Selective adsorption for removing sulfur: a potential ultra-deep desulfurization approach of jet fuels
Yuesong
Shen
ab,
Peiwen
Li
*b,
Xinhai
Xu
b and
Hong
Liu
b
aCollege of Materials Science and Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing University of Technology, Nanjing, 210009, PR China. E-mail: sys-njut@163.com
bDepartment of Aerospace and Mechanical Engineering, The University of Arizona, Tucson, AZ 85721-0119, USA. E-mail: peiwen@email.arizona.edu
First published on 12th December 2011
Abstract
Jet fuels are strategic fuels widely used in airplanes. Through appropriate reforming and shifting processing, jet fuels can be converted into syngas, which is a suitable fuel to solid oxide fuel cells for many auxiliary and backup power units. Integrated micro fuel processors in combination with solid oxide fuel cell (SOFC) stacks using jet fuels have been viewed as attractive portable power sources. Because the sulfur in jet fuels easily causes catalyst poisoning for fuel processing reactions and the electrochemical reactions in fuel cells, ultra-deep sulfur removal in jet fuels and many other hydrocarbon fuels has become a very important and active research subject worldwide in the last 15 years. Amongst the state-of-the-art technologies, selective adsorption for removing sulfur (SARS) is emerged to be very attractive. SARS has been regarded as the most promising approach because it obtains ultra-deep desulfurization efficiency at ambient temperature and atmospheric pressure without hydrogen consumption. In this paper, we survey the current status and prospect of the SARS technology for jet fuels, and will discuss some important issues remaining for the SARS technology in the future. The final goal of this survey is to find/innovate a promising method for jet fuel desulfurization, which is most suitable for supplying fuels to solid oxide fuel cell auxiliary and backup power units.
 Yuesong Shen | Yuesong Shen received his PhD degree at Nanjing University of Technology in 2010. He is an assistant professor at Nanjing University of Technology since 2010 to present. He is currently a postdoctoral research associate at The University of Arizona, USA. His research interests include catalytic materials for environmental protection, catalytic materials for new energy conversion, flue gas denitrification and ceramic-based structural and functional materials. He won the National Natural Science Foundation of China in 2011. |
 Peiwen Li | Peiwen Li earned his Ph.D. (1995) degree in energy and power engineering area from Xi’an Jiaotong University, China. He is currently an assistant professor in the Department of Aerospace and Mechanical Engineering at the University of Arizona, USA. He is interested in heat transfer enhancement in industrial processes, turbulence drag reduction, gas turbine cooling technologies, the multi-physics transport phenomena in fuel cells and electrolysis cells, and concentrated solar thermal power systems. Professor Li is an active member of the American Society of Mechanical Engineers. He is also a reviewer of papers for more than 10 technical Journals. |
1. Introduction
High energy efficiency and energy density, together with rapid refueling capability, renders fuel cells highly attractive for portable power generation.1–2 Of the various types of fuel cells, solid-oxide fuel cells (SOFCs) are energy convertors with a high energy efficiency, low use of noble metal catalyst, and low environmental impact.3–4 SOFCs enable direct use of higher hydrocarbons,5–7 but have not been seriously considered for portable applications because of the thermal management difficulties at small scales, and slow start-up and poor thermal cyclability.1 Essentially, SOFCs can take hydrogen-enriched synthesis gas (syngas) as an efficient ultra-clean fuel, which can be effectively converted from jet fuels. Therefore, jet fuels are particularly attractive as logistic fuels for some portable power applications based on SOFCs.8 As a consequence, reformers such as steam reformers, catalytic partial oxidation (CPO) reformers, and autothermal reformers (ATR) 9–13 are typically employed in the reforming systems to convert jet fuels to hydrogen-enriched syngases.14 An integrated micro fuel processor in combination with a SOFC stack using jet fuels has been viewed as an attractive portable power source as an alternative to the package of conventional batteries needed for portable electronic operations for several days.8 However, jet fuels usually contain various organic sulfur compounds with total sulfur content ranging from 300 to 3000 parts per million by weight (ppmw); sulfur in the fuel can poison the fuel processing catalysts such as reforming and water-shift catalysts. Even trace of sulfur in the processed fuel can poison the anode catalysts in fuel cells. Hence, sulfur must be reduced to below 1 ppm for most fuel cells, perhaps even below 60 ppb for proton exchange membrane fuel cells.8,15–18 Additionally, to reduce sulfur pollution, stringent emission regulations, rigorous emission control standards are being imposed or raised on hydrocarbon fuels. Therefore, ultra-deep desulfurization of jet fuels without environmental impact has been a very important challenge for syngas production in application of SOFCs.Current desulfurization technologies mainly include hydrodesulfurization (HDS), oxidative desulfurization (ODS), extractive desulfurization, catalytic cracking desulfurization, biodesulfurization, adsorptive desulfurization (physical adsorption desulfurization, activated adsorption desulfurization), ultrasound-assisted oxidative desulfurization (UAOD), selective adsorption for removing sulfur (SARS), etc.17–20 Targeting for providing fuels (H2 and CO) in solid oxide fuel cells for auxiliary power units, the jet fuel desulfurization technology of particular interest in this study must be simple and convenient, possess no separation process. It also must be able to obtain sufficient ultradeep desulfurization efficiency at room temperature and atmospheric pressure without hydrogen consumption.
At present, HDS is a conventional approach that is being employed by the refineries to produce low-sulfur gasoline and diesel in order to meet environmental regulations.17–19 However, the current HDS technology is not applicable for reducing sulfur content in logistic fuels to a level for a portable fuel cell application. On the other hand, the HDS process needs to work at higher temperature and higher pressure using hydrogen gas, which is not suitable for the on-board or on-site desulfurization for fuel cell applications. Therefore, some alternative non-hydrodesulfurization techniques such as ODS, extractive desulfurization, catalytic cracking desulfurization, biodesulfurization, adsorptive desulfurization and son on, are being explored in recent years to produce ultra-low-sulfur fuels. Amongst various alternative desulfurization approaches regarding ultra-deep adsorption capacity of sulfur, selectivity for sulfur compounds, and working temperatures, the UAOD21–22 and SARS23 are considered to be more promising as they react at ambient temperatures under atmospheric pressure without hydrogen consumption. However, expensive peroxides20,24 and regenerated catalysts, phase-transfer agents, and adsorbents are issues have to be addressed for scaling up purposes in the UOAD process.25
Song and coworkers10,23,26–28 in Pennsylvania State University have been exploring the new PSU-SARS process for deep desulfurization by using different solid adsorbents. In comparison with the HDS process, the PSU-SARS in jet fuels have some significant advantages:18 (a) selective adsorption is able to remove sulfur in jet fuels to the level for a portable fuel cell system; (b) the process is usually conducted at ambient temperature and atmospheric pressure, resulting in more energy efficiency and cost efficiency; (c) most of these processes don't need to use hydrogen gas, which is the most costly item in HDS; (d) the SARS process provides a clean fuel that meets the new emission control standards with minimum harm to catalytic converters. However, the challenge to the approach is to attract and selectively adsorb sulfur compounds onto the surface of the solid adsorbent but leave the aromatic and olefinic hydrocarbons as well as the open-chain and cyclic paraffinic hydrocarbons untouched.23 Thus, a key point in developing a successful SARS process is to develop an adsorbent that has high sulfur-adsorption capacity and high selectivity for sulfur compounds, and is facile to be generated.
In this paper we focus our attention on the present status and prospect of SARS of jet fuels for application in SOFCs. In the following, after a brief description of the SARS chemistry and physical process, the current research progress of SARS adsorbents is reviewed. The mechanism of the SARS reactions is then summarized. Finally the improvement of absorbents and future prospect of the SARS process are discussed based on our study.
2. The chemistry and physical process of SARS
SARS is a new approach for ultradeep desulfurization by selective adsorption for removing sulfur using adsorbents at ambient temperatures and atmospheric pressure without using hydrogen.23 The chemistry of SARS process is illustrated in Fig. 1 and 2. Adsorbents are usually comprised of transition metals supported on base oxides. Ni/ZnO and Ni/Al2O3–SiO2 are prototypical formulations most often in literature and patents.19,29-31 As shown in Fig. 1 and 2, Ni functions as adsorptive desulfurization sites.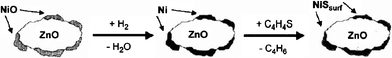
 | ||
| Fig. 2 Schematic illustration of SARS of 1-benzothiophene with Ni/Al2O3–SiO2 (Adapted from Ref. 19). | ||
Pretreatment of the Ni-based adsorbents with H2 gas:
| NiO + H2 → Ni + H2O | (1) |
Selective adsorption for removing sulfur from thiophene (C4H4S) and 1-benzothiophene (C8H6S):
| Ni + C4H4S → NiSsurf + C4H6 | (2) |
| Ni + C8H6S → NiSsurf + C8H8 | (3) |
Fig. 3 shows the technological process of SARS. Selective adsorptive desulfurization is performed on a fixed-bed flow apparatus. The adsorbent with different particle sizes is packed in custom-made stainless steel columns with different lengths and diameters. The packed columns are placed in a multichannel convection oven. The temperature of the oven is measured with a digital temperature display. To ensure that the adsorbents such as supported reduced metals are in the reduced form, the adsorbent bed must be pretreated with reducing atmosphere at a designed rising temperature. For an example of reducing schedule, the Ni-based adsorbent was pretreated with H2 gas at a flow rate of 50–60 ml min−1 at ambient pressure, heated slowly (2 °C min−1) up to 230 °C, and kept at this temperature for about 1 h in H2 flow.8 The oven temperature is then decreased to the desired adsorption temperature. The flow of reducing atmosphere is stopped and jet fuel is passed through the adsorption bed by use of an HPLC pump or advection pump at the desired flow rate.
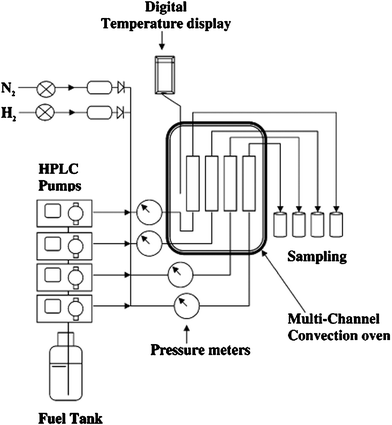 | ||
| Fig. 3 Schematic representation of adsorption desulfurization system designed for the PSU-SARS study.8 | ||
3. SARS adsorbents
As the technological core of the SARS process, the absorbent has been attracting wide attention. A wide variety of adsorbents such as ion-exchanged zeolites, supported metals, metal oxides, activated carbons, and ionic liquids are being evaluated for the SARS processes under ambient conditions. In this section, we summarized the developments of the above-mentioned adsorbents.3.1 Zeolites-based adsorbents
Zeolites have attracted much attention for desulfurization of liquid fuels due to their big surface area, polarity, surface acidity and basicity, and size-selective adsorption property. Especially the Y zeolites are the most interesting and attractive. Yang and coworkers32–35 have prepared a serial of ions (containing Cu+, Ag+, Ni2+, Zn2+ and Na+) exchanged zeolites for selective adsorption desulfurization of diesel, gasoline, and jet fuels. They found the π-complexation-based sorbents desulfurization performance decrease as follows: Cu+Y > Ag+Y > Ni2+Y > Zn2+Y > Na+Y. While Bhandari et al.36 obtained the zeolites desulfurization performance decrease as follows: Ni2+Y > Cu+Y > Fe2+Y > Zn2+Y > Na+Y. Xue et al.37 reported the zeolites desulfurization performance decrease as follows: Ce4+Y > Ag+Y > Cu+Y > Na+Y. Zhang et al.38 observed the other zeolites-based adsorbents desulfurization performance decrease as follows: AgY ≈ CuZnY > ZnNdY > ZnY > CuY > NiNdY = NaY. From comparative analysis of the above results, it can be seen that the same two adsorbents even obtain the converse desulfurization performance, indicating that the desulfurization performance is not merely determined by the chemical composition of the adsorbents.Song and coworkers27 synthesized a serial of transition metal ion-exchanged Y zeolites (with Cu, Ni, Zn, Pd, and Ce ions) for removing sulfur from Jet fuels. They found that Ce-exchanged Y zeolites exhibited better adsorption capacity of sulfur and high selectivity for sulfur compounds as compared to the selectivity of aromatics, and the sulfur compounds are adsorbed over Ce-exchanged Y zeolites via direct sulfur–adsorbent (S–M) interaction rather than via π-complexation. Li et al.39 further revealed that Ce(IV)Y adsorbed thiophene not only through physical adsorption, but largely via direct S–M (δ) interaction. Zhang et al.38 further confirmed that the desulfurization efficiency follows the order direct adsorption > oxidation–adsorption > direct oxidation.
Selective adsorption of sulfur compounds and aromatic compounds in a hexadecane on commercial zeolites, including NaY, USY, HY and 13X, has been investigated by adsorption and flow calorimetry techniques.40 Among the investigated zeolites, NaY has the highest saturation sorption capacity for the sulfur compounds. A linear correlation between the heat of adsorption and the amount of S adsorbed was found for NaY. Competitive adsorption using a mixture of anthracene, DBT and quinoline indicates that NaY selectively adsorbs quinoline while anthracene and DBT have similar affinity to NaY. It suggests that NaY is difficult to adsorptively separate sulfur compounds from aromatic hydrocarbons with the same number of the aromatic rings.
Yang and coworkers33 further studied the effect of ion exchange technique on desulfurization performance. The investigated techniques include liquid-phase ion exchange (LPIE), vapor-phase ion exchange (VPIE), and solid-state ion exchange (SSIE) techniques. They found that the sorbent capacities for total sulfur removal followed the order Cu(I)–Y(VPIE) > Ni(II)–Y(SSIE) > Ni(II)–X(LPIE) > Zn(II)–X(LPIE) > Zn(II)–Y(LPIE). The best sorbent, Cu(I)–Y(VPIE), has breakthrough adsorption capacity of 0.395 mmol-S g−1 of sorbent for commercial jet fuel (364.1 ppmw S).
Takahashi et al.34 found that the sorbent capacities for thiophene at the low pressure followed the order Cu–Y and Ag–Y ≫ Na–ZSM–5 > activated carbon > Na–Y > modified alumina and H–USY. The separation factors of thiophene over benzene (at low concentrations of thiophene) calculated from pure component adsorption isotherms exhibited the trend Ag–Y > Na–ZSM–5 > Cu–Y ≈ activated carbon ≫ Na–Y ≫ H–USY ≈ modified activated alumina. The above results illustrate that the different composition of the tested real fuel (such as different fuel additives) significantly affects the desulfurization performance, and the adsorption capacities of metal ion exchanged zeolites is higher than those of activated carbon and modified activated alumina.
3.2 Supported metals
Nickel adsorbent supported on silica-alumina (Ni/SiO2–Al2O3) exhibited an excellent performance in SARS from jet fuels.17 Sulfur compounds in this approach are selectively removed by a direct sulfur–adsorbent interaction rather than π-complexation.18 The adsorptive desulfurization of fractionated light JP-8 over the Ni/SiO2–Al2O3 adsorbent having particle sizes between 0.15 and 0.25 mm offered a sulfur breakthrough adsorption capacity of about 11.5 mg of S g−1 of adsorbent without developing any significant pressure drop across the beds. Kim et al.41 found that the sorption selectivity of the nickel-based sorbent for various compounds at room temperature increases in the order of Nap ≈ 1-MNap < 4, 6-DMDBT < DBT < quinoline < indole, as shown in Fig. 4. But the Nickel-based adsorbents usually have much higher capacity for removing sulfur at high temperature, such as 200 °C. 42 On the other hand, storage and pretreatment of the reduced metals under reducing atmosphere are important issues for scale-up techniques in desulfurization.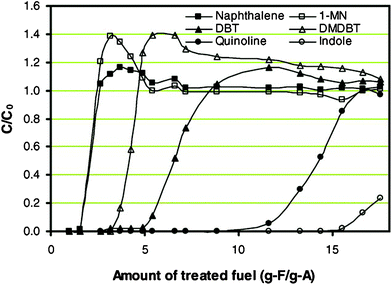
3.3 Metal oxides
Current technologies using metal oxide-based sorbents can reduce sulfur concentration from several thousand ppmv to sub-ppmv levels.43 However, this type of desulfurization is able to obtain high efficiency only at high temperatures. ZnO14 and ZnO–based sorbents such as ZnO stabilized by Fe2O344–45 and TiO246–47 are widely employed in desulfurization applications at higher temperatures. ZnO-based sorbents have low structural stability at high temperatures, because of the reduction to volatile zinc, as well as zinc migration and agglomeration occurred on the sorbent particle surface. Thus, the improvement of thermal stability of ZnO has become an important issue. Zhang et al.48 reported that the desulfurization performance of Zn–Fe–O/Al2O3 sorbent was definitely improved by the addition of cerium at low temperatures ranging from 35–90 °C, and the spent adsorbent is easy to be regenerated. Li et al.49 also confirmed that the desulfurization activity of nanosized Ce–ZnO was greatly improved by doping Ce, because cerium additive decreased particle size of the nanosized ZnO. However, in the current authors' opinion, the promotional effect may also be a result of direct interaction between Ce and thiophene sulfur. This is because that ceria possesses high oxygen storage capacity and perfect redox properties, and also sulfur compounds have more affinity to oxidation than their analogue hydrocarbons in diesel fuels. Therefore, there might be a consequence of high conversions of sulfides to sulfones and sulfoxides. These substances have different polarity compared to sulfide that can be used for selective removal of organic sulfur compounds with the solid adsorbent. Furthermore, as a result of big size effect of cerium ion, cerium enhances the thermal and structural stability of the nanosized ZnO. As mentioned above, it seems that the desulfurization performance of complex metal oxides is much better than that of single metal oxide sorbent.Activated alumina has good adsorptive properties and has been used for selective removal of sulfur compounds from fuels. Srivastav et al.50 found that the surface carbon-oxygen functional groups of alumina were effective in the adsorption of DBT. But Kim et al.41 observed that the activated alumina is not very successful for separating the thiophenic compounds from aromatics. They found that the adsorptive selectivity of the activated alumina increases only in the order of Nap ≈ 1-MNap < 4,6-DMDBT ≈ DBT ≪ indole < quinoline, as shown in Fig. 5. Therefore, low temperature desulfurization performance and high selectivity for sulfur compounds of metal oxides will be an interesting research topic in the future SARS.

3.4 Activated carbons
Activated carbons, as cheap porous materials with very high surface areas, large pore volume and rich surface functional groups, have been widely used in deodorization, decolorization, purification of drinking water, treatment of waste water, and adsorption and separation of various organic and inorganic chemicals.18,51 Activated carbons are also found to be effective in adsorptive desulfurization of liquid hydrocarbon fuels. Kim et al.41 found that the adsorption capacities based on the adsorbent weight at room temperature increase in the order of activated alumina < Ni/SiO2–Al2O3 < activated carbon for total sulfur as shown in Fig. 6, indicating that the activated carbon is the best adsorbent for total sulfur removal at room temperatures. They also observed that the activated carbon shows the highest adsorption capacity and selectivity for sulfur compounds, especially for the sulfur compounds with methyl groups, such as 4,6-DMBT. The adsorption selectivity trend for selectivity increases in the order Nap < 1–MNap < DBT < 4,6–DBT < quinoline < indole, as shown in Fig. 7. Zhou et al.52 studied the effects of structural and surface properties of carbon materials on the adsorption of benzothiophene (BT), dibenzothiophene (DBT), 4-methyldibenzothiophene (4-MDBT) and 4,6-dimethyl-dibenzothiophene (4,6-DMDBT) in the presence of 10 wt% of aromatics in liquid alkanes that simulate sulfur compounds in diesel fuels. They found that different carbon materials have significantly different sulfur-adsorption capacities and selectivities that depend not only on textural structure but also on surface functional groups. The oxygen-containing functional groups on the surface appear to play an important role in increasing sulfur-adsorption capacity. The adsorption-selectivity trend of the carbon materials for various compounds increases in the order of BT < naphthalene < 2-methylnaphthalene < DBT < 4-MDBT < 4,6-DMDBT. This selectivity trend for sulfur compounds is dramatically different and almost opposite from that for adsorption over nickel-based adsorbents. The high-adsorption capacity and selectivity for methyl DBTs indicate that certain activated carbons are promising adsorbents for SARS as a new approach to ultra-deep desulfurization of diesel fuels at room temperature.

The total sulfur adsorption amounts (normalized per adsorbent weight) were obtained after solving the following equation:35,53–54
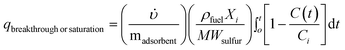 | (4) |
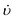 is the feed volumetric flow rate (mL min−1), ρfuel is the fuel density (g mL−1) at room temperature, Xi is the total sulfur fraction (by weight) in the feed, Ci is the total sulfur concentration in the feed (ppmw S), madsorbent is the weight of the sorbent bed (g), MWsulfur the molecular weight of sulfur, C(t) the effluent total sulfur concentration (ppmw S) at time t (min). The breakthrough adsorption amounts were obtained at the point where the fuel total sulfur concentration was less than approximately 1 ppmw S.
is the feed volumetric flow rate (mL min−1), ρfuel is the fuel density (g mL−1) at room temperature, Xi is the total sulfur fraction (by weight) in the feed, Ci is the total sulfur concentration in the feed (ppmw S), madsorbent is the weight of the sorbent bed (g), MWsulfur the molecular weight of sulfur, C(t) the effluent total sulfur concentration (ppmw S) at time t (min). The breakthrough adsorption amounts were obtained at the point where the fuel total sulfur concentration was less than approximately 1 ppmw S.
To facilitate the quantitative analysis and discuss the adsorption selectivity of carbon materials for each compound, a selectivity factor was used, which is defined as52
 | (5) |
3.5 Ionic liquids
Ionic liquids have been examined for possible applications related to green chemical processes, such as liquid/liquid extraction, gas separations, electrochemistry and catalysis.55–63 These liquids are easy to handle because of non-volatility, non-flammability, and high thermal stability. As novel liquid adsorbents, ionic liquids broaden the concept of PSU-SARS which is limited base on solid adsorbents. The ionic liquids have been applied for selective sulfur removal from fuels in recent years, because the ionic liquids are easy to be regenerated from adsorbed S-containing compounds by distillation or by dissolution in water, where ionic liquids are air- and moisture-stable at low temperature and non-corrosive. Hence, the ionic liquids can be used in multiple cycles for the removal of S-containing compounds from fuels.Zhang et al.64 reported that the ionic liquids, 1-ethyl-3-methylimidazolium tetrafluoroborate, 1-butyl-3-methylimidazolium hexafluorophosphate and 1-butyl-3-methylimidazolium tetrafluoroborate were effective for the selective removal of sulfur-containing compounds from transportation fuels such as gasoline at room temperature. They found the local structure of the ionic liquids appears to have a significant effect on their interaction with the aromatic compounds. S-containing compounds with a C5 aromatic ring are favorably absorbed over C6 aromatics, while S-containing non-aromatic compounds are poorly absorbed. As shown in Fig. 8, the absorption capacity follows the general order thiophene ≫ methylthiophene > toluene ≫ trimethylbezene > isobutylthiol, hexane, 2-methylpentane, methylcyclopentane. It appears that adsorption is favored for molecules with higher density of aromatic π electrons. The absorption capacity of the ionic liquids for S-containing compounds is sensitive to the structure of both the anion and cation of the ionic liquids, which is manifested by the significant inhibiting effect of methyl group substitution on the aromatic ring.
 | ||
| Fig. 8 Absorption capacities of EMIM+BF4−, BMIM+PF6− and BMIM+BF4−.64 | ||
As stated above, different adsorbents exhibit different adsorption capacities and selectivities. In general, the adsorption performance of adsorbents usually depends on chemical composition and surface physical and chemical properties of adsorbents, such as active sites and their density, surface area, pore size and distribution, functional groups and so on, which are functions of chemical components and their compatibility, the preparation process, and activation conditions. A fundamental understanding of the effect of surface physical and chemical properties on adsorption capacity and selectivity will be important for designing and developing more efficient adsorbents for selective adsorptive desulfurization.
As the different adsorbents are suitable for selective removal of sulfur compounds from different hydrocarbon species, a jet fuel usually includes many coexisting sulfur-containing hydrocarbon species; therefore a combination of two or more adsorbents used might be more efficient for a practical ultradeep SARS process. The size and composition of a given adsorbent bed depend on the composition of the fuel and servicing period required in the particular application. According to the operating temperatures, the reported selective adsorptive desulfurization processes can also be classed into high temperature process (>600 °C), medium temperature process (150–550 °C), and ambient temperature process.18 The high and medium temperature processes involve a chemical reaction or absorption, and the adsorbents include metal oxides and reduced metals. The ambient temperature process usually involves physical or chemical adsorption on the surface, and the adsorbents include zeolites, activated carbons, ionic liquids and other mesoporous materials.
At present, researches on the adsorbents for ultra-deep desulfurization of real jet fuels are still not systematic as shown in Table 1. Especially, the desulfurization performance at low temperature, such as room temperature, is not ideal. Low temperature desulfurization performance of adsorbents should be the key for its wide application in SARS of jet fuels in the near future. Hence, in developing a successful SARS process for ultra-deep desulfurization of jet fuels, the major challenge is to develop a novel efficient, environmentally friendly and cheap adsorbent that has higher sulfur adsorption capacity and selectivity at low temperature under atmospheric pressure without hydrogen consumption, and is easy for regeneration. Anyway, more efforts should be focused on improving the room temperature desulfurization performance of adsorbents in SARS of jet fuels for portable power applications in the near future.
| Adsorbent | Fuel description | Sulfur in fuel (ppmw) | Adsorption condition | Breakthrough capacity (mg-S g−1) |
|---|---|---|---|---|
| Cu(I)–Y(VPIE)33 | Jet fuel | 364 | Room temperature | 12.6 |
| Zn(II)–Y(LPIE)33 | Jet fuel | 364 | Room temperature | 1.4 |
| Zn(II)–X(LPIE)33 | Jet fuel | 364 | Room temperature | 2.8 |
| CuCl/AC65 | JP-5 | 1172 | Room temperature LHSV: 2.3 h−1 | 1.0 |
| PdCl2/Al2O365 | JP-5 | 1172 | Room temperature LHSV: 2.3 h−1 | 2.1 |
| PdCl2/AC65 | JP-5 | 1172 | Room temperature LHSV: 2.3 h−1 | 3.2 |
| Ni/SiO2–Al2O35 | Light JP-8 | 380 | 220 °C, LHSV: 2.4 h−1 | 13.5 |
| KYNiE-366 | Light JP-8 | 380 | 80 °C | 4.5 |
| KYNi8IWI66 | Light JP-8 | 380 | 80 °C | 2.4 |
| NiY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 2 |
| CuY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 0.3 |
| ZnY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 0 |
| CeY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 2.7 |
| PdY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 2.6 |
| HY–Zeolite67 | JP-8 | 736 | 80 °C for 4–5 h | 1.3 |
| Ce–Y27 | JP-8 | 750 | 80 °C | 4.5 |
4. Mechanism of SARS reaction
4.1 π-complexation
For industrial adsorption processes, chemical complexation bonds are generally stronger than van der Waals interactions, yet weak enough to be broken by traditional engineering means such as increasing temperature and/or decreasing pressure.68 Therefore, a tremendous opportunity exists for developing new sorbents by using weak chemical bonds, including various forms of complexation bonds.32,34 On the basis of the principles of π-complexation, Yang and coworkers have been developing a number of new sorbents for many applications, including desulfurization,32,34,69–76 olefin/paraffin, diene/olefin, and aromatics/aliphatics separations.77–86 For desulfurization, they prepared transition metal ion exchanged zeolites to selectively remove organosulfur molecules from different fuels like diesel, gasoline and jet fuels.Yang and coworkers72 found that the methyl groups in 4-methyldibenzothiophene (4-MDBT) and 4,6-dimethyldibenzothiophene (4,6-DMDBT) create a steric effect that hinders the capacity of HDS catalysts to chemisorb the sulfur atoms as depicted in Fig. 9(A).33 However, such steric hindrance is not present for adsorption by π-complexation. Fig. 9(B) depicts the mechanism involved during π-complexation between 4,6-DMDBT and a copper(I) cation. In the complexation mechanism, the cation can form the usual σ bonds with their empty s-orbitals and, in addition, their d-orbitals can back-donate electron density to the antibonding π-orbitals (π*) of the sulfur rings (see Fig. 10).33,87,88 Experimental data and molecular orbital (MO) calculations have shown that π-complexation with cuprous ions is stronger with organo-sulfur molecules (i.e., thiophenic molecules) than with aromatics without sulfur (e.g., benzene).34
 | ||
| Fig. 9 Schematic representation for desulfurization of 4,6-dimethyl-dibenzothiophene with molybdenum-based (A) and copper(I)-based (B) adsorbents. Case (B) corresponds to π-complexation.33 | ||
 | ||
| Fig. 10 Copper ions occupying faujasite six-ring window sites (A); σ–donation of π–electrons of thiophene to the 4 s orbital of copper(I) (B); d-π* back-donation of electrons from 3d orbitals of copper(I) to π* orbitals of thiophene (C). Here 3d represents dxy, dyz, or dxz, or three of the five 3d orbitals.33,87,88 | ||
From the above results, it can be seen that the charge-compensating anion has a very strong effect on the π-complexation by the cation for the sorbents.54,74 But the effect of the substrate on the metal salt is much weaker. For the metal salt, a geometric effect is the dominant reason for the strong synergistic effect rather than any electronic effect. Yang and coworkers54 further studied a novel adsorbent of PdCl2/AC for desulfurization of a model jet fuel, and proposed a theory about edge sites: there were a large number of peripheral sites on the edges of the supported metal salt which was well dispersed on the carbon support, and the edge sites provided an ideal combination of sites for the benzothiophene molecule: PdCl2 for the thiophene ring and carbon for the benzene ring. Adsorption of a substituted methylbenzothiophene molecule on such an edge site is depicted in Fig. 11. In this depiction, benzene ring is adsorbed strongly on the surface of carbon, while the thiophene ring is bonded to the adjacent Pd2+ site by π-complexation. By binding cooperatively in this manner, stronger adsorption and hence higher adsorbed amounts were achieved.
 | ||
| Fig. 11 Depiction of synergistic effect in adsorption of methylbenzothiophene on PdCl2/AC.54 | ||
4.2 Direct sulfur–adsorbent (S–M) interaction
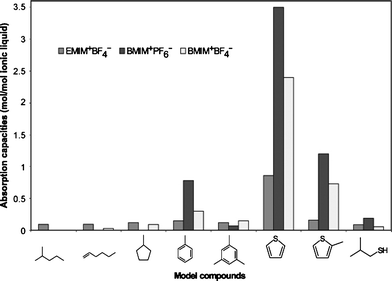
Song and coworkers8,89–91 found that the π-complexation could not explain the reason why the modified zeolites were able to effectively adsorb trace of sulfides from gasoline with many aromatics and olefins. For exploring selective adsorption, they are interested in the coordination geometries of thiophene in organometallic complexes. Fig. 12 shows eight coordination configurations of thiophene in organometallic complexes.92–95 Among the coordination configurations, η1S and S–μ3 are the two specific configurations where thiophene coordinates directly with the metal through sulfur-metal interaction, suggesting there are likely adsorbents that are able to adsorb the thiophenic compounds selectively through η1S or S–μ3 –bonding. Four π-complexation configurations of η1C, η2, η4 and η5 are formed via interactions between thiophene and metal species through one or more C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds. Two configurations of η4, S–μ2 and η4, S–μ3 involve both π-complexes and direct S–M bonds. As illustrated above, it seems that the selectivity for sulfur compounds of the absorbent should be improved greatly if thiophene coordinates with the metal species merely through direct S–M interaction.
C double bonds. Two configurations of η4, S–μ2 and η4, S–μ3 involve both π-complexes and direct S–M bonds. As illustrated above, it seems that the selectivity for sulfur compounds of the absorbent should be improved greatly if thiophene coordinates with the metal species merely through direct S–M interaction.
Song and coworkers' experiment27 revealed that Ce-exchanged Y zeolites exhibited higher selectivity for sulfur compounds as compared to the selectivity of aromatics in the adsorptive desulfurization of a mode jet fuel and a real JP-8 fuel, for which a comparative study indicated that the sulfur compounds are adsorbed over Ce-exchanged Y zeolites via direct sulfur-adsorbent (S–M) interaction rather than via π-complexation. Meanwhile, they also performed a computational analysis for the electronic properties by using molecular orbital package from CAChe software system (MOPAC).23 The calculated results showed that the highest occupied molecular orbital (HOMO) of thiophene, benzothiophene and dibenzothiophene is located more on the sulfur atom, whereas the HOMO of alkyl benzenes and naphthalene on the conjugated six-member ring. These results suggest that it may be possible to achieve preferential adsorption by interaction of sulfur atom with certain metal species through the interaction of HOMO on sulfur with lowest unoccupied molecular orbital (LUMO) on metal species.
Song and coworkers27 further observed that the selectivity for 2-methy benzothiophene (2-MBT) was higher in the static adsorption studies, while the adsorption selectivity decreased in the order of 5-methyl benzothiophene (5-MBT) > benzothiophene > 2-MBT under dynamic conditions. This trend was correlated to the electron density on sulfur atoms derived from computer-aided molecular orbital calculations.27 The electron density isosurfaces of BT and 2,3,7-TMBT are shown in Fig. 13.8 The computer simulation clearly demonstrates the existence of steric hindrance due to the presence of methyl groups at 2 and 7 positions of the BT ring. The electrostatic potential color-mapped on the electron density with values at the color boundaries for the seven molecules was calculated and the results are shown in Fig. 14.41 It shows clearly that the negative electrostatic potential are dominantly located on the two sides of the molecular plane (except quinoline), and the value of the negative electrostatic potential increases in the order of Nap < 1-MNap < DBT < 4,6-DMDBT < indole < dihydroindole. Evidently, the methyl group at the aromatic ring enhances the negative electrostatic potential on the two sides of the molecular plane because the methyl group is an electron donor to the aromatic ring.
 | ||
| Fig. 13 Electron density isosurfaces corresponding to molecular structures of benzothiophene and 2,3,7-trimethylbenzothiophene by molecular simulation with MOPAC.8 | ||
 | ||
| Fig. 14 Electrostatic potential on electron density for the examined compounds.41 | ||
4.3 Active sites for adsorption
According to the π-complexation mechanism, the π-complexation adsorbents exhibit low selectivity for sulfur compounds as a result of competitive adsorption of aromatic compounds, while according to the adsorption mechanism of direct S–M interaction, the adsorbents possess high selectivity for sulfur compounds, but the steric hindrance make them difficult to remove sulfur from DMDBT etc. However, the contradiction is not irresolvable. On the one hand, thiophene has two lone pairs of electrons on the sulfur atom; one pair lies on the six-electron π system and the other lies in the plane of the ring. Thiophene can act either as an n-type donor by donating the lone pairs of electrons that lie in the plane of the ring to the adsorbent (direct S–M σ bond) or as a π-type donor by utilizing the delocalized π electrons of the aromatic ring (π bond) to form a π-type complex with the metal ions.27 On the other hand, the adsorption capacity and selectivity of adsorbents can be further improved by modifying various types of surface active sites for adsorption, such as Lewis acid sites, useful functional groups, electronic defect centers, micro-structural defects and so on. Seredych et al.96 synthesized a serial of novel mesoporous carbons with highly dispersed copper, cobalt, and iron for the reactive adsorption of dibenzothiophene. The small metal content (less than 1%) and its chelation in the precursor polymers ensure a high dispersion of metallic centers. The materials obtained are mainly mesoporous but differ significantly in their porosity and surface chemistry, which is linked to the effect of template constraints and chemistry and the kind of metal and is related to the differences in the carbonization mechanism. The high capacity of the investigated carbons in adsorption of DBT from hexane is up to 130 mg S g−1, which were linked to the high volume of mesopores and specific interactions of DBT with surface acidic groups and strong interactions of metals with dibenzothiophene via S–M σ bonds or, in the case of copper, via interaction of metals with disturbed π electrons of aromatic rings of DBT.According to Lewis acid–base theory, most thiophene sulfur compounds in jet fuels are Lewis base, which are easy to be adsorbed at Lewis acid sites. Hence, it can select materials that can form Lewis acid sites to selective adsorb thiophene sulfur compounds with the lone pair electrons in jet fuels. The Lewis acid–base adsorption mechanism is the interaction between the acid sites on the surface of adsorbent and thiophene derivatives. Xue et al.37 confirmed that AgY–zeolite shows stronger adsorption of the soft bases TP and 1-BTP than CuY–zeolite because Ag+ is a softer acid than Cu+, and Ag+ and Cu+ can combine with TP and 1-BTP mainly by π bonds and form π-type complexes. Ce4+ is a hard acid and prefers to combine with TP and 1-BTP by the direct S–M σ bond rather than by the π bond.
5. Prospect of SARS process
5.1 Improvement of SARS adsorbents
Among various alternative adsorbents, supported reduced metals are considered to be more promising for SARS process because of much higher sulfur-adsorption capacity and selectivity for sulfur compounds in comparison of other adsorbents.8 Representative Ni/SiO2–Al2O3 exhibits an excellent performance in removing sulfur from jet fuels.97 However, nickel-based adsorbents have some drawbacks:41,52 (a) desulfurization is merely able to obtain high efficiency at high temperature, and the supported metals possess low thermal stability at high temperature; (b) the adsorptive selectivity of nickel-based adsorbents for alkyl dibenzothiophenes with one or two alkyl groups at the 4 and/or 6 positions, which are major sulfur compounds in commercial diesel fuel, is reduced significantly because of the steric hindrance of the alkyl groups; (c) the reduced metal is easy to be oxidized and would require careful passivation treatment during storage and transport, which is an issue for scaleup techniques in desulfurization; (d) the regeneration of spent nickel-based adsorbents requires reduction by using hydrogen gas at high temperature. Although zeolites, carbon materials, and activated alumina possess perfect adsorption capacity at low temperature, even at room temperature, they have low selectivity for sulfur compounds from jet fuels, because the desulfurization efficiency is dramatically dependant on the physical adsorption. Herein a combination of reduced metals modified porous materials may be promising for ultradeep desulfurization of jet fuels at room temperature under atmospheric pressure.Currently, selection of adsorbent active components and the effect of chemical composition on desulfurization performance have already been investigated maturely. It has already obtained many different desulfurization orders in sulfur-adsorption capacity and selectivity for different adsorbents. However, it can be even observed that different scientists obtain the converse active orders from the same two adsorbents in SARS of the same fuels. Thus, with only studies to the effect of chemical composition on desulfurization performance it is insufficient to fully reveal the nature of selective adsorptive desulfurization by adsorbents, and thereby difficult to summarize the general action rules. In our previous work,98 we found that even the same chemical component in different structure exhibits different performance, such as TiO2 for denitrification, the TiO2–anatase exhibits more than 90% denitrification efficiency in NH3–SCR of NO, but the TiO2–rutile possesses less than 15% denitrification efficiency. Therefore, chemical composition is only the basic factor for affecting material performance, whereas material structure is also a very important factor. Once the chemical composition of adsorbents is chosen, materials structure can significantly affect the physical and chemical properties. However, the effect of material structure on desulfurization performance of the absorbents has rarely been systematically studied so far, which severely restricts the development and application of novel adsorbents with features of high efficiency, environmental benign and low cost.
Adsorptive performance of adsorbents not only depends on the solid acidity, unbond d electrons and empty hybrid orbital of the active components, but also on the microstructure and structural defects of the solid adsorbents, such as the crystal structure, solid solution, grain boundary stress, non-stoichiometric defects, pore structure, dispersion and refinement of grains, and uneven surface atomic steps, kinks, impurities, dangling bonds and so on, which can cause a variety of chemical defects to provide reactants with chemical adsorption energy or increase the interface and active sites for chemical adsorption. Furthermore, the geometry and surface state of the active components changing can induce the variation of chemical adsorption energy, and thus cause changes in adsorption capacity and selectivity. Adsorbent supports can also affect the adsorptive properties of adsorbent active components; particularly increase the solid acid sites. Moreover, support can act as structural promoter to increase the surface area, prevent sintering of the active components and improve the structural stability of the active component; or act as electronic regulators to adjust electronic structure (combined-state), surface properties and crystal structure of the active components, thereby enhance the activity of the adsorbents.
The specific structure of an adsorbent is generally determined by its synthesis and preparation. Shan et al.99–100 found that ultrasound was able to improve ion exchange degree, reduce the ion exchange time and impregnation time, decrease metal particle size and increase the metal dispersion on the adsorbent surface, which contributed better desulfurization performance. It illustrates that the perfect desulfurization performance of adsorbent in SARS process can be further obtained by grafting novel preparation technology. Recently, Liu and coworkers101 developed a strategy for the fabrication of novel π-complexation adsorbents by grafting Cu(I)-containing molecular precursor onto β-cyclodextrin (CD). Isolated Cu(I) sites are successfully generated on CD (Fig. 15A). The resulting materials provide a molecular-level dispersion of Cu(I) (denoted as Cu–CD). Such an extreme disperse state of Cu(I) is quite different from conventional ones (e.g. CuCl), and can avoid the possible aggregation of active species that usually occurs in Cu(I)-containing oxides and salts. They demonstrated that Cu–CD materials exhibit excellent adsorptive desulfurization capacity in thiophene capture through the π-complexation mechanism, which is much better than CuCl supported on CD via the conventional thermal dispersion method (Cu/CD).
 | ||
| Fig. 15 Electrostatic potential on electron density for the examined compounds.101 | ||
As stated above, study on material structure and exploring novel preparation methods of adsorbents will be the key in developing new adsorbents of high performance. In view of the significant synergistic effect between the carbon substrate and the supported π-complexation sorbent,54 it is inferred that a high sulfur-adsorption capacity at low temperature could be due to the geometric effect. In comparison with activated carbons and zeolites, it is promising to obtain perfect low temperature desulfurization performance by designing and synthesizing a serial of novel mesoporous materials with better merits of bigger specific surface, more active sites and their density, more useful functional groups, more structural defect centers and perfect size-selective adsorption property. According to the adsorption mechanism of direct sulfur-adsorbent (S–M) interaction, it is also promising to obtain perfect selectivity of sulfur compounds at low temperature by ionic modifications. i.e., via specific ions to modify the above mesoporous materials into novel adsorbents for ultradeep desulfurization, which is under research in the authors' team.
5.2 Future prospect of SARS technology
The limited situated adsorption capacity and regeneration of the adsorbents are important issues to be considered for scaleup techniques in refineries.22 This requires us to find solutions when an adsorbent reaches its saturated adsorption in the SARS process and lost further desulfurization capability, or if adsorption alone can not technologically reach the ultradeep desulfurization levels for jet fuels.Some new technology may speed up the desulfurization of hydrocarbon fuels; for example using ultrasound. Gunnerman et al.102 invented a novel ultrasound-assisted desulfurization of fossil fuels in the presence of dialkyl earths, they found that the emerging mixture separates spontaneously into aqueous and organic phases under ultrasound, from which the organic phase is readily isolated as the desulfurized fossil fuel. For another example, acidic alumina shows promising results as an adsorbent in the UAOD process,22 the sulfur concentration in JP-8 jet fuel can be reduced from the original 850 ppm to 1 ppm at an oxidation time of 10 min at ambient temperature and atmospheric pressure. It is encouraging that we may further improve the desulfurization performance via ultrasound technologies when the situated capacity of an adsorbent still can not reach the desired ultra low sulfur level. To prove this hypothesis the authors' team is exploring for ultradeep desulfurization by ultrasound-assisted selective adsorptive desulfurization (UASAD) using a novel mesoporous solid adsorbent at room temperature and atmospheric pressure without using hydrogen. The adsorption capacity of sulfur and selectivity for sulfur compounds in real jet fuels in the UASAD process will be studied.
Besides improving desulfurization performance, ultrasound is also found to be effective for adsorbent regeneration. Yang and coworkers103–104 reported that ultrasound can effectively desorb strongly-adsorbed molecules, in desorbing phenol from activated carbon and polymeric resin. They also found that ultrasound was an effective technique for regenerating spent CuCl/Al2O3 sorbent at room temperature.35 The saturated PdCl2/AC sorbent could be effectively desorbed by the ultrasound technique, as shown in Fig. 16. It shows the comparison results of desorption with ultrasound and without ultrasound, both at 50 °C. The amount of sulfur desorbed was higher with ultrasound, 65 wt% desorption vs. 45 wt% desorption without ultrasound. Anyway, when an adsorbent reaches its saturated adsorption in SARS of jet fuels, quick regeneration of the adsorbent by new technology should be an important and interesting topic for future study.
 | ||
| Fig. 16 Amount of total sulfur desorbed (in percent, gS/g sorbent) from spent PdCl2/AC sorbent without ultrasound at 20 °C (▾), 50 °C (■) and with ultrasound at 50 °C (•) in a static system with 30 wt% benzene and 70 wt% n-octane.54 | ||
6. Conclusion
In summary, the desulfurization performance of SARS has been improved and understood step by step. As the most promising ultradeep desulfurization approach for jet fuels applied in SOFCs, SARS is ready to play a big role. However, there still are some remaining issues: (a) the limited saturated adsorption capacity and regeneration of the adsorbents make the SARS process difficult to put into practice in large scale application; (b) a systematic study to the effects of material structure and structural defects on the adsorption performance is not seen yet, which restricts the improvement of adsorbent desulfurization performance; (c) the current studies on the mechanism of the SARS reaction are still focused on some specific types of sorbents, and a universal theory for all the adsorbents is still not available. It is very promising that cross-discipline research methods, as surveyed in this study, and grafting of novel techniques to some known approaches may create new efficient SARS processes. We expect that with our significant efforts and study, an attractive ultradeep desulfurization method could be widely applied in practical devices in the near future.Acknowledgements
The support from the Office of Naval Research of the USA and the University of Tennessee SimCenter under the contract #8500011366 is gratefully acknowledged.References
- Z. P. Shao, S. M. Haile, J. Ahn, P. D. Ronney, Z. L. Zhan and S. A. Barnett, Nature, 2005, 435, 795–798 CrossRef CAS.
- A. Heinzel, C. Hebling, M. Muller, M. Zedda and C. Muller, J. Power Sources, 2002, 105, 250–255 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS.
- Z. P. Shao, C. M. Zhang, W. Wang, C. Su, W. Zhou, Z. H. Zhu, H. J. Park and C. Kwak, Angew. Chem., 2011, 123, 1832–1837 CrossRef.
- E. Perry Murray, T. Tsai and S. A. Barnett, Nature, 1999, 400, 649–651 CrossRef.
- S. Park, J. M. Vohs and R. J. Gorte, Nature, 2000, 404, 265–267 CrossRef CAS.
- S. W. Tao and J. T. S. Irvine, Nat. Mater., 2003, 2, 320–323 CrossRef CAS.
- S. Velu, X. L. Ma, C. S. Song, M. Namazian, S. Sethuraman and G. Venkataraman, Energy Fuels, 2005, 19, 1116–1125 CrossRef CAS.
- A. Ersoz, H. Olgun and S. Ozdogan, J. Power Sources, 2006, 154, 67–73 CrossRef CAS.
- C. S. Song, Catal. Today, 2002, 77, 17–50 CrossRef CAS.
- D. Shekhawat, D. A. Berry, T. H. Gardner, D. J. Haynes and J. J. Spivey, J. Power Sources, 2007, 168, 477–483 CrossRef CAS.
- S. Roychoudhury, M. Lyubovsky, D. Walsh, D. Chu and E. Kallio, J. Power Sources, 2006, 160, 510–513 CrossRef CAS.
- A. Lindermeir, S. Kah, S. Kavurucu and M. Muehlner, Appl. Catal., B, 2007, 70, 488–497 CrossRef CAS.
- H. Y. Yang, R. Sothen, D. R. Cahela and B. J. Tatarchuk, Ind. Eng. Chem. Res., 2008, 47, 10064–10070 CrossRef CAS.
- T. Fukunaga, H. Katsuno, H. Matsumoto, O. Takahashi and Y. Akai, Catal. Today, 2003, 84, 197–200 CrossRef CAS.
- J. Zheng, J. J. Strohm, M. Hoehn and C. S. Song, Prepr.sAm. Chem. Soc., Div. Pet. Chem., 2004, 49, 21 CAS.
- C. S. Song, Catal. Today, 2003, 86, 211–263 CrossRef CAS.
- K. Liu, C.S. Song, S. Velu, Hydrogen and Syngas Production and Purification Technologies. John Wiley & Sons, Inc.: Hoboken, New Jersey, 2009 Search PubMed.
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- H. Rang, J. Kann and V. Oja, Oil Shale, 2006, 23, 164–176 CAS.
- O. Etemadi and T. F. Yen, Prepr. Pap. Am. Chem. Soc., DiV. Fuel Chem., 2006, 51, 820 CAS.
- O. Etemadi and T. F. Yen, Energy Fuels, 2007, 21, 2250–2257 CrossRef CAS.
- X.L. Ma, L. Sun and C. S. Song, Catal. Today, 2002, 77, 107–116 CrossRef CAS.
- R. D. Smetana, S. Herbstman, T. C. Mead, US Pat., 1971, 3,595, 778 Search PubMed.
- M.-W. Wan, S.-S. Cheng and T. F. Yen, Appl. Catal., A, 2007, 52, 198–199 Search PubMed.
- C. S. Song and X. L. Ma, Appl. Catal., B, 2003, 41, 207–238 CrossRef CAS.
- S. Velu, X. L. Ma and C. S. Song, Ind. Eng. Chem. Res., 2003, 42, 5293–5304 CrossRef CAS.
- S. Watanabe, S. Velu, X. L. Ma and C. S. Song, Prepr. Pap. Am. Chem. Soc., Div. Fuel. Chem., 2003, 48, 695 CAS.
- K. Tawara, T. Nishimura and H. Iwanami, J. Jpn. Pet. Inst., 2000, 43, 114–120 CrossRef CAS.
- K. Tawara, T. Nishimura, H. Iwanami, T. Nishimoto and T. Hasuike, Ind. Eng. Chem. Res., 2001, 40, 2367–2370 CrossRef CAS.
- E. Ito and J. A. R. van Veen, Catal. Today, 2006, 116, 446–460 CrossRef CAS.
- R. T. Yang, A. Takahashi and F. H. Yang, Ind. Eng. Chem. Res., 2001, 40, 6236–6239 CrossRef CAS.
- A. J. Hernandez-Maldonado, F. H. Yang, G. Qi and R. T. Yang, Appl. Catal., B, 2005, 56, 111–126 CrossRef CAS.
- A. Takahashi, H. Yang and R. T. Yang, Ind. Eng. Chem. Res., 2002, 41, 2487–2496 CrossRef CAS.
- A. J. Hernandez-Maldonado, G. S. Qi and R.T. Yang, Appl. Catal., B, 2005, 61, 212–218 CrossRef CAS.
- V. M. Bhandari, C. H. Ko, J. G. Park, S. S. Han, S. H. Cho and J. N. Kim, Chem. Eng. Sci., 2006, 61, 2599–2608 CrossRef CAS.
- M. Xue, R. Chitrakar, K. Sakane, T. Hirotsu, K. Ooi, Y. Yoshimura, Q. Feng and N. Sumida, J. Colloid Interface Sci., 2005, 285, 487–492 CrossRef CAS.
- Z. Y. Zhang, T. B. Shi, C. Z. Jia, W. J. Ji, Y. Chen and M. Y. He, Appl. Catal., B, 2008, 82, 1–10 CrossRef CAS.
- X. Q. Li, K. Tang, L. H. Duan, F. F. Li and L. J. Song, J. Mol. Catal. A: Chem., 2007, 15, 1–5 CrossRef.
- T. T. Ng F., A. Rahman, T. Ohasi and M. Jiang, Appl. Catal., B, 2005, 56, 127–136 CrossRef.
- J. H. Kim, X. L. Ma, A. N. Zhou and C. S. Song, Catal. Today, 2006, 111, 74–83 CrossRef CAS.
- P. Zeuthen, K. G. Knudsen and D. D. Whitehurst, Catal. Today, 2001, 65, 307–314 CrossRef CAS.
- S. S. Tamhankar, M. Bagajewicz, G. R. Gavalas, P. K. Sharma and M. Flytzani-Stephanopoulos, Ind. Eng. Chem. Process Des. Dev., 1986, 25, 429–437 CAS.
- S. K. Gangwal, S. M. Harkins, J. M. Stogner and S. J. Bossart, Environ. Prog., 1989, 8, 26–34 CrossRef CAS.
- R. Gupta, S. K. Gangwal and S. C. Jain, Energy Fuels, 1992, 6, 21–27 CrossRef CAS.
- S. Lew, K. Jothimurugesan and M. Flytzani-Stephanopoulos, Ind. Eng. Chem. Res., 1989, 28, 535–541 CrossRef CAS.
- M. C. Woods, S. K. Gangwal, K. Jothimurugesan and D. P. Harrison, Ind. Eng. Chem. Res., 1990, 29, 1160–1167 CrossRef CAS.
- J. C. Zhang, L. F. Song, J. Y. Hu, S. L. Ong, W. J. Ng, L. Y. Lee, Y. H. Wang, J. G. Zhao and R. Y. Ma, Energy Convers. Manage., 2005, 46, 1–9 CrossRef CAS.
- F. Li, B. Yan, J. Zhang, A. X. Jiang, C. H. Shao, X. J. Kong and X. Wang, J. Rare Earths, 2007, 25, 306–310 CrossRef.
- A. Srivastav and V. C. Srivastava, J. Hazard. Mater., 2009, 170, 1133–1140 CrossRef CAS.
- Y. J. Jin, H. L. Song and L. J. Chou, Chem. Ind. Eng. Process, 2009, 28, 1540–1545 CAS.
- A. N. Zhou, X. L. Ma and C. S. Song, J. Phys. Chem. B, 2006, 110, 4699–4707 CrossRef CAS.
- H. Tamon, K. Kitamura and M. Okazaki, AIChE J., 1996, 42, 422–430 CrossRef CAS.
- Y. Wang and R. T. Yang, Langmuir, 2007, 23, 3825–3831 CrossRef CAS.
- K. A. Howard, H. L. Mitchell, R. H. Waghore, US Pat., 1982, 4,359, 596 Search PubMed.
- D. R. Boate, M. J. Zaworoktko, US Pat., 1993, 5,220,106 Search PubMed.
- F. G. Sherif, L. Shyyu, C. C. Greco, US Pat., 1998, 5,824, 832 Search PubMed.
- V. R. Koch, C. Nanjundian. R. T. Carlin, US Pat., 1998, 5,827, 602 Search PubMed.
- S. M. Silvu, P. A. Z. Suarcz and R. F. de Souza, Polym. Bull., 1998, 40, 401–405 CrossRef.
- A. L. Carmichael, D. M. Haddleton, S. A. F. Bon and K. R. Seddon, Chem. Commun., 2000, 1237–1238 RSC.
- R. T. Carlin and J. S. Wilkes, J. Mol. Catal., 1990, 63, 125–129 CrossRef CAS.
- M. Goledzinowski and V. I. Birss, Ind. Eng. Chem. Res., 1993, 32, 1795–1797 CrossRef CAS.
- J. A. Boon, J. A. Levisky, J. L. Pflug and J. S. Wilkes, J. Org. Chem., 1986, 51, 480–483 CrossRef CAS.
- S. G. Zhang and Z. C. Zhang, Green Chem., 2002, 4, 376–379 RSC.
- Y. H. Wang, F. H. Yang, R. T. Yang, J. M. Heinzel and R. T. Nickens, Ind. Eng. Chem. Res., 2006, 45, 7649–7655 CrossRef CAS.
- S. Velu, C. S. Song, M. H. Engelhard and Y. H. Chin, Ind. Eng. Chem. Res., 2005, 44, 5740–5749 CrossRef CAS.
- S. Velu, X. L. Ma and C. S. Song, Fuel Chem. Division Preprints, 2002, 47, 447–448 CAS.
- C. J. King, Separation Process Based on Reversible Chemical Complexation. In Handbook of Separation Process Technology, Rousseau, R.W., Ed.; Wiley: New York, 1987; Chapter 15 Search PubMed.
- A. J. Hernández-Maldonado and R. T. Yang, J. Am. Chem. Soc., 2004, 126, 992–993 CrossRef.
- A. J. Hernández-Maldonado and R. T. Yang, Ind. Eng. Chem. Res., 2004, 43, 1081–1089 CrossRef.
- A. J. Hernández-Maldonado and R. T. Yang, Ind. Eng. Chem. Res., 2003, 42, 3103–3110 CrossRef.
- A. J. Hernández-Maldonado and R. T. Yang, AIChE J., 2004, 50, 791–801 CrossRef.
- A. J. Hernández-Maldonado, S. D. Stamatis, R. T. Yang, A. Z. He and W. Cannella, Ind. Eng. Chem. Res., 2004, 43, 769–776 CrossRef.
- R.T. Yang, Adsorbents: Fundamentals and Applications, Wiley, New York, 2003 Search PubMed.
- R. T. Yang, A. Takahashi, F. H. Yang and A. Hernandez-Maldonado, US and Foreign Patent Applications Filed, September, 2002 Search PubMed.
- R. T. Yang, A. J. Hernández-Maldonado and F. H. Yang, Science, 2003, 301, 79–101 CrossRef CAS.
- R. T. Yang and E. S. Kikkinides, AIChE J., 1995, 41, 509–517 CrossRef CAS.
- S. U. Rege, J. Padin and R. T. Yang, AIChE J., 1998, 44, 799–809 CrossRef CAS.
- H. Y. Huang, J. Padin and R. T. Yang, Ind. Eng. Chem. Res., 1999, 38, 2720–2725 CrossRef CAS.
- H. Y. Huang, J. Padin and R. T. Yang, J. Phys. Chem. B, 1999, 103, 3206–3212 CrossRef CAS.
- J. Padin and R. T. Yang, Ind. Eng. Chem. Res., 1997, 36, 4224–4230 CrossRef CAS.
- J. Padin, R. T. Yang and C. L. Munson, Ind. Eng. Chem. Res., 1999, 38, 3614–3621 CrossRef CAS.
- J. Padin and R. T. Yang, Chem. Eng. Sci., 2000, 55, 2607–2616 CrossRef CAS.
- A. Jayaraman, R. T. Yang, C. L. Munson and D. Chinn, Ind. Eng. Chem. Res., 2001, 40, 4370–4376 CrossRef CAS.
- A. Takahashi, F. H. Yang and R. T. Yang, Ind. Eng. Chem. Res., 2000, 39, 3856–3867 CrossRef CAS.
- A. Takahashi, R. T. Yang, C. L. Munson and D. Chinn, Langmuir, 2001, 17, 8405–8413 CrossRef CAS.
- A. J. Hernández-Maldonado and R. T. Yang, J. Am. Chem. Soc., 2004, 126, 992–993 CrossRef.
- A. J. Hernández-Maldonado and R. T. Yang, Ind. Eng. Chem. Res., 2004, 43, 6142–6149 CrossRef.
- S. Velu, X. L. Ma and C. S. Song, Pre. Pr., 2003, 48, 58–59 CAS.
- A. N. Zhou, X. L. Ma and C. S. Song, Pre. Pr., 2004, 49, 329–332 CAS.
- X. L. Ma, V. Subramani and C. S. Song, Appl. Catal., B, 2005, 56, 137–147 CrossRef CAS.
- C. Potrin, J.-M. Bregalt and J.-M. Manoli, J. Chem. Soc., Chem. Commun., 1980, 664–665 Search PubMed.
- D. L. Hughes, R. L. Richards and C. Shortman, J. Chem. Soc., Chem. Commun., 1986, 1731–1732 RSC.
- R. A. Sanchez-Delgado, J. Mol. Catal., 1994, 86, 287–307 CrossRef CAS.
- R. J. Angelici, Bull. Soc. Chim. Beld., 1995, 104, 268 Search PubMed.
- M. Seredych and T. J. Bandosz, Langmuir, 2007, 23, 6033–6041 CrossRef CAS.
- X. L. Ma, S. Velu, L. Sun, C. S. Song, N. Mehdi and S. Siva, Prepr. Pap.sAm. Chem. Soc., Div. Fuel Chem., 2003, 48, 688 CAS.
- Y. S. Shen, J.L. Wang, S. M. Zhu and T. Qiu, Rare Metal Mater. Eng., 2010, 39, 814–819 CAS.
- J. H. Shan, X. Q. Liu and R. Cui, J. Chem. Eng. Chin. Univer., 2008, 22, 839–843 CAS.
- J. H. Shan, X. Q. Liu and R. Cui, Chem. Ind. Eng. Prog., 2008, 27, 1065–1069 CAS.
- X. L. Song, L. B. Sun, G. S. He and X. Q. Liu, Chem. Commun., 2011, 47, 650–652 RSC.
- W. G. Rudolf, N. V. Reno, US Pat., 2002, 6,827,844, B2 Search PubMed.
- S. U. Rege, R. T. Yang and C. A. Cain, AIChE J., 1998, 44, 1519–1528 CrossRef CAS.
- B. S. Schueller and R. T. Yang, Ind. Eng. Chem. Res., 2001, 40, 4912–4918 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |