
- J. Hassoun, S. Panero, P. Simon, P. L. Taberna and B. Scrosati, Adv. Mater., 2007, 19, 1632 CrossRef CAS.
- L. Ji, A. J. Medford and X. Zhang, J. Mater. Chem., 2009, 19, 5593 RSC.
- L. Ji, A. J. Medford and X. Zhang, Polymer, 2009, 50, 605 CrossRef CAS.
- L. Ji, K.-H. Jung, A. J. Medford and X. Zhang, J. Mater. Chem., 2009, 19, 4992 RSC.
- O. Toprakci, L. Ji, Z. Lin, H. A. K. Toprakci and X. Zhang, J. Power Sources, 2011, 196, 7692 CrossRef CAS.
- J. Kim, Y. C. Kim, W. Ahn and C. Y. Kim, Polym. Eng. Sci., 1993, 33, 1452 CAS.
- L. Ji, Z. Lin, Y. Li, S. Li, Y. Liang, O. Toprakci, Q. Shi and X. Zhang, Polymer, 2010, 51, 4368 CrossRef CAS.
- M. A. Mohamed, S. A. Halawy and M. M. Ebrahim, J. Anal. Appl. Pyrolysis, 1993, 27, 109 CrossRef CAS.
- J. Hong, G. Guo and K. Zhang, J. Anal. Appl. Pyrolysis, 2006, 77, 111 CrossRef CAS.
- J. C. De Jesus, I. González, A. Quevedo and T. Puerta, J. Mol. Catal. A: Chem., 2005, 228, 283 CrossRef CAS.
- G. Hussein, A. Nohman and K. Attyia, J. Therm. Anal., 1994, 42, 1155 CrossRef CAS.
- H. Wu, R. Zhang, X. Liu, D. Lin and W. Pan, Chem. Mater., 2007, 19, 3506 CrossRef CAS.
- N. A. M. Barakat, B. Kim and H. Y. Kim, J. Phys. Chem. C, 2008, 113, 531 Search PubMed.
- I. M. Afanasov, O. N. Shornikova, V. V. Avdeev, O. I. Lebedev, G. V. Tendeloo and A. T. Matveev, Carbon, 2009, 47, 513 CrossRef CAS.
- Z. Bai, H. Chen, B. Li and W. Li, Int. J. Hydrogen Energy, 2007, 32, 32 CrossRef CAS.
- L. M. Gandia and M. Montes, J. Catal., 1994, 145, 276 CrossRef CAS.
- M. S. A. Rahaman, A. F. Ismail and A. Mustafa, Polym. Degrad. Stab., 2007, 92, 1421 CrossRef CAS.
- A. P. Grosvenor, M. C. Biesinger, R. S. C. Smart and N. S. McIntyre, Surf. Sci., 2006, 600, 1771 CrossRef CAS.
- X. X. Wang, J. N. Wang, H. Chang and Y. F. Zhang, Adv. Funct. Mater., 2007, 17, 3613 CrossRef CAS.
- Y. J. Mai, J. P. Tu, X. H. Xia, C. D. Gu and X. L. Wang, J. Power Sources, 2011, 196, 6388 CrossRef CAS.
- H. Liu, G. Wang, J. Liu, S. Qiao and H. Ahn, J. Mater. Chem., 2011, 21, 3046 RSC.
- X. Liu, Y.-S. Hu, J.-O. Müller, R. Schlögl, J. Maier and D. S. Su, ChemSusChem, 2010, 3, 261 CrossRef CAS.
- Y.-S. Hu, X. Liu, J.-O. Müller, R. Schlögl, J. Maier and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 210 CrossRef CAS.
- L. Ji, Z. Lin, A. J. Medford and X. Zhang, Carbon, 2009, 47, 3346 CrossRef CAS.
- M. M. Rahman, S.-L. Chou, C. Zhong, J.-Z. Wang, D. Wexler and H.-K. Liu, Solid State Ionics, 2010, 180, 1646 CrossRef CAS.
- M. D. Fleischauer, M. N. Obrovac, J. D. McGraw, R. A. Dunlap, J. M. Topple and J. R. Dahn, J. Electrochem. Soc., 2006, 153, A484 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00676b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |