DOI:
10.1039/C1RA00616A
(Paper)
RSC Adv., 2012,
2, 1656-1662
Nuclear transport facilitated by the interaction between nuclear pores and carbohydrates†
Received
19th August 2011
, Accepted 15th November 2011
First published on 22nd December 2011
Abstract
The nuclear pores located on the nuclear envelope work as a selective barrier to nuclear import. The transport of large cargo molecules (larger than ∼40 kD) into the nucleus generally requires the aid of nuclear transport proteins, such as importins, which bind to nuclear localization signal (NLS) sequences in the cargo molecules and transport these molecules into the nucleus through the nuclear pore. In our previous paper, we showed that maltotriose(Glc3)-displaying quantum dots (QDs) can pass through the nuclear pore in digitonin-treated HeLa cells. The aim of this study is to clarify the mechanism of the nuclear import of maltooligosaccharide-displaying QDs (maltooligo-QDs). We prepared maltopentaose(Glc5) and maltoheptaose(Glc7)-QDs in addition to Glc3-QDs. The effect of the number of glucose units in the maltooligosaccharide on the rate of nuclear import of QDs has been explored by confocal laser scanning microscopy (CLSM). Further, we analyzed that the direct interactions of maltooligo-QDs to an internal protein in the nuclear pore (Nucleoporin62) using a surface plasmon resonance (SPR) system. We found that maltooligo-QDs with more than three glucose units (Glc3, Glc5 and Glc7-QDs) rapidly entered the cellular nucleus, and had a higher affinity to nucleoporin62 than those of PEG and Glc1-QDs. These data indicate that the interaction between nuclear pores and carbohydrates is the driving force behind the nuclear import of maltooligo-QDs.
Introduction
Nanoparticle permeation of biological barriers, such as the cellular membrane, nuclear envelope and blood–brain barrier, is an important theme in the development of new medicines, diagnostic techniques and drug delivery systems (DDSs).1–5 For drugs targeting cells, there are two major biological barriers: one is the plasma membrane and the other is the nuclear envelope. Permeation of the plasma membrane has been extensively studied by a large number of research groups and various molecules for the surface modification of nanoparticles have been explored, such as cationic molecules (including cell-penetrating peptides)6,7 and ligands specific to cell surface receptors.8,9 On the other hand, despite the fact that the nuclear-targeting is also crucial to development of DDSs, the number of methods available for the nanoparticle permeation into the nucleus is limited. On the nuclear envelope, the nuclear pore complex (NPC) forms a 25–30 nm pore and works as a robust biological barrier to nuclear-targeting drug delivery (Fig. 1).10,11 The inside of the pore is filled with gel-like proteins, called nucleoporins (Nups).12,13 This permeability barrier allows the free passage of only small molecules, and the transit of macromolecules (larger than ∼40 kD) into the nucleus generally requires the aid of nuclear transport proteins, such as importin, which bind to nuclear localization signals (NLSs).14–16 Previous studies on the nuclear import of nanoparticles strongly rely on NLS modifications on the nanoparticle surface.2,17–19 However, when the cargo molecules are highly negatively charged, such as DNA, the NLSs do not work efficiently.20,21 Furthermore, since importins themselves are accumulated under certain sets of cellular conditions,22 it might be difficult to regulate the nuclear import of NLS-modified nanoparticles. Therefore, a new approach to the transport of nanoparticles into the nucleus without the aid of a natural transporter is in high demand. Monsigny and co-workers reported that the carbohydrate (glucose or mannose)-modification of proteins facilitates their nuclear import.23–25 In general, carbohydrates are neutral, highly water soluble and biocompatible, which makes them suitable for the modification of target molecules. We have reported that maltotriose (Glc3: three glucose units)-displaying quantum dots (Glc3-QDs) easily diffuse into the nucleus in digitonin-permeabilized cells.26,27 However, carbohydrate-dependent nuclear import based on a chemical approach has not yet been explored and the mechanism is still unclear.
 |
| | Fig. 1 Schematic illustration of nuclear import through the nuclear pore. Nucleoporins (Nups) are internal proteins of the nuclear pore complex (NPC) that work as a selective barrier to nuclear import. | |
We aim to clarify how the maltooligosaccharide modification of nanoparticles allows rapid passage through the NPC barrier and then optimize the nuclear import of QDs through the modulation of the length of the attached oligosaccharide. We prepared maltooligo-QDs, in which the length of the oligosaccharide varied from 1 to 7 glucose units (Glc1, Glc3, Glc5 and Glc7) (Scheme 1). The direct interactions between maltooligo-QDs and nucleoporin62 (Nup62), which is a component of the nuclear pore, were monitored using a surface plasmon resonance (SPR) method. Surprisingly, Glc3-, Glc5-and Glc7-QDs efficiently penetrated into the nucleus and showed high affinities to the Nup62. In contrast, PEG (no carbohydrate) and Glc1-QDs were not imported into the nucleus and had low affinities to Nup62. To the best of our knowledge, this is the first report to disclose the interaction between nuclear pores and carbohydrates and this property is thought to have potential benefits for the further design of molecular drugs targeting the nucleus.
Experimental
General information
Succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate (SMCC) was purchased from Pierce. Texas-Red BSA was purchased from Invitrogen. The SV40 large T-antigen NLS peptide (NH2–PKKKRKVC–CO2H) was obtained from Greiner Bio-one in an HPLC purified form. Dulbecco's modified Eagle's medium (DMEM) was purchased from Sigma. Fetal bovine serum, penicillin, and streptomycin were purchased from Gibco. Digitonin was purchased from Fluka. All other chemical reagents were purchased from Wako Pure Chemical Industries, Tokyo Chemical Industry Company and Aldrich and used as received. MALDI-TOF MS spectra were measured with a Voyager-DE STR-H spectrometer (Applied Biosystems) using 2,5-dihydroxybenzoic acid as the matrix. NMR spectra were measured with a JEOL 400 MHz spectrometer. Fluorescence and differential interference contrast (DIC) images were obtained with a confocal-laser scanning microscope (Olympus FV-300). The experiment for Fig. 2 were done under the same condition including laser power [power level; 10 (range; 0–100) in FV-300 software (OLYMPUS)] and gain of photomultiplier [gain level; PMT 550 (range; 0–1000) in FV-300 software (OLYMPUS)] on the same day, and the saturation of intensity means a saturation of the microscope detectors. Microinjection was carried out by using a semi-automatic injection system (Eppendorf FemtoJet) attached to Eppendorf micromanipulator (Eppendorf InjectMan NI 2) mounted on an inverted confocal-laser scanning microscope. The image analysis was carried out by Image-Pro Plus (MediaCybernetics). SPR signals were measured with SPR-670M (MORITEX). Stokes diameter of QDs were measured with DelsaNano (Beckman Coulter).
 |
| | Fig. 2 (A) DIC and fluorescence images of HeLa cells incubated with PEG-, Glc1-, Glc3-, Glc5- and Glc7-QDs for 5, 10, 30 min after the addition of the QD solution. (B) Time courses of fluorescence intensity in the nucleus. The gray, red, blue, orange and purple lines correspond to PEG-, Glc1-, Glc3-, Glc5-and Glc7-QDs, respectively. | |
Preparation of maltooligo-QDs and their characterizations
The maltooligosaccharide derivatives were synthesized according to the method of Penadés28 with some modifications (see details in ESI†). These compounds were immobilized on CdTe/ZnS QDs via surface exchange reaction from original trioctylphosphine oxide (TOPO)/tributylphosphine (TBP) to PEG- or maltooligosaccharide derivatives. Briefly, the TOPO/TBP-coated CdTe/ZnS QDs were dissolved in MeOH and added to a methanol solution of PEG and maltooligosaccharide derivatives (final conc. 40 mM). After sonication, the mixture was left to stand in the dark for 12 h at room temperature. The excess maltooligosaccharide derivatives were removed by dialysis and repeated centrifugal filtrations to yield PEG- and maltooligo-QDs. The number of ligands on the QDs was determined using ICP-OES (Shimazu) according to the method of Yang.29 The Stokes diameter of the maltooligosaccharide-displaying QDs in water was determined from CONTIN size distribution using dynamic light scattering (DLS).
Digitonin-permeabilization of HeLa cells
HeLa cells were grown in a monolayer in Dulbecco's modified Eagle's culture medium supplemented with 10% fetal bovine serum (FBS), 500 units/ml penicillin, and 500 μg ml−1streptomycin. The cultures were kept at 37 °C in a humidified incubator under a 5% CO2 atmosphere. Cell permeabilization using digitonin was performed as described by Adam et al.30 Briefly, HeLa cells (ca. 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per dish; dish diameter 35 mm) were seeded on glass-based dishes and grown for 2 days. The cells were incubated for 5 min in an ice-cold solution of 32.5 mM digitonin-dissolved import buffer (20 mM HEPES, pH 7.3, 110 mM potassium acetate, 5 mM sodium acetate, 2 mM magnesium acetate, 0.5 mM EGTA, 2 mM DTT) and then washed twice with cold import buffer. They were then incubated for 10 min in an import buffer containing a protease inhibitor cocktail (at a final concentration of 1 mg ml−1 Pepstatin A, 1 mg ml−1Aprotinin, 1 mg ml−1Leupeptin) at 37 °C. After the addition the QD solution (at a final concentration of 100 nM) to the permeabilized cells, the dishes were immediately examined using a CLMS and a fluorescence microscope.
000 cells per dish; dish diameter 35 mm) were seeded on glass-based dishes and grown for 2 days. The cells were incubated for 5 min in an ice-cold solution of 32.5 mM digitonin-dissolved import buffer (20 mM HEPES, pH 7.3, 110 mM potassium acetate, 5 mM sodium acetate, 2 mM magnesium acetate, 0.5 mM EGTA, 2 mM DTT) and then washed twice with cold import buffer. They were then incubated for 10 min in an import buffer containing a protease inhibitor cocktail (at a final concentration of 1 mg ml−1 Pepstatin A, 1 mg ml−1Aprotinin, 1 mg ml−1Leupeptin) at 37 °C. After the addition the QD solution (at a final concentration of 100 nM) to the permeabilized cells, the dishes were immediately examined using a CLMS and a fluorescence microscope.
Construction and expression of importin β, Nup62N and mutated Nup62N
The expression and purification of mouse importin β was described in the previous report.31 The amino acid sequence of Nup62N used in this study is as follows, where the amino acid residues described in boldface correspond to FG repeat motifs (5 FxFG motifs and 1 FG motif): MSGFNFGGTGAPAGGFTFGTAKTATTTPATGFSFSASGTGTGGFNFGTPSQPAATTPSTSLFSLATQTSTTQTPGFNFGTTPASGGTGFSLGISTPKLSLSSTAATPATANTGSFGLGSSTLTNAISGASTSSQGTAPTGFVFGSSTTSAPSTGTTGFSFTSGSASQPGASGFNIGSV.
cDNA encoding the amino-terminal fragment of rat Nup62 (amino acid number 1–178) was amplified by PCR using a set of primers carrying XhoI and NotI (forward: 5′-cccctcgagctatgagtgggtttaactttggaggcacc-3′, reverse: 3′-cctggagcctccggcttcaacattggctctgttaggcggccgctgg-5′). The PCR product was digested with Xho I and Not I, and then ligated into a pGEX-6P2 vector (GE Healthcare) to generate a fusion protein with glutathione S-transferase (GST). A site-specific biotinylation sequence (MSGLNDIFEAQKIEWHE) was inserted into the carboxyl terminus of Nup62N by ligating a double-stranded DNA oligo (the nucleotide sequence of the coding strand is 5′-gcggccgcccatgtccggcctgaacgacatcttcgaggctcagaaaatcgaatggcacgaaaagcttgcggccgc-3′) into the Not I site. We prepared the mutated Nup62N, in which 11 phenylalanines (position: 4, 6, 16, 18, 44, 46, 76, 78, 115, 141 and 143) were replaced by alanines. To substitute the phenylalanines with alanines, PCR-based site-directed mutagenesis was performed using the following primers; for F4A and F6A: 5′-cccctcgagctatgagtgggGCGaacGCGggaggcaccggggct-3′, for F16A and F18A: 5′-cctgctggcggcGCGacaGCGgggaccgcaaagactgcg-3′, for F44A and F46A: 5′-ggcaccggagggGCGaatGCGgggactccctcccagcc-3′, for F76A and F78A: 5′-cagaccccaggaGCGaacGCGggaacaacacctgcttctggggg-3′, for F115A: 5′-aacactggcagcGCGgggcttggcagcagtactcttaccaatg-3′, and for F141A and F143A: 5′-acagcccccactggcGCGgtcGCGggctcttctaccacctctgctccg-3′.
Recombinant proteins were expressed in E. coli strain AVB101, which expresses biotin ligase (Avidity LLC.). The biotin ligase covalently attaches the biotin moiety to the amino group of the lysine residue within the recognition sequence.32 The cells were grown at 37 °C in LB medium until the O.D.600 reached 0.6, and then the expression of the recombinant protein was induced by incubation with 1 mM IPTG at 20 °C for 3 h in the presence of d-biotin (final 50 μM). The cells were collected by centrifugation at 5000 g for 10 min at 4 °C and resuspended in lysis buffer (50 mM Tris–HCl, pH 8.3, 500 mM NaCl, 1 mM EDTA, 1 mM PMSF and 2 mM DTT). The cells were sonicated for 2 min, and treated with 5 μg ml−1DNase I for 30 min on ice, and then centrifuged at 12000 g for 15 min. The supernatant was collected and incubated with glutathione-sepharose 4B beads (GE Healthcare) for 4 h at 4 °C. The beads were washed with the lysis buffer, and then incubated with site-specific protease (PreScision, GE) in the cleavage buffer (50 mM Tris–HCl, pH 7.0, 150 mM NaCl, 1 mM EGTA, 2 mM DTT) at 4 °C for 6 h to separate the Nup62N fragment from GST moiety. The supernatant was collected and dialyzed against the transport buffer (20 mM HEPES-KOH (pH 7.3), 110 mM CH3COOK, 2 mM (CH3COO)2Mg, 5 mM CH3COONa, 0.5 mM EGTA).
Binding analysis of maltooligo-QDs to Nup62N using a SPR system
A 4,4′-dithio dibutyric aid (DDA) SAM was formed on a gold sensor chip in ethanol (10 μM) and reacted with EDC (25 mg in 1 ml H2O)–NHS (15 mg/9 ml in 1,4-Dioxane) for 30 min. The sensor chip was set up in the SPR system, and streptavidin (50 μg ml−1) was injected for 4 min and immobilized on the sensor chip via an amino-coupling procedure. The import buffer was used as the running buffer at a flow rate of 15 μl min−1. BSA (0.5 μg ml−1) was injected for 4 min as a blocking reagent and HCl (10 mM) was injected for 4 min as a washing reagent. Subsequently, biotinated Nup62N (wild type) (0.05 μM) or Nup62N-AG (mutant) (0.05 μM) were immobilized using avidin-biotin recognition. After immobilization, concentration series (10 nM – 1 μM) of importin β and PEG-, Glc1-, Glc3-, Glc5-and Glc7-QDs and dextran 60000 were injected for 4 min increments to obtain the kinetic parameters to Nup62N or Nup62N-AG. We then calculated the association rate constant (kon) and dissociation rate constant (koff) on the assumption that the ligand-analyte interaction proceeded at 1:1 binding.
Preparation of nuclear localization signal (NLS)-attached BSA
Texas-Red BSA (TR-BSA, Invitrogen) was dissolved in PBS (pH 7.2), then the TR-BSA solution (5 mg ml−1, 100 μl) was added to succinimidyltrans-4-(maleimidylmethyl)cyclohexane-1-carboxylate (SMCC) in DMF (100 mM, 10 μl) and the mixture was reacted for 1 h at room temperature. Subsequently, the unreacted SMCC was removed by Sephadex G25. SMCC modified TR-BSA was added to an NLS peptide (NH2–PKKKRKVC–CO2H) solution (40 mM, 16 μl) and reacted overnight at room temperature. NLS-BSA was then purified by dialysis.
Microinjection of Glc3-QDs or NLS-attached BSA into HeLa cells
HeLa cells were grown in a monolayer in Dulbecco's modified Eagle's culture medium supplemented with 10% fetal bovine serum (FBS), 500 units/ml penicillin, and 500 μg ml−1streptomycin. The cultures were kept at 37 °C in a humidified incubator under a 5% CO2 atmosphere. HeLa cells (ca. 25000 cells per dish; dish diameter 35 mm) were seeded on glass-based dishes and grown for 2 days. Cytoplasmic injections (30 μM QDs or NLS-BSA in import buffer) were performed under an injection pressure of 60 hPa and a maintenance pressure of 40 hPa, with an injection time of 0.1 s. After microinjection, the dishes were immediately examined using a CLSM.
Results and discussion
Effect of oligosaccharide length on the nuclear import of QDs in digitonin-permeabilized HeLa cells
The Stokes diameters of the maltooligo-QDs in water and the numbers of surface ligands were determined using DLS and ICP-OES, respectively (Table 1). The average Stokes diameter of the PEG-QDs was 5.7 nm and the diameter increased with increases in the maltooligosaccharide unit number. The ligand number for the PEG-QDs was 178 and those for the maltooligo-QDs were lower at 71–94, probably due to the bulkiness of the carbohydrate moieties. These QDs were used in subsequent experiments. HeLa cells were first treated with digitonin, which causes partial damage to the cellular membrane (but not the nuclear envelope), thereby increasing the permeability of QDs into the cytosol.30 This assay has often been used in biochemical studies to estimate the accurate efficiency of nuclear import of proteins.33,34 The maltooligo-QDs and PEG-QDs (100 nM) were added to the digitonin-treated HeLa cells, and nuclear import was then traced by CLSM (Fig. 2A). Glc3-, Glc5-and Glc7-QDs entered the cellular nucleus within 5 min, whereas PEG- and Glc1-QDs were retained in the cytosol even after 30 min. The nuclear import of maltooligo-QDs was observed for more than 85% cells (see wide view in Fig. S2, ESI†). There is a clear threshold between the nuclear import of QDs with one and those with three glucose residues. All QDs were well-suspended without aggregation in buffered solution for at least 7 days. We qualitatively compared the hydrophobicity of each type of QD by HPLC on a phenyl sepharose column (Fig. S1, ESI†). Fig. S1 indicates that the hydrophobicity was increased with a decrease in the number of glucose units on the QDs. This means that the hydrophobicity of the QDs is not a driving force behind the nuclear import. Average fluorescence intensities in the nucleus were plotted as a function of time (Fig. 2B). As these intensities are not quantitative values, we will limit our discussion to the relative order of QD permeation rates that can be estimated from the initial slopes of these lines.
Interestingly, Glc5-QDs showed the fastest diffusion into the nucleus. If this were a case of simple passive diffusion, smaller particles, such as PEG- and Glc1-QDs (diameters; 5.7 and 7.9 nm), would pass more rapidly through the NPC than Glc3-, Glc5- or Glc7-QDs (diameters; 10.7, 11.3 and 12.2 nm, respectively). Therefore, our results suggest that the maltooligosaccharide residues preferentially interact with proteins in the NPC, which is called "facilitated diffusion". In fact, image analyses of CLSM images indicates that Glc3-QDs were temporarily trapped within the nuclear envelope before transit into the nucleus, whereas PEG-QDs were not bound to the nuclear envelope (Fig. 3). Although the lag time is different, the slope of the increase in fluorescence intensity for each cell was almost the same. We could not explain the differences in lag time between nuclei, but suspect that they depend on minute differences in the physical chemical properties of the nuclear pore or cell cycles. As this was also observed for Glc5- and Glc7-QDs, binding to the nuclear pore appears to be necessary for nuclear import.
 |
| | Fig. 3 Time courses of fluorescence intensity along the white lines shown after the addition of (A) PEG-QDs and (B) Glc3-QDs. | |
Table 1 Ligand numbers on single quantum dots (QDs) and Stokes diameter of PEG- and maltooligo-QDs determined by ICP-OES and DLS, respectively. Error represents the standard deviation for three replicates
| Ligand |
Ligand/nanoparticle |
Stokes diameter/nm |
| PEG |
178 ± 3 |
5.7 ± 0.1 |
|
Glc1 |
88 ± 8 |
7.9 ± 0.3 |
|
Glc3 |
87 ± 16 |
10.7 ± 0.4 |
|
Glc5 |
71 ± 20 |
11.3 ± 0.1 |
|
Glc7 |
94 ± 19 |
12.2 ± 0.1 |
To exclude the possibility that they damaged the nuclear pore/membrane, we tested the nuclear import of Glc3-QDs in the presence of dye-conjugated BSA using a digitonin assay.26 Glc3-QDs, but not dye-conjugated BSA, entered into the nucleus of the HeLa cells, indicating that the nuclear envelope was left intact. In fact, when Glc3-QDs and NLS-BSA were simultaneously added to digitonin-treated cells without the addition of cytosolic factors, the nuclear import of only the Glc3-QDs was observed. This data support the conclusion that the importin-mediated mechanism does not play a role in the assay (Fig S3, ESI†).
Kinetic analysis of the binding of maltooligo-QDs to nuclear pore protein (Nup62N)
Based on CLSM data showing the temporary entrapment of Glc3-QDs, we hypothesize that maltooligo-QDs may interact directly with the nuclear pore proteins. The direct interactions between maltooligo-QDs and the internal proteins of the NPC were investigated using an SPR method. In this study, we used biotin-modified Nup62N (178 amino acid residues from the N–terminus of native Nup62), which is well known as a component of the selective barrier of NPC35 and binding site for importin β.31,36 Nup62N was immobilized on sensor chips via an avidin–biotin interaction, and the affinities of a series of maltooligo-QDs were measured (Fig. 4A). As a positive control, injection of importin β (50 nM) into Nup62N produced a marked SPR response (Fig. 4B), with the dissociation constant (Kd) estimated to be 300 nM, which is in good agreement with those in previous reports.37 Next, the injection of Glc3-, Glc5- and Glc7-QDs (50 nM) onto a Nup62N-immobilized chip was clearly observed to cause a large increase in SPR signals, whereas the injection of PEG- and Glc1-QDs resulted in only weak binding (Fig. 4C). This trend is in good agreement with the CLSM data observed in Fig. 2. For SPR experiments, Glc3-QDs showed the highest affinity to Nup62N among the tested QDs. However, the digitonin assay in Fig. 2 indicates that Glc5-QDs have the highest permeation rate. This means that the binding affinity to Nup62N is not perfectly parallel to the permeation rate into the nucleus, and rather kinetic parameters (discussed below) are important in explaining the permeation rate observed in Fig. 2. Further, since the Nup62N used in this study is not modified with O–GlcNAc, as in mammalian cells, this might explain the difference in the order of the affinity of maltooligo-QDs to Nup62N and the observed permeation rate. Interestingly, dextran (Molecular weight: 60 kDa), a polymer in which glucose molecules are linked by α-1,6-glucosyl bonds, did not bind to the Nup62N-immobilized surface (Fig. S4, ESI†). This implies that it is important to immobilize the oligosaccharides in a defined geometry to gain a high affinity.
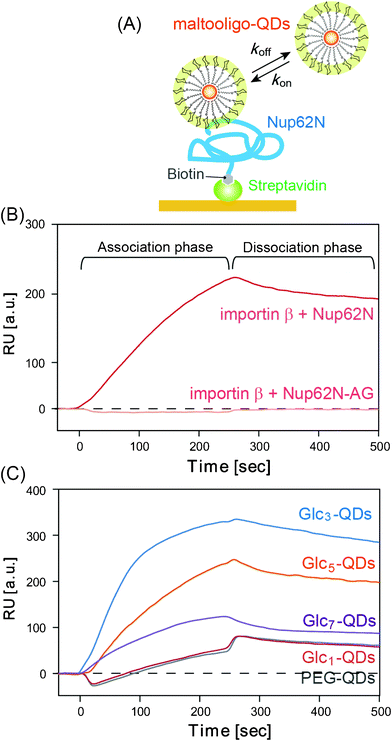 |
| | Fig. 4
SPR analysis of the binding of importin β and maltooligo-QDs to Nup62. (A) Schematic representation of the interaction of maltooligo-QDs and the Nup62N-immobilized sensor chip. (B) SPR sensorgrams of the interactions between importin β (50 nM) and Nup62N and Nup62N-AG. (C) Sensorgrams of the interactions between maltooligo-QDs (50 nM) and Nup62N. The importins or QDs were injected at time 0. | |
The association and dissociation rate constants (kon and koff) could be dominant factors in determining the permeation rate of maltooligo-QDs, and these constants are summarized for each QD in Fig. 5. The association rate constants of maltooligo-QDs (Glc3, Glc5 and Glc7) were clearly higher than those of Glc1-QDs and PEG-QDs. Particularly, the kon value of the Glc3-QDs was 78 times higher than that of the PEG-QDs (Glc3-QDs; 38 × 104, PEG-QDs; 0.49 × 104M−1s−1). Since the kon value reflects the adsorption rate into the nuclear pore protein, these data mean that the selective permeation of maltooligo-QDs above that of monosaccharide-displaying QDs is determined at the early adsorption into the nuclear pore. In contrast, there was a slight increase in koff value as the number of glucose residues increased within the same order of magnitude. Since the koff value reflects the dissociation rate from the nuclear pore protein, the large koff values characteristic of Glc5- and Glc7-QDs might allow their rapid transportation into the nucleus. In general, the permeation rate through the nuclear pore depends on the combination of adsorption (kon) and dissociation (koff) rates. QDs with high kon value are more readily adsorbed into the nuclear pore, whereas QDs with high koff values are more readily transported from the nuclear pore to the nucleus. Therefore, the balance between kon and koff values determines the permeation rate into the nucleus through the nuclear pore. Glc3-QDs had the highest kon value, whereas Glc7-QDs had the highest koff value, with the Glc5-QDs having moderate kon and koff values. This means that Glc3-QD is adsorbed easily, but is not readily transported from the nuclear pore to the nucleus. The case of Glc7-QDs is exactly opposite to that of Glc3-QDs, and we consider that Glc5-QDs have the best combination of kon and koff values for the passage through the nuclear pore, leading to the highest permeation rate (see Fig. 2). The affinity between the karyopherin complex and Nup1p is quite high (c.a. 103 times higher)38,39 compared to those between maltooligo-QDs and Nup62. The association rate constants (kon) of maltooligo-QDs are 2–3 orders of magnitude less than those of karyopherin, and the dissociation rate constants (koff) are one order of magnitude less than those of karyopherin. This means that the karyopherin complex is able to accomplish efficient nuclear import with high kon and koff values. Based on these kinetic parameters, maltooligo-QDs can be expected to be inefficiently imported in comparison with natural karyopherins; however, as they have kinetic values equivalent to importin β, the affinities of the maltooligo-QDs might be sufficient to allow their passage through the nuclear pore.
 |
| | Fig. 5 Kinetic parameters of importin β and maltooligo-QDs to Nup62N. (A) kon value (B) koff value. Errors represent the standard deviation for three replicates. | |
Since the nuclear pore proteins are not originally carbohydrate-binding proteins (lectins), the molecular based-driving force behind this affinity was then explored by the amino acid mutation of Nup62N. Nup62N has 6 phenylalanine-glycine (FG) motifs (5 FxFG motifs and 1 FG motif; see experimental section), and it is well known that the FG motif is the binding site for importin β.31,36 We created a mutant in which all 11 phenylalanine residues in the 6 FG motifs were replaced with alanine residues, referred to as Nup62N-AG in this paper. The binding of importin β to Nup62N was drastically decreased by this mutation (Fig. 4B), showing that our SPR system is a good model system of the nuclear pore. Interestingly, for maltooligo-QDs, there was no decrease in binding affinities to Nup62N-AG (Table 2). This indicates that the FG motif does not contribute to the binding of maltooligo-QDs and Nup62 has different binding sites for maltooligo-QDs from those of natural transporters.
Table 2 Dissociation constant of importin β and maltooligo-QDs to Nup62N and Nup62N-AG (mutant) determined by SPR measurements
| |
K
d (10−9M) |
|
Analyte
|
Nup62N |
Nup62N-AG |
|
The binding of importin β to Nup62N-AG (mutant) was too small to determine these kinetic parameters at the tested range of concentrations (∼100 nM). Errors represent the standard deviation for three replicates.
|
| importin β |
300 ± 22 |
n.d.a |
|
Glc3-QD
|
16 ± 5.1 |
12 ± 3.0 |
|
Glc5-QD
|
75 ± 15 |
66 ± 15 |
|
Glc7-QD
|
190 ± 34 |
200 ± 13 |
Observation of nuclear import of Glc3-QDs in live cells
Our previous work on nuclear imports and the above experiments were performed using digitonin-treated cells. Finally, we confirmed the nuclear import of maltooligo-QDs in living HeLa cells using microinjection techniques by comparing the permeation rate of Glc3-QDs with NLS-attached bovine serum albumin (BSA). NLS-BSAs were visualized by modification with Texas Red dye (Fig. 6). Glc3-QDs were diffused into the nucleus soon after microinjection and reached equilibrium between the cytoplasm and nucleus, rather than displaying preferential accumulation in the nucleus. This indicates that the maltooligosaccharide-dependent nuclear import pathway basically operates via passive diffusion. PEG-QDs were retained in the cytoplasm even after 120 min. The nuclear pore works as a robust barrier that macromolecules (>40 kD) cannot pass through. However, based on the SPR results, it is considered that maltooligo-QDs circumvent this by their affinity to the nuclear pore without the need for any natural transport system. For NLS-BSAs, their concentration in the nucleus after 15 min was higher than that in the cytoplasm, resulting in their accumulation in the nucleus. This shows that NLS-containing proteins are actively transported by importin into the nucleus and accumulated in nucleus by the Ran gradient.
 |
| | Fig. 6
DIC and fluorescence images of living HeLa cells after microinjection of Glc3-QDs, PEG-QDs and NLS-BSA. | |
Conclusion
We found that maltooligo-QDs with more than three glucose units (Glc3, Glc5 and Glc7–QDs), rapidly entered the cellular nucleus. As natural transporters (e.g., importins) have a greater hydrophobicity than other cytoplasmic proteins, hydrophobic interactions mimicking those of the transporters were believed to be crucial for interaction with the nuclear pore.40 Hanover and co-workers reported that O-GlcNAc and further galactose modification is not related to the assembly of nuclear import or events relating the nuclear pore.41–43 Based on these reports, it is clear that nuclear pore proteins are not originally carbohydrate-binding proteins (natural lectins). However, we found that the nuclear pore proteins have an affinity to maltooligo-QDs, and this interaction is one of the driving forces for the nuclear import of maltooligo-QDs. This implies that the multiple molecular interactions, such as hydrogen bonds, which occurred on the surface of QDs, might be essential to produce the specific affinity. Recently, our collaborators demonstrated that maltotriose residues can work as efficient gene delivery carriers in vivo.44 Our finding of facilitated diffusion of maltooligo-QDs expands the possibility of carbohydrate-driven nuclear transport due to high water solubility, neutral charge and high biocompatibility of carbohydrates. Maltooligosaccharide modification could be an effective approach to nuclear targeting for not only various nanoparticles, but also for a variety of molecules, including DNA, RNA and proteins.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research from the MEXT, Japan. S. S. thanks the JSPS for a Grant-in-Aid for JSPS Fellows.
References
- V. Sokolova and M. Epple, Angew. Chem., Int. Ed., 2008, 47, 1382–1395 CrossRef CAS.
- B. Kang, M. A. Mackey and M. A. EI-Sayed, J. Am. Chem. Soc., 2010, 132, 1517–1519 CrossRef CAS.
- S. Bhaskar, F. Tian, T. Stoegar, W. Kreyling, F. M. Fuente, V. Grazu, P. Borm, G. Estrada, V. Ntziachristos and D. Razansky, Part. Fibre Toxicol., 2010, 7, 3 CrossRef.
- A. Verma, O. Uzun, Y. Hu, H.-S. Han, N. Watson, S. Chen, D. J. Irvine and F. Stellacci, Nat. Mater., 2008, 7, 588–595 CrossRef CAS.
- H.-L. Liu, M.-Y. Hua, H.-W. Yang, C.-Y. Huang, P.-C. Chu, J.-S. Wu, I.-C. Tseng, J.-J. Wang, T.-C. Yen, P.-Y. Chen and K.-C. Wei, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 15205–15210 CrossRef CAS.
- H. Yukawa, Y. Kagami, M. Watanabe, K. Oishi, Y. Miyamoto, Y. Okamoto, M. Tokeshi, N. Kaji, H. Noguchi, K. Ono, M. Sawada, Y. Bada, N. Hamajima and S. Hayashi, Biomaterials, 2010, 31, 4094–4103 CrossRef CAS.
- L. Sun, D. Liu and Z. Wang, Langmuir, 2008, 24, 10293–10297 CrossRef CAS.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- R. Kikkeri, B. Lepenies, A. Adibekian, P. Laurino and P. H. Seeberger, J. Am. Chem. Soc., 2009, 131, 2110–2112 CrossRef CAS.
- R. S. Faustin, T. J. Nelson, A. Terzic and C. Perez-Terzic, Clin. Pharmacol. Ther., 2007, 81, 880–886 CrossRef.
- A. M. Miller and D. A. Dean, Adv. Drug Delivery Rev., 2009, 61, 603–613 CrossRef CAS.
- S. Frey, R. P. Richter and D. Gorlich, Science, 2006, 314, 815–817 CrossRef CAS.
- S. Frey and D. Gorlich, Cell, 2007, 130, 512–523 CrossRef CAS.
- L. J. Terry, E. B. Shows and S. R. Wente, Science, 2007, 318, 1412–1416 CrossRef CAS.
- C. Dingwall and R. A. Laskey, Trends Biochem. Sci., 1991, 16, 478–481 CrossRef CAS.
- A. Lange, R. E. Mills, C. J. Lange, M. Stewart, S. E. Devine and A. H. Corbett, J. Biol. Chem., 2007, 282, 5101–5105 CrossRef CAS.
- F. Chen and D. Gerion, Nano Lett., 2004, 4, 1827–1832 CrossRef CAS.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS.
- N. Pante and M. Kann, Mol. Biol. Cell, 2002, 13, 425–434 CrossRef CAS.
- T. Nagasaki, T. Myohoji, T. Tachibana, S. Futaki and S. Tamagaki, Bioconjugate Chem., 2003, 14, 282–286 CrossRef CAS.
- M. Tanimoto, H. Kamiya, N. Minakawa, A. Matsuda and H. Harashima, Bioconjugate Chem., 2003, 14, 1197–1202 CrossRef CAS.
- Y. Miyamoto, T. Saiwaki, J. Yamashita, Y. Yasuda, I. Kotera, S. Shibata, M. Shigeta, Y. Hiraoka, T. Haraguchi and Y. Yoneda, J. Cell Biol., 2004, 5, 617–623 CrossRef.
- M. Monsigny, C. Rondanino, E. Duverger, I. Fajac and A.-C. Roche, Biochim. Biophys. Acta, Gen. Subj., 2004, 673, 94–103 CrossRef.
- C. Rondanino, M.-T. Bousser, M. Monsigny and A.-C. Roche, Glycobiology, 2003, 7, 509–519 CrossRef.
- E. Duverger, C. Pellerin-Mendes, R. Mayer, A.-C. Roche and M. Monsigny, J. Cell Sci., 1995, 108, 1325–1332 CAS.
- K. Niikura, S. Sekiguchi, T. Nishio, T. Masuda, H. Akita, Y. Matsuo, K. Kogure, H. Harashima and K. Ijiro, ChemBioChem, 2008, 9, 2623–2627 CrossRef CAS.
- K. Niikura, K. Ijiro and S. Sekiguchi, Trends Glycosci. Glycotechnol., 2009, 21, 335–344 CrossRef CAS.
- A. G. Barrientos, J. M. Fuente, T. C. Rojas, A. Fernandez and S. Penades, Chem.–Eur. J., 2003, 9, 1909–1921 CrossRef CAS.
- Y. Yang, Y.-T. Zhao, T.-T. Yan, M. Yu, Y.-L. Sha, Z.-H. Zhao and Z.-J. Li, Tetrahedron Lett., 2010, 51, 4182–4185 CrossRef CAS.
- S. A. Adam, R. S. Marr and L. Gerace, J. Cell Biol., 1990, 111, 807–816 CrossRef CAS.
- S. Otsuka, S. Iwasaka, Y. Yoneda, K. Takeyasu and S. H. Yoshimura, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16101–16106 CrossRef CAS.
- I. Chen, M. Howarth, W. Lin and A. Y. Ting, Nat. Methods, 2005, 2, 99–104 CrossRef CAS.
- H. Lorenz, D. W. Hailey and J. Lippincott-Schwartz, Nat. Methods, 2006, 3, 205–210 CrossRef CAS.
- C. E. Hindley, F. J. Lawrence and D. A. Matthews, Traffic, 2007, 8, 1313–1322 CrossRef CAS.
- T. Jovanovic-Talisman, J. Tetenbaum-Novatt, A. S. McKenney, A. Zilman, R. Peters, M. P. Rout and B. T. Chait, Nature, 2009, 457, 1023–1027 CrossRef CAS.
- S. M. Liu and M. Stewart, J. Mol. Biol., 2005, 349, 515–525 CrossRef CAS.
- N. B. Eisele, S. Frey, J. Piehler, D. Gorlich and R. P. Richter, EMBO Rep., 2010, 11, 366–372 CrossRef CAS.
- D. Gilchrist, B. Mykytka and M. Rexach, J. Biol. Chem., 2002, 277, 18161–18172 CrossRef CAS.
- S. S. Patel, B. J. Belmont, J. M. Sante and M. F. Rexach, Cell, 2007, 129, 83–96 CrossRef CAS.
- B. Naim, D. Zbaida, S. Dagan, R. Kapon and Z. Reich, EMBO J., 2009, 28, 2697–2705 CrossRef CAS.
- M. W. Miller and J. A. Hanover, J. Bol. Chem., 1994, 269, 9289–9297 CAS.
- J. A. Hanover, FASEB J., 2001, 15, 1865–1876 CrossRef CAS.
- W. A. Lubas, M. Smith, C. M. Starr and J. A. Hanover, Biochemistry, 1995, 34, 1686–1694 CrossRef CAS.
- H. Akita, T. Masuda, T. Nishio, K. Niikura, K. Ijiro and H. Harashima, Mol. Pharmaceutics, 2011, 8, 1436–1442 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures of maltooligosaccharide derivatives and TOPO/TBP-stabilized CdTe/ZnS QDs. HPLC analysis of maltooligo-QDs. See DOI: 10.1039/c1ra00616a |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 