Electrosynthesis, characterization and electrocatalytic properties of Pt–Sn/CCE towards oxidation of formic acid
Biuck
Habibi
* and
Nasrin
Delnavaz
Electroanalytical Chemistry Laboratory, Faculty of Sciences, Department of Chemistry, Azarbaijan University of Tarbiat Moallem, Tabriz, Iran. E-mail: B.Habibi@azaruniv.edu; Fax: +98-412 4327541
First published on 22nd December 2011
Abstract
Bimetallic platinum-tin nanoparticles were co-deposited on a carbon-ceramic electrode (CCE) surface using a simple one-step electrochemical process. The obtained catalyst (Pt–Sn/CCE) was characterized by scanning electron microscopy, energy-dispersive X-ray spectroscopy, X-ray diffraction and cyclic voltammetry. The electrocatalytic activity of the Pt–Sn/CCE towards the electrooxidation of formic acid (FA) was evaluated by cyclic voltammetry in 0.1 M H2SO4 solution. It was found that the Pt–Sn/CCE was catalytically more active than the Pt nanoparticles (alone) supported on the same substrate. The obtained results also showed that the presence of Sn greatly enhances the activity of the Pt towards the electrooxidation of FA. Moreover, it contributes to reduce the amount of the noble metal in the anode of direct formic acid fuel cells (DFAFCs), which remains one of the challenges to make the technology of DFAFCs economically viable.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) are believed to be the best among the different types of fuel cells being developed for transportation vehicles and portable electronic devices because of their high efficiency and low pollutant emission.1–4 One of the main problems in PEMFCs is the development of anodic materials with high electroactivity towards the oxidation of methanol and formic acid (FA). It is well known that platinum (Pt) is an effective electrocatalyst for these oxidations.5,6 Unfortunately, Pt is highly sensitive to CO poisoning; the catalyst surface is progressively poisoned by the adsorbed CO (COads), which is formed as a result of the stepwise dehydrogenation of these oxidants in oxidation reaction.7,8 Poisoning COads species can be oxidatively removed from the Pt surface through a Langmuir–Hinshelwood type surface reaction with neighboring OHads species electrosorbed from water at more positive potentials.9 As a result, alloying of Pt with oxophilic metals enables electrochemical dissociation of water on oxophilic metal sites at more negative potentials compared with pure Pt and, therefore, allows the electrocatalytic oxidation of COads at lower anodic overpotentials. Among these alloys, bimetallic Pt–Sn catalysts have been demonstrated to exhibit attractive electroactivity. Pt–Sn alloy catalysts demonstrate a superior activity toward electrooxidation of these oxidants and derived residues and have been intensively investigated and show high activity for methanol10–25 and FA16,19–28 oxidation. Therefore, these catalysts deserve closer attention as promising candidates for possible applications as anode materials in low-temperature PEMFCs, including direct formic acid fuel cells (DFAFCs). On the other hand, it is observed that the support of catalysts has important effects on the dispersion and size of metals or metal alloy nanoparticles and therefore directly influences the electrocatalytic activity and stability of these electrocatalysts.29 A support with large surface area, good conductivity and strong adsorption of metals can enhance the dispersion of electrocatalysts and increase the utilization and efficiency of the precious electrocatalysts. Therefore, the physico-chemical characteristics and surface chemistry of substrate influence properties of supported catalysts.30,31 Carbon materials are of special interest due to their outstanding properties, such as their tunable shape, size, porosity, chemical stability, corrosion resistance, low cost, good thermal resistance and electrical conductivity. The combination of all these characteristics has promoted the use of these materials as electrocatalyst supports.Sol–gel technology has aroused great interest in the design and application of electrochemical sensors and electroanalysis due to its simplicity, stability, physical rigidity, transparency, porosity, permeability, versatility and flexibility in the preparation procedure.32–34 Now, the sol–gel process conducted in the presence of graphite powder was proposed for the fabrication of a carbon–ceramic electrode (CCE) as a new kind of carbon substrate in electrochemical systems.32–39 Considering the stability, permeability, simplicity, easy production, good porosity, and the especially low cost of CCEs, it is one of the best materials that can be used as catalyst support in fuel cells.40–43
In this paper, the Pt–Sn nanoparticles are co-deposited by a simple electrochemical procedure on CCE to produce carbon-ceramic supported Pt–Sn nanoparticles catalyst (Pt–Sn/CCE), aiming to have a less expensive electrocatalyst in the DFAFCs. The physicochemical properties and electrochemical activities of the Pt–Sn nanoparticles for FA oxidation are investigated. It was found that Pt–Sn/CCE is catalytically more active than Pt nanoparticles (alone) supported on the CCE and smooth Pt.
2. Experimental
2.1. Chemicals
Methyltrimethoxysilane (MTMOS), formic acid, methanol, H2PtCl6·5H2O, SnCl2·2H2O, HCl, H2SO4 and graphite powder were obtained from Merck or Fluka. All solutions were prepared with double distilled water.2.2. Procedure of Pt–Sn/CCE preparation
The sol–gel processing method was used for fabricating CCE according to the following procedure: An amount of 0.9 ml MTMOS was mixed with 0.6 ml methanol. After addition of 0.6 ml HCl 0.1 M as catalyst, the mixture was magnetically stirred (for about 15 min) until it produced a clear and homogeneous solution. Then, 0.3 g graphite powder was added and the mixture was shaken for another 5 min. Subsequently, the homogenized mixture was firmly packed into a Teflon tube (with 3 mm inner diameter and 10 mm length) and dried for at least 24 h at room temperature. A copper wire was inserted through the other end to set up electric contact. The electrode surface was polished with emery paper grade 1500 and rinsed with water. Electrochemical deposition method (potential one step deposition) was used for preparation of the Pt–Sn/CCE electrocatalyst. The Pt–Sn nanoparticles were electrodeposited on the CCE surface from an aqueous solution of 0.1 M H2SO4 containing H2PtCl6·5H2O (5 × 10−3 M) and SnCl2·2H2O (5 × 10−3 M) at −0.2 V versus the saturated calomel electrode (SCE) at 25 °C.2.3. Instrumentation
The electrochemical experiments were carried out using an AUTOLAB PGSTAT-100 (potentiostat/galvanostat) and GEPS software equipped with a USB electrochemical interface was used for electrochemical experiments. A conventional three electrode cell was used at room temperature. A disc of Pt–Sn/CCE (3 mm diameter) was used as a working electrode. An SCE and a platinum wire were used as the reference and auxiliary electrodes, respectively. JULABO thermostat was used to control cell temperature at 25 °C. X-ray Diffraction (XRD) of the Pt–Sn nanoparticles was studied using a Brucker AXF (D8 Advance) X-ray power diffractometer with a Cu-Kα radiation source (λ = 0.154056 nm) generated at 40 kV and 35 mA. Scanning electron microscopy (SEM) was performed with a LEO 440i Oxford instrument. Chemical composition analyses of the catalysts were carried out by an energy dispersive X-ray (EDX) analyzer attached to the same device.3. Results and discussion
3.1. Physical characteristics of the Pt–Sn/CCE catalyst
In order to perform the surface characterization of the Pt–Sn/CCE catalyst, the micrograph of bare CCE and Pt–Sn/CCE surface was investigated by SEM and the corresponding results are shown in Fig. 1. Image A in Fig. 1 shows the structure of the bare CCE surface immediately after polishing with emery paper, grade 1500. As seen in this image, the surface of bare CCE is dense and scaly and has a high porosity. Fig. 1B shows the SEM micrograph of the CCE surface after Pt–Sn nanoparticle co-deposition. As seen, the Pt–Sn nanoparticles are spherical and separate and the surface coverage is almost uniform without any holes and deficiencies. Most likely, the particulate structures of Pt–Sn in this image are not the individual Pt–Sn crystallites. They are most probably, a collection of ball consists the aggregates of alloy crystallite. | ||
| Fig. 1 SEM images of (A) CCE surface immediately after polishing and (B) same electrode after Pt–Sn nanoparticles deposition (Pt–Sn/CCE). | ||
The elemental analysis of the Pt–Sn catalyst prepared by electrochemical deposition over the CCE was done by EDX. The EDX spectrum of Pt–Sn/CCE is shown in Fig. 2A and the calculated chemical compositions of the catalyst are listed in Table 1. The bare CCE data (Fig. 2B) is also shown for comparison. It is evident that Pt and Sn are present on the CCE support, indicating that the H2PtCl6 and SnCl2 precursors can be co-reduced to their respective metal phases by an electro co-deposition method from an aqueous solution of H2SO4. According to the composition values in Table 1, the ratio of Pt![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn in the Pt–Sn/CCE catalyst is quite close to the ratio of their salts in the plating solution.
:Sn in the Pt–Sn/CCE catalyst is quite close to the ratio of their salts in the plating solution.
 | ||
| Fig. 2 EDX data for the bare CCE (A) and Pt–Sn nanoparticles modified CCE (B). | ||
The crystallographic structure and chemical characterization of the present catalyst on the CCE was determined by the XRD method. For clarity, the XRD pattern of the bare CCE is also reported. The XRD pattern of the Pt–Sn catalyst shown in Fig. 3 presents the main characteristic peaks of the face-centered cubic (fcc) crystalline Pt at 2θ = 41.5, 46.8 and 68.3, namely, the planes (111), (200) and (220). The diffraction peaks appearing at 2θ = 35.6, 62.8 and 66.5 can be attributed to the 101, 112 and 202 indices of the tetragonal phase of β-Sn. As can be seen in the Fig. 3, no peaks for Sn oxides were found.25,44–47 On the other hand, the sharp diffraction peak at 54.82° which were observed in both of Fig. 3 and its inset belonged to carbon matrix (006).40
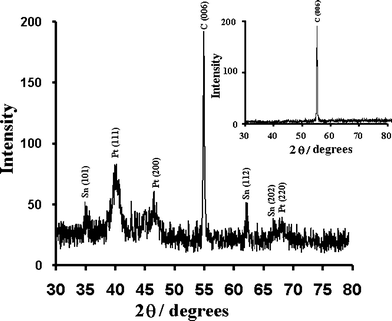 | ||
| Fig. 3 XRD pattern of the Pt–Sn electrocatalyst. Inset is the XRD pattern of bare CCE. | ||
3.2. Electrochemical characteristics of the Pt–Sn/CCE catalyst
The electrochemical behavior of the Pt–Sn/CCE catalyst was investigated using cyclic voltammetry at a scan rate of 50 mV s−1 in 0.1 M H2SO4 aqueous solution within the potential range of −0.3 and 1.3 V. Fig. 4 shows the cyclic voltammograms (CVs) of the Pt–Sn/CCE (thick line), Pt/CCE (thin line), smooth Pt (inset, curve 1) and bare CCE (inset, curve 2). These CVs can be separated into three regions: | ||
| Fig. 4 CVs of Pt–Sn/CCE (thick line) and Pt/CCE (thin line) in 0.1M H2SO4 at a scan rate of 50 mV s−1. The inset is CVs of smooth Pt (1)and bare CCE (2)in the same conditions. | ||
I) Hydrogen adsorption/desorption region
Typical Pt-peaks48 for the hydrogen under-potential deposition (Hudp) and the oxidation of hydrogen (Hoh) are present on the Pt–Sn/CCE but they become ill-shaped compared with Pt/CCE and smooth Pt. Also, in comparison with the unalloyed Pt modified CCE (Pt/CCE)42 (Fig. 4, thin line), both peaks for hydrogen adsorption and desorption of Pt–Sn/CCE decrease. Indeed, the actual surface area of the Pt–Sn/CCE is equivalent to the number of Pt sites available for hydrogen adsorption/desorption. In calculating the adsorption charge, i.e. the integrated area under the peaks, we assume that the double layer capacitance is constant across the entire potential range. The hydrogen adsorption charge (QaH) was calculated as 0.021 mC cm−2 for smooth Pt and 0.806 mC cm−2 for Pt–Sn/CCE, respectively. Therefore, the actual active surface area (Ar) of Pt–Sn/CCE can be obtained from charge for hydrogen adsorption as: Ar = QaH/Q0 = 3.8 cm2 (Q0 has been commonly taken as 210 μC/real cm2). These results show that the real surface area of the Pt–Sn/CCE is over 38 times larger than that of polycrystalline Pt. On the other hand, the adsorption and desorption peaks of hydrogen are not doublets. Indeed, the higher the Sn contents in the bimetallic nanoparticles, the lower the peak resolution in the hydrogen adsorption/desorption region on Pt.
On the other hand, the decrease of the hydrogen peak was linear with the increase in the amount of Sn on the Pt–Sn/CCE. Therefore, the contribution of the Sn atoms (θSn) deposited together with Pt atoms on the CCE surface was calculated by the following equation:49,50
| θSn = QbH − QaH/QbH | (1) |
Q aH is the electric charge of Pt–Sn/CCE evaluated from the CV in the hydrogen region charge between −0.30 and + 0.10 V in Fig. 4. QbH is the electric charge measured from Pt/CCE in the same region on the CV program. Under the deposition conditions, a stable Sn contribution (0.48) can be easily achieved. Under this ratio, the electrocatalyst has the best electrocatalytic activity. Compared with surface composition analysis by EDX (Table 1), there is good agreement between the two independent results.
II) Double-layer region
Comparison of double-layer region in CVs of Pt–Sn/CCE and Pt/CCE shows that the current is increased when Sn is added to Pt to form Pt–Sn. As this double-layer region corresponds to the potential domain for which water can be activated, water activation can take place at lower potentials on Pt–Sn surfaces than on smooth Pt.
III) Formation and reduction of the Pt oxide region
Inspection of this region on the CVs of the Pt–Sn/CCE and Pt/CCE shows that entering of the second metal (Sn) results in the decrease of the Pt character of the surface. The formation of the Pt oxides on the Pt–Sn catalyst decreases and consequently their current and reduction in the backward scan also decreases. The reduced changes in the formation and reduction currents of Pt oxide increase with the increasing amount of the second metal (Sn) in the Pt–Sn catalysts. On the other hand, the performance of cyclic voltammetry with the different ranges of potential (potential limits), over which the formation and reduction of surface oxides occur on Pt–Sn/CCE, show that the amplitude of the different CVs remained constant. In addition, after successive sweeps of cyclic voltammograms (20 cycles) between −0.3 and 1.3 V, the current of the different CVs in whole potential region remained constant, indicating stability of the deposited Pt–Sn nanoparticles on the CCE.
Curve 2 of inset Fig. 4, shows the CV of the bare CCE in the potential range −0.3 to 1.3 V in 0.1 M H2SO4 solution. Within the above mentioned potential range, there are no observable redox peaks at the CCE. In order to perform surface characterization of the bare CCE, the surface area of CCE was investigated by CV using Fe(CN)63−/4− as the probe ion and the Randles–Sevcik equation. The electrochemical area of CCE (geometric area ≈ 0.07 cm2), was 0.162 cm2.37,38
3.3. The electrocatalytic activity of Pt–Sn/CCE in FA oxidation
The electrocatalytic activity of the Pt–Sn/CCE in the oxidation of FA was evaluated using cyclic voltammetric experiments. Fig. 5 shows the CVs of the Pt–Sn/CCE, Pt/CCE (inset A) and the smooth Pt (inset B) in a 0.5 M FA + 0.1 M H2SO4 solution. During the forward scan of the CV of smooth Pt, the first two anodic peaks (at potentials of 0.1 and 0.5 V, respectively) result from FA oxidation, while the third peak at 1.06 V can be attributed to CO oxidation and FA oxidation on sites that were previously blocked by CO.51 There is a different pattern on the Pt–Sn/CCE; the electrooxidation of FA shows only one main peak in both forward and reverse scan and the oxidation currents of FA on the Pt–Sn/CCE are significantly higher than them on the smooth Pt. The CV of the FA oxidation on the Pt–Sn/CCE indicates that the reaction commences in the hydrogen region and proceeds rapidly to the positive direction. At potentials higher than −0.290 V, the reaction accelerates and the maximum rate (Ipf) is reached at ca. 0.69 V (Epf). An increase in current at potentials more than ca. 0.96 V is the result of oxygen evolution. The oxidation current in the forward scan is attributed to FA oxidation and the formation of Pt-adsorbed carbonaceous intermediates, including CO and CO2. | ||
| Fig. 5 CV of 0.5M FA on the Pt–Sn/CCE in 0.1M H2SO4 at a scan rate of 50 mV s−1. Insets are the CV of 0.5M FA in the same conditions on the Pt/CCE (A) and on the smooth Pt (B), respectively. | ||
The mechanism of FA oxidation on the Pt electrode in acid solution is reasonably well established as the so-called dual pathway mechanism, originally proposed by Capon and Parsons.52 In the direct pathway, the adsorption of FA leads to formation of an adsorbed intermediate, which then further oxidizes to CO2. This pathway requires free Pt sites (2).
| Pt(site) + HCOOH → Pt(site) + CO2 + 2H+ + 2e− | (2) |
There is also an indirect path involving a stable intermediate that blocks the surface and inhibits further adsorption of FA molecules. This poisoning species is generally accepted as being COads, as evidenced by in situ IR spectrocopy.53 Its production is given by reaction (3).
| Pt(site) + HCOOH → PtCOads + H2O | (3) |
It is removed by reaction with an oxygen-containing species to produce CO2. The nature of the oxygen-containing species is very controversial. Adsorbed hydroxide appears at about 0.5 V through water dissociation at a Pt electrode.
| Pt(site) + H2O → PtOHads + H+ + e− | (4) |
Pt oxide can be formed at potentials above about 0.75 V [reaction (5)] and is assumed to be the oxygen-containing species at these potentials.
| Pt(site) + H2O → PtO + 2H+ + 2e− | (5) |
As a result, the removal of the COads is written as either reaction (6) or (7), depending on whether the potential is below or above about 0.75V, respectively.53,54
| PtCOads + PtOHads → 2Pt(site) + CO2 + H+ + e− | (6) |
| PtCOads + PtO → 2Pt(site) + CO2 | (7) |
While the net reaction is same in both pathways, the water activation reaction is rather difficult. The OHads formed by the dissociation of water molecules on the Pt surface aids in removing the adsorbed surface poison COads by oxidizing it to CO2(6). But this process is indeed very intricate because a higher potential is required for the water activation (>0.5 V) on Pt surfaces. Consequently, the electrode surface will be blocked by larger numbers of COads species, thereby hindering the further adsorption of other FA molecules on the electrode surface. This drawback will decrease the number of FA molecules that can be oxidized as the surface poison COads remains on the electrode surface for a longer time, occupying active catalyst sites and thereby reducing the overall activity of FA oxidation. Hence the rate of FA oxidation primarily depends on the amount of COads removed from the electrode surface.
In comparison with the Pt nanoparticle modified CCE42 (inset A of Fig. 5), smooth Pt (inset B of Fig. 5) and even the Pd electrode;55 the Pt–Sn nanoparticles modified CCE showed an enhanced rate of oxidation of FA. Three aspects from the semi-qualitative analysis of CVs of FA oxidation on the Pt–Sn/CCE and on the Pt/CCE42 can be determined (Table 2). Firstly, at the Pt–Sn/CCE surface the peak potential for FA oxidation is shifted negatively by 121 mV with respect to Pt/CCE. Secondly, the anodic current of FA oxidation at the Pt–Sn/CCE is dramatically higher than these for the Pt/CCE. The thirdly, the ratio of Ipf/Ipb on the Pt–Sn/CCE was higher than these of Pt/CCE. These behaviors clearly show that the poisoning effect is markedly decreased by the presence of Sn, which is in agreement with results described previously.56
Based on the above deduction about the electrooxidation of FA, the ratio of the forward peak current (Ipf) to the backward peak current (Ipb) reflects the ratio of the amount of FA oxidized to CO2 to the amount of CO. Hence the ratio of Ipf to Ipb, Ipf/Ipb, can be used to describe the catalyst tolerance to the accumulation of carbonaceous species. Basically, a high Ipf/Ipb value represents a relatively complete oxidation of FA, producing CO2, while a low Ipf/Ipb ratio indicates poor oxidation of FA to CO2 during the anodic scan and excessive accumulation of carbonaceous residues on the catalyst surface.57 In other words, this ratio essentially reflects the fraction of the catalyst surface that is not poisoned by CO adsorption and can be used to measure the catalyst tolerance to COads poisoning.58 As can be seen in Table 3, the ratio of Ipf/Ipb for Pt–Sn/CCE in FA oxidation is more than 2 times higher than those of Pt/CCE and Pt, which indicates that more intermediate carbonaceous species were oxidized to CO2 in the forward scan on Pt–Sn/CCE surface than those on Pt/CCE and Pt surfaces.
| Catalyst type | I f (mA) | Onset potential (V vs. SCE) | I f/Ib at 1st cycle (dimensionless) | % (If/Ib)Pt–Sn/CCE/(If/Ib)Pt/CCE |
|---|---|---|---|---|
| Pt | 1.8 | 0.0 | 0.49 | — |
| Pt–Sn/CCE (80:20) |
2.1 | −0.20 | 0.56 | 14.3 |
| Pt–Sn/CCE (60:40) |
3.5 | −0.23 | 0.86 | 75.5 |
| Pt–Sn/CCE (50:50) |
4.4 | −0.30 | 1.05 | 114.28 |
| Pt–Sn/CCE (40:60) |
4 | −0.28 | 0.94 | 91.8 |
| Pt–Sn/CCE (20:80) |
2.7 | −0.26 | 0.70 | 42.8 |
The enhanced activity of the Pt–Sn nanoparticle modified CCE in the electrooxidation of FA can be attributed to the following reasons. As already mentioned; the oxidation of FA on Pt proceeds via two pathways. It is clear that, on the Pt–Sn nanoparticle modified electrode, the presence of Sn excites the reaction to proceed through a direct pathway mechanism or dehydrogenation mechanism wherein the reaction proceeds without any poisonous intermediate (High Ipf/Ipb for Pt–Sn nanoparticles). Therefore, an enhanced oxidation rate towards the electrooxidation of FA is observed due to the fact that Sn can increase the rate of FA via a direct CO2 pathway.59 Also, the presence of Sn causes the neighbor free Pt sites in the Pt–Sn/CCE surface minimize and therefore the amount of poisonous intermediate, COads, which adsorbed to Pt sites reduced.23,60
On the other hand, the presence of promoter (Sn atoms) acts as a supplier of oxygen atom:61
| Sn + H2O → SnOHads + H+ + e− | (8) |
The removal of the COads then is written as reaction (9):
| SnOHads + PtCOads → CO2 + Pt + Sn + H+ + e− | (9) |
The idea is that the Sn atoms are more easily oxidized than Pt and thus are able to oxidize the FA adsorbate at a lower potential. Indeed, the promoters act via a so-called bifunctional mechanism.62,63 Another important factor to be taken into account towards FA oxidation on the Pt–Sn/CCE is the role of second element (Sn) to modify the electronic properties of Pt by contributing d-electron density.64
In the backward potential scan (Fig. 5), a small reduction peak (IPt) appears due to the reduction of Pt oxide to Pt, as shown in reaction (10).
| PtO + 2H+ + e− → Pt + H2O | (10) |
And then a very steep increase of the reaction rate at ca. 0.37 V develops and a maximum current (Ipb) is observed at ca. 0.27 V (Epb). The backward oxidation peak can be attributed to the additional oxidation of the adsorbed CO to CO2:
| PtCOads +H2O → Pt + CO2 + 2H+ +2e− | (11) |
The obtained results show that the final potential (potential limits) in the cyclic voltammetric method (CVs not shown) can also affect the ratio of Ipf/Ipb in the electrooxidation of FA on the Pt–Sn/CCE. With an increase in the final potential, the Ipf remains constant, Ipb decreases, and the ratio of Ipf/Ipb increases (Fig. 6A, B and C). In fact, increasing the final potential accelerates the formation of Pt oxide via reaction (5). Acceleration of PtO formation causes the acceleration of reaction (7), the decrease of COads and the consequent decrease of Ipb. This phenomenon again shows that the ratio of Ipf/Ipb is a sign of electrocatalytic and promoter activity of the catalyst towards the oxidation of the poisonous intermediate COads. As can be seen in Fig. 6A, the final potential has no effect on Ipf, in other words, by increasing the final potential the conversion of metal to metal oxides is accelerated and as a result, an increase in the reduction peak of the Pt oxide to Pt happens. In case of peaks potential, it can be seen (Fig. 6B) that the potential of the FA oxidation peak in the forward scan (Epf) remains invariable, while the potential of oxidation peak in the backwards scan (Epb) shifts positively and consequently, the difference between Epf–Epb increases by increasing the final potential (Fig. 6C).
 | ||
| Fig. 6 (A) Plot of anodic peak current in the forward (Ipf) (♦) and backwards scan (Ipb) (■), (B) Variation of anodic peak potential in forward (♦) and backward (■) and (C) plot of Ipf/Ipb and difference between Epf–Epb in the electrooxidation of 0.5M FA on the Pt–Sn/CCE as a function of the final potential in the cyclic voltammetric method at a scan rate of 50 mV s−1. | ||
In order to optimize the contents of the electro co-deposited Pt–Sn nanoparticles on CCE for FA oxidation, solutions with various Pt/Sn (salts of these metals) molar ratios were chosen. The metal loading was controlled at 400 μg cm−2. The CV curve of FA oxidation on the six Pt–Sn/CCE samples are drawn (not shown) and compared with together. The peak current in the forward scan, the onset potential and the ratio of Ipf/Ipb are three key parameters used to evaluate the electrocatalytic activity of different Pt–Sn/CCE electrocatalysts in the electrooxidation of FA. Table 2 shows that the currents of FA oxidation in the forward scan are significantly enhanced after the introduction of second metal, e.g., Pt (100:0) < Pt–Sn (80:20) < Pt–Sn (70:30) < Pt–Sn (60:40) < Pt–Sn (50:50). Obviously, Pt–Sn (50:50) exhibits the best electrocatalytic performance in FA electrooxidation. Additionally, it should be noted that the onset potential of FA oxidation depends on the content of the alloy (Table 3). It is also generally recognized that the onset potential can be an indicator in determining the electrochemical activity.58 This sequence reveals that the Pt–Sn catalyst prepared from the solution of Pt and Sn (50:50) shows a fairly good electrocatalytic capability in the FA oxidation. Another important index, Ipf/Ipb ratio, which evaluates the oxidation activity of FA is shown in Table 3. We observe from Table 3 that all bimetallic Pt–Sn catalysts exhibit high Ipf/Ipb values by at least 14–114% higher than these in Pt/CCE catalyst. Such a high current ratio shows the most of the intermediate carbonaceous species can be oxidized to CO2 in the forward scan on Pt–Sn catalysts.58 The corresponding value of the Pt–Sn/CCE (50:50) catalyst is an indication of reduced COads poisoning when compared with that in the Pt/CCE catalyst. The results indicate that the Pt–Sn nanoparticles electro co-deposited from a Pt–Sn = 1:1 atomic ratio solution, exhibit the highest electrocatalytic activity, i.e. the lowest onset potential (−0.30 V), the highest peak current (1.3 mA/Cm2) and highest ratio of Ipf/Ipb (1.05). So the combination of optimal Pt–Sn nanoparticles with excellent catalytic activity and CCE support with high electrochemical accessible surface areas and stability can become a promising electrocatalyst for FA oxidation in DFAFCs.
More accurate inspection of the CV profile within the potential ranges of −0.3 V to 0.0 V in Fig. 5 compared with the CV profile at the Hudp region in FA free electrolyte (Fig. 4) shows that the hydrogen adsorption/desorption peaks not observed in the presence of FA, imply that FA is adsorbed preferentially on the electrode surface within those potentials.65–67 It should be noted that, for low concentrations of FA in the first sweep (not shown), the hydrogen adsorption/desorption peaks are still show that FA does not completely prevent hydrogen adsorption.
For the investigation of the transport characteristics of FA on the Pt–Sn/CCE, the influence of the scan rate (υ) in the electrooxidation of FA on the Pt–Sn/CCE was investigated (Fig. 7). The anodic peak currents are linearly proportional to υ1/2 (inset of Fig. 7) which suggests that the electrocatalytic oxidation of the FA on the Pt–Sn/CCE is a diffusion-controlled process.
 | ||
| Fig. 7 Effect of scan rate on 0.5M FA electrooxidation obtained on the Pt–Sn/CCE in 0.1M H2SO4. Scan rates are shown on CVs. The inset shows the dependence of the anodic peak currents on the square root of the scan rates. | ||
Finally, in order to further assess the activity of the Pt–Sn/CCE, the peak potential and peak current were determined as a function of bulk concentration of FA. The obtained results show that the peak potential is shifted positively with the FA concentration, but remains fairly constant within a range of 60 mV over the whole change in concentration; meanwhile, the anodic current in the forward scan increases linearly as the FA concentration increases. In the case of the highest concentration employed (1.2 M), the anodic current for FA oxidation reaches the value 6.4 mA.
 | ||
| Fig. 8 Plot of anodic peak current in forward scan (Ipf) in the electrooxidation of 0.5M FA as a function of the scan number by cyclic voltammetry at a scan rate of 50 mV s−1 (Long-term stability of Pt–Sn/CCE in 0.1M H2SO4 and 0.5M FA). | ||
3.4. Long-term stability of the Pt–Sn/CCE
Practically, long-term stability of the anodic material in the fuel cells is important. The long-term stability of Pt–Sn/CCE was examined in 0.1 M H2SO4 solution containing 0.5 M FA (Fig. 8). It can be observed that the anodic current decreases with an increase in the scan number at the initial stage. i.e. the anodic current starts to decrease until it reaches 40 cycles and remains constant afterwards. The peak current of the 500th cycle is about 85% that of the first scan. After 500 cycles, the Pt–Sn catalyst still has the high Ipf/Ipb value of 0.72, which is higher than that in the Pt/CCE catalyst (Ipf/Ipb ratio: 0.49). The corresponding value of the Pt–Sn catalyst is an indication of reduced COads poisoning compared with that in the Pt nanoparticle catalyst. Moreover, the cycle stability of electrocatalysts for FA oxidation is the crucial factor in the practical application of DFAFCs.4 In general, the loss of the catalytic activity after a successive number of scans may result from the consumption of FA during the CV scan. It may also be due to poisoning and the structure change of the Pt–Sn nanoparticles as a result of the perturbation of the potentials during the scanning in aqueous solutions, especially in the presence of the organic compound. The diffusion process occurring between the surface of the electrode and the bulk solution might be another factor. With an increase in scan numbers, FA gradually diffuses from the bulk solution to the surface of the electrode. After the long-term CV experiments, the Pt–Sn/CCE was stored in water for a week and then the FA oxidation was carried out again by the CV. This process revealed that the excellent electrocatalytic activity the FA oxidation was still observable.4. Conclusion
I. The Pt–Sn nanoparticles can be co-deposited on the CCE using a simple electrochemical process to form a stable bimetallic electrocatalyst.II. The Pt–Sn/CCE electrocatalyst presents similar CV profiles to Pt/CCE and smooth Pt in 0.1 M H2SO4 and 0.5 M FA + 0.1 M H2SO4 solutions. However, the Pt–Sn/CCE exhibits a very high FA oxidation current. These results indicate that Pt–Sn/CCE exhibits high electroactivity for the oxidation of FA.
III. The high electrocatalytic activity of Pt–Sn/CCE towards the oxidation of FA may be directly related to effect of the Sn atoms in the electrocatalytic activity of Pt and its large surface electroactive area (the real surface area of the Pt–Sn/CCE is over 38 times larger than that of smooth Pt).
IV. The prepared modified electrode exhibits satisfactory stability and reproducibility when stored in ambient conditions or continues cycling, which makes it attractive as an anode in the DFAFCs and other practical applications.
Acknowledgements
The authors gratefully acknowledge the research council of Azarbaijan University of Tarbiat Moallem for financial support.References
- B. Rajesh, T. K. Ravindranathan, H. J. Bonard, X. N. Mathieu and B. Viswanathan, Electrochem. Solid-State Lett., 2004, 7, A404 CrossRef CAS.
- T. D. Jarvi, S. Sriramulu and E. M.Stuve, Colloids Surf., A, 1998, 134, 145 CrossRef CAS.
- B. Rajesh and Z. Piotr, Nature, 2006, 443, 63 CrossRef.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. M. Lu, Y. M. Zhu and Y. N. Xia, Science, 2009, 324, 1302 CrossRef CAS.
- E. Antolini, R. C. Salgado Jose and E. R. Gonzalez, Appl. Catal., B, 2006, 63, 137 CrossRef CAS.
- T. Iwasita, X. H. Xia and E. Herrero, Langmuir, 1996, 12, 4260 CrossRef CAS.
- S. Wasmus and A. Kuver, J. Electroanal. Chem., 1999, 461, 14 CrossRef CAS.
- F. Melissa Mrozek, L. Hai and M. J. Weaver, Langmuir, 2000, 16, 8463 CrossRef.
- A. A. Mikhailova, A. A. Pasynskii, V. A. Grinberg, Yu. A. Velikodnyi and O. A. Khazova, Russ. J. Electrochem., 2010, 46, 26 CrossRef CAS.
- Y.-X. Bai, J.-J. Wu, X.-P. Qiu, J.-S. Wang, W.-T. Zhu and L.-Q. Chen, Acta Chimica Sinica., 2006, 64, 527 CAS.
- Y.-G. Guo, J.-S. m Hu, H.-M. Zhang, H.-P. Liang, L.-J. Wan and C.-L. Bai, Adv. Mater., 2005, 17, 746 CrossRef CAS.
- M. A. Abdel Rahim, M. W. Khalil and H. B. Hassan, J. Appl. Electrochem., 2000, 30, 1151 CrossRef CAS.
- Z. Wei, H. Guo and Z. Tang, J. Power Sources, 1996, 58, 239 CrossRef CAS.
- A. S. Arico, V. Antonucci, N. Giordano, A. K. Shukla, M. K. Ravikumar, A. Roy, S. R. Barman and D. D. Sarma, J. Power Sources, 1994, 50, 295 CrossRef CAS.
- A. N. Haner and P. N. Ross, J. Phys. Chem., 1991, 95, 3740 CrossRef CAS.
- M. A. Spitsyn, A. N. Zhuchkov, V. N. Andreev and V. E. Kazarinov, Soviet Electrochem., 1988, 24, 640 Search PubMed.
- A. Katayama, J. Phys. Chem., 1980, 84, 376 CrossRef CAS.
- M. M. P. Janssen and J. Moolhuysen, J. Catal., 1977, 46, 289 CrossRef CAS.
- M. M. P. Janssen and J. Moolhuysen, Electrochim. Acta, 1976, 21, 861 CrossRef CAS.
- W. T. Napporn, H. Laborde, J.-M. Léger and C. Lamy, J. Electroanal. Chem., 1996, 404, 153 CrossRef.
- S. A. Campbell and R. Parsons, J. Chem. Soc., Faraday Trans., 1992, 88, 833 RSC.
- M. J. González, C. T. Hable and M. S. Wrighton, J. Phys. Chem. B, 1998, 102, 9881 CrossRef.
- G. Stalnionis, L. Tamasauskaite-Tamasiunaite, V. Pautieniene and Z. Jusys, J. Solid State Electrochem., 2004, 8, 900 CrossRef CAS.
- A. Kelaidopoulou, E. Abelidou and G. Kokkinidis, J. Appl. Electrochem., 1999, 29, 1255 CrossRef CAS.
- Q. Yi, J. Zhang, A. Chen, X. Liu, G. Xu and Z. Zhou, J. Appl. Electrochem., 2008, 38, 695 CrossRef CAS.
- Z. Liu and X. Zhang, Electrochem. Commun., 2009, 11, 1667 CrossRef CAS.
- M. Shibata, O. Takahashi and S. Motoo, J. Electroanal. Chem., 1988, 249, 253 CrossRef CAS.
- X. H. Xia, Electrochim. Acta, 1999, 45, 1057 CrossRef CAS.
- B. Wu, D. Hu, Y. Kuang, B. Liu and X. Zhang, Angew. Chem., Int. Ed., 2009, 48, 4751 CrossRef CAS.
- J. M. Solar, C. Leon y Leon, K. Osseo-Asare and R. Radovic, Carbon, 1990, 28, 369 CrossRef CAS.
- M. A. Fraga, E. Jordão, M. J. Mendes, M. M. A. Freitas, J. L. Faria and J. L. Figueiredo, J. Catal., 2002, 209, 355 CrossRef CAS.
- M. Tsionsky, G. Gun, V. Glezer and O. Lev, Anal. Chem., 1994, 66, 1747 CrossRef CAS.
- O. Lev, Z. Wu, S. Bharathi, V. Glezer, A. Modestov, J. Gun, L. Rabinovich and S. Sampth, Chem. Mater., 1997, 9, 2354 CrossRef CAS.
- J. Wang, Anal. Chim. Acta, 1999, 399, 21 CrossRef CAS.
- X. Kong, J. Du, K. Wang, J. Ni, N. Xu and Z. Wu, Int. J. Hydrogen Energy, 2010, 35, 8088 CrossRef CAS.
- G. Oskam and P. C. Searson, J. Phys. Chem. B, 1998, 102, 2464 CrossRef CAS.
- B. Habibi, H. Phezhhan and M. H. Pournaghi-Azar, J. Iran. Chem. Soc., 2010, 7, S103 CAS.
- B. Habibi, H. Phezhhan and M. H. Pournaghi-Azar, Microchim. Acta, 2010, 169, 313 CrossRef CAS.
- B. Habibi and M. H. Pournaghi-Azar, Electrochim. Acta, 2010, 55, 5492 CrossRef CAS.
- H. Razmi, Es. Habibi and H. Heidari, Electrochim. Acta, 2008, 53, 8178 CrossRef CAS.
- H. Razmi and Es. Habibi, J. Solid State Electrochem., 2009, 13, 1897 CrossRef CAS.
- B. Habibi and N. Delnavaz, Int. J. Hydrogen Energy, 2010, 35, 8831 CrossRef CAS.
- B. Habibi and N. Delnavaz, Int. J. Hydrogen Energy, 2011, 36, 9581 CrossRef CAS.
- G. Siné, G. Fóti and Ch. Comninellis, J. Electroanal. Chem., 2006, 595, 115 CrossRef.
- W. Zhou, Z. Zhou, S. Song, W. Li, G. Sun, P. Tsiakaras and Q. Xin, Appl. Catal., B, 2003, 46, 273 CrossRef CAS.
- J. Araña, P. Ramirez de la Piscina, J. Llorca, J. Sales and N. Homs, Chem. Mater., 1998, 10, 1333 CrossRef.
- V. A. Grinberg, N. A. Mayorova and A. A. Pasynskii, Russ. J. Electrochem., 2009, 45, 1321 CrossRef CAS.
- J. Iniesta, J. Gonzá lez-García, J. Fernández, V. Montiel and A. Aldaz, J. Mater. Chem., 1999, 9, 3141 RSC.
- E. Lamy-Pitara and J. Barvier, Appl. Catal., A, 1997, 149, 49 CrossRef CAS.
- Y. Du, B. Su, N. Zhang and C. Wang, Appl. Surf. Sci., 2008, 255, 2641 CrossRef CAS.
- R. S. Jayashree, J. S. Spendelow, J. Yeom, C. Rastogi, M. A. Shannon and P. J. A. Kenis, Electrochim. Acta, 2005, 50, 4674 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 44, 1 CrossRef CAS.
- B. Beden, C. Lamy, N. R. D. Tacconi and A. J. Arvia, Electrochim. Acta, 1990, 35, 691 CrossRef CAS.
- P. Strasser, M. Eiswirth and G. Ertl, J. Chem. Phys., 1997, 107, 991 CrossRef CAS.
- X. Wang, W. Wang, Z. Qi, C. Zhao, H. Ji and Z. Zhang, Electrochem. Commun., 2009, 11, 1896 CrossRef CAS.
- W. T. Napporn, J.-M. LeÂger and C. Lamy, J. Electroanal. Chem., 1996, 408, 141 CrossRef.
- Z. L. Liu, X. Y. Ling and X. D. Su, J. Phys. Chem. B, 2004, 108, 8234 CrossRef CAS.
- W. Chen, J. Kim, S. Sue and S. Chen, Langmuir, 2007, 23, 11303 CrossRef CAS.
- T. Frelink, W. Visscher and J. A. R. van Veen, Surf. Sci., 1995, 335, 353 CrossRef CAS.
- G. Stalnionis, L. Tamašauskaitė-Tamašiūnaitė, V. Pautienienė, A. Sudavičius and Z. Jusys, J. Solid State Electrochem., 2004, 8, 892 CrossRef CAS.
- F. L. S. Purgato, P. Olivi, J.-M. Léger, A. R. de Andrade, G. Tremiliosi-Filho, E. R. Gonzalez, C. Lamy and K. B. Kokoh, J. Electroanal. Chem., 2009, 628, 81 CrossRef CAS.
- A. Capon and R. Parsons, J. Electroanal. Chem., 1973, 45, 205 CrossRef CAS.
- M. Watanabe and S. Motoo, J. Electroanal. Chem., 1975, 60, 267 CrossRef CAS.
- B. Gurau, R. Viswanathan, R. Liu, T. J. Lafrenz, K. L. Ley, E. S. Smotkin, E. Reddington, A. Sapienza, B. C. Chan and T. E. M. S. Sarangapani, J. Phys. Chem. B, 1998, 102, 9997 CrossRef CAS.
- H. Kita and H.-W. Lei, J. Electroanal. Chem., 1995, 388, 167 CrossRef.
- C. Lamy, J. M. Leger, J. Clavilier and R. Parsons, J. Electroanal. Chem., 1983, 150, 71 CrossRef CAS.
- R. R. Adzic, A. V. Tripkovic and N. M. Markovic, J. Electroanal. Chem., 1983, 150, 79 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |