Size-tunable Ag nanoparticles immobilized in electrospun nanofibers: synthesis, characterization, and application for catalytic reduction of 4-nitrophenol†
Shili
Xiao
*a,
Weilin
Xu
a,
Hui
Ma
b and
Xu
Fang
b
aKey Laboratory of Green Processing and Functional Textiles of New Textile Materials, Ministry of Education, Wuhan Textile University, Wuhan, 430073, People's Republic of China. E-mail: xshili@yahoo.com
bCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, People's Republic of China
First published on 3rd November 2011
Abstract
We report a facile method to immobilizing silver nanoparticles (AgNPs) into electrospun polymer nanofibers. In this approach, water-stable polyacrylic acid (PAA)/polyvinyl alcohol (PVA) nanofibers, fabricated by electrospinning a 10 wt% PAA/PVA solution and subsequent heating treatment at 145 °C for 30 min, were used as nanoreactors to complex Ag(I) ions through binding with carboxyl groups of PAA for the subsequent reductive formation of AgNPs. The as-prepared AgNP-immobilized nanofibers are thoroughly characterized by scanning electronic microscopy, transmission electron microscopy, energy dispersive spectroscopy, selected area electron diffraction, Fourier transform infrared spectroscopy, and thermogravimetric analysis. Moreover, the effect of AgNO3 solution concentration on the morphology of hybrid nanofibers, Ag content, the size of AgNPs, and catalytic activity of hybrid nanofibrous mats are systematically investigated. We show that spherical AgNPs are uniformly distributed along the cross-section of nanofibers. X-Ray diffraction indicates that the formed AgNPs in the nanofibers are crystalline. The AgNP-immobilized nanofibrous mats exhibit superior catalytic reduction capacity to 4-nitrophenol with efficiency approaching 100% within 30 min and excellent reusability. Furthermore, the size and spacial distribution of AgNPs can be tuned by varying the AgNO3 solution concentration, thus manipulating the catalytic activity of AgNP-immobilized nanofibrous mats. The strategy to immobilizing and manipulating the size of the AgNPs within polymer nanofibers may be extended to other particle systems for various applications in catalysis, energy, sensing, photonic and biomedical applications.
Introduction
Noble metal nanoparticles are of scientific and technological interest because of their unique optical, electronic and quantum size-related properties that are not present in their bulk conterparts.1–5 These novel properties enable their use in catalysis,2,4,6 sensors,7,8 biology detection1 and electronic devices.9 Among noble metal nanoparticles, silver nanoparticles (AgNPs) have been intensively studied for their important applications for antimicrobials,10–12 catalysis,2,13,14 biosensing,15,16 imaging,17 and photoelectronics.18 The remarkable properties of Ag nanoparticles mainly stem from quantum confinement effects and their large surface areas relative to volume, hence it is crucial to develop effective methods for the preparation of nanoparticles with well-controlled shape and size. Various methods have been employed to produce more dispersible and stable Ag nanoparticles, or to immobilize the nanoparticles onto microsized particles or into polymeric films without compromising their surface reactivity.12,19–25 For instance, Dadosh19 successfully synthesized uniform Ag nanoparticles with diameters ranging from 18–30 nm through varying the concentration of tannic acid, a reducing agent. Yin and co-workers26 used poly(N-vinylpyrrolidone) (PVP) as the stabilizer to sythesize size-controlled spherical silver nanoparticles through the electroreduction method. It showed that PVP could greatly promote Ag particle formation rate and significantly reduce the agglomeration of AgNPs, thereby resulting in monodisperse Ag nanoparticles. Lu14 demonstrated that well-dispersed Ag nanoparticles with a size less than 10 nm could be formed in situ on the surface of polystyrene core–poly(acylic acid) polyelectrolyte brush particles by photoemulsion polymerization of Ag-acrylate under UV irradiation. The Ag nanoparticles thus synthesized displayed good catalytic reduction capability to 4-nitrophenol. In addition, to manipulate and process these nanoparticles technologically into useful forms, Cohen and coworkers24 immobilized the Ag nanoparticles into the multilayered polyelectrolyte films produced by layer-by-layer assembly technology and systematically investigated the effect of synthesizing parameters such as the number of layers, pH, and the metal concentration on the nanoparticle size. However, for the best reaction activity of Ag nanoparticles, continuous supporting media with high surface to volume ratio is still required.Electrospinning has attracted much attention as a simple and versatile technique capable of generating continuous nanofibers with novel properties including high surface-to-volume ratio, high aspect ratio, and pore size as non-wowen fabrics in the past decade.27–29 Plenty of organic, inorganic, and organic–inorganic hybrid nanostructured fiber-based materials have been fabricated and found intriguing applications in tissue engineering scaffords,30,31 wound dressing,32,33 sensors,34 catalyts,35,36 and environmental remediation29,37,38etc. Generating nanoparticle-containing nanofibrous mats is expected to form nanoparticle-based nanocatalyst systems. By utilizing electrospinning, polymer–silver composite nanomaterials have been demonstrated by directly electrospinning polymer solutions containing Ag nanoparticles39 or by electrospinning polymer–Ag salts mixtures and subsequent Ag ion reduction through UV irradiation,33 or heat treatment.40 The former is simple but it is difficult to obtain well-dispersed nanoparticles due to easy aggregation of nanoparticles. Although tailored size and size distribution of silver nanoparticles can be synthesized by varying the processing parameters using the latter method, the electrospinning process might be too complicated. Therefore, the preparation of Ag nanoparticle functionalized nanofibers by an easy way remains a great challenge.
In our previous studies we reported a facile method to synthesize and immobilize zero-valent iron nanoparticles (ZVI NPs), an agent widely used in environmental remediation, into water-stable electrospun polyacrylic acid (PAA)/polyvinyl alcohol (PVA) nanofirous mats.38 In this approach, a PAA/PVA nanofibrous mat was used as a nanoreactor to complex ferrous ions in aqueous solution through an electrostatic interaction with carboxylic acid groups of PAA. Then, ZVI NPs can be synthesized and immobilized into the PAA/PVA nanofibers by subsequent chemical reduction. The composite nanofibrous mats containing ZVI NPs exhibited a superior decoloration capability to various model dyes in printing and dyeing wastewater and excellent reactivity for the dechlorination of trichloroethylene.37,38 Therefore, we anticipate that the carboxylic acid group that resides in the PAA polymer can also allow for the binding of silver ions to generate Ag-containing nanofibers for catalytic applications.
In this present study, freshly prepared electrospun PAA/PVA nanofibrous mats were first crosslinked via heat treatment at 145 °C to form water-stable fibrous mats. Then, Ag nanoparticles were synthesized and immobilized into the PAA/PVA nanofibers by chemical reduction of the Ag(I) ions complexed with the water-stable nanofibrous mats. Scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), energy dispersive spectroscopy (EDS), Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD) and thermogravimetric analysis (TGA) were utilized to characterize the morphology and composition of Ag NP-containing nanofibers. In addition, the effect of AgNO3 concentration on the Ag content, Ag particle size and spacial distribution in nanofibers were systematically investigated. Otherwise, the catalytic capacity of Ag-immobilized polymer nanofibers synthesized with various AgNO3 solution concentrations was investigated using 4-nitrophenol as a model. We show that the developed Ag NP-immobilized polymer nanofibrous mats display excellent catalytic efficiency to transform 4-nitrophenol to 4-aminophenol with high reusability. To our best knowledge, this is the first report related to the use of functional groups of nanofibers as a nanoreactor to complex Ag(I) ions for subsequent Ag nanoparticle formation.
Experimental
Materials
PAA (average Mw = 240![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 25% in water) was obtained from Aldrich. PVA (88% hydrolyzed, average Mw = 88000) and sodium borohydride were from J&K Chemical®. Silver nitrate (AgNO3) was purchased from Sinopharm Chemical Reagen Co., Ltd. 4-Nitrophenol was analytical grade and used without further purification. Water used in all experiments was purified using a Milli-Q Plus 185 water purification system (Millipore, Bedford, MA) with resistivity higher than 18 MΩ cm.
000, 25% in water) was obtained from Aldrich. PVA (88% hydrolyzed, average Mw = 88000) and sodium borohydride were from J&K Chemical®. Silver nitrate (AgNO3) was purchased from Sinopharm Chemical Reagen Co., Ltd. 4-Nitrophenol was analytical grade and used without further purification. Water used in all experiments was purified using a Milli-Q Plus 185 water purification system (Millipore, Bedford, MA) with resistivity higher than 18 MΩ cm.
Fabrication of Ag NP-immobilized polymer nanofibers
The procedure to fabricate the electrospun nanofibers and to subsequently immobilize AgNPs into the electrospun nanofibers is similar to our previous report.37,38 In brief, PAA/PVA nanofibers were firstly fabricated by electrospinning 10 wt% PAA/PVA mixture solution. Specifically, a PVA solution was prepared by dissolving PVA powder into water at 80 °C for 3 h under magnetic stirring, and then the solution was cooled down to room temperature before use. PAA and PVA solutions were mixed together under magnetic stirring overnight with a mass ratio of PAA/PVA equivalent to 1:1 to achieve a homogeneous solution. A measured amount of polymer solution was pumped into a syringe with a needle having an inner diameter of 0.8 mm. The flow rate was controlled by a syringe pump (JZB-1800, Jian Yuan Medical Technology Co., Ltd., China) at 0.5 mL h−1. The high voltage power supplier (BGG40/2, Institute of Beijing High Voltage Technologe, China) was connected to the tip of the needle by a high-voltage insulating wire with two clamps at the end. An aluminum board was grounded and used as the collector. The electrospinning setup can be found in our previous report.41 The electrospinning voltage was fixed at 16.8 kV, and the collection distance was set at 25 cm. Under the above electrospinning conditions, a polymer jet was formed, elongated and ultimately deposited onto the aluminum foil, resulting in the nanofibrous mats. Freshly prepared PAA/PVA nanofibrous mats were crosslinked upon heating treatment at 145 °C for 30 min to produce water-stable nanofibrous mats. Then, equivalent water-stable PAA/PVA nanofibrous mats were respectively immersed into AgNO3 solutions with different concentrations. In aqueous solution, a fraction of carboxyl groups of PAA remained protonated in water-stable PAA/PVA nanofibers. Therefore, the acid protons can be subsequently exchanged for cationic silver ions. Upon chemical reducing Ag(I) ions that complexed with nanofibers using sodium borohydride solution (0.2 M) for half an hour, zero-valent silver nanoparticles are formed and immobilized into the nanofibers, similar to that reported in the literature.24,42,43 The formed AgNP-containing nanofibrous mats were rinsed with water 3 times, followed by vacuum drying at room temperature for 24 h, and stored in a desiccator before use.
Characterization
Morphologies of AgNP-containing nanofibrous mats were observed using a scanning electron microscope (JSM-5600LV, JEOL Ltd., Japan) with an operating voltage of 15 kV. Prior to SEM observation, the samples were sputter-coated with Pt films with a thickness of 10 nm. The elemental composition of samples was analyzed using an X-ray energy dispersive spectroscopy (EDS) detector (IE 300X, Oxford, UK) attached to the SEM. To observe the morphology and the distribution of AgNPs in the nanofibers, AgNP-immobilized nanofibrous mats were firstly embedded in epoxy resin and then were cut into ultrathin sections with ultramicrotome equipped with a diamond knife. The cross-sectional image of the fibers containing AgNPs was imaged with transmission electron microscopy (TEM) (JEM2100, JEOL, Ltd., Japan) under an acceleration voltage of 200 kV. Selected area electron diffraction (SAED) was performed to analyze the crystal structure of the formed AgNPs. 300 randomly selected nanofibers or AgNPs in different SEM or TEM images were measured using imageJ 1.40G (http://rsb.info.nih.gov/ij/download.html) for each sample in order to acquire the diameter or size distribution histograms. X-Ray diffraction (XRD) was performed at 25 °C (D/Max-2550 PC, RIGAKU, Japan) in reflection mode using Cu Kα radiation. Fourier transform infrared (FTIR) spectra were recorded using a Nicolet 5700 spectrometer (Thermo Nicolet Corporation, US) at the wavenumber range 4000–500 cm−1 at ambient conditions. The silver content in the samples was determined by thermogravimetric analysis (TGA) using a TG 209 F1 (NETZSCH Instruments Co., Ltd., Germany) thermogravimetric anlyzer with a heating rate of 10 °C min−1 in air.Catalytic reduction of 4-nitrophenol
In order to evaluate the catalytic efficiency and reusability of AgNP-containing nanofibrous mats, a catalytic reduction experiment of 4-nitrophenol was performed according to the procedure reported in the literature.14 A mixed solution containing water (19.5 mL), 4-nitrophenol (20 mM, 0.25 mL) and sodium borohydride solution (5 M, 0.25 mL) was first prepared in a 30 mL glass beaker at room temperature, and then a given amount of the composite nanofibrous mats containing AgNPs was added into the mixture to reach a final Ag concentration of 0.2 g L−1, followed by gentle magnetic stirring. At each time interval, 0.5 mL of the aqueous solution was withdrawn and diluted to 1.5 mL for the analysis of reduction efficiency using a Lambda-25 UV-vis spectrometer (Perkin-Elmer, United States). For comparison, the control experiment was also carried out under similar conditions using the water-stable PAA/PVA nanofibrous mats without AgNPs. The catalytic reduction efficiency of 4-nitrophenol was calculated according to Eq. 1: | (1) |
Results and discussion
Under the selected electrospinning parameters reported in our previous work, smooth and uniform PAA/PVA nanofibers were fabricated with a mean diameter of 170 ± 27 nm.38 Then, heating treatment was introduced at 145 °C for 30 min to render the PAA/PVA nanofibrous mats water-stable. The water-stable PAA/PVA nanofibrous mats were immersed into the Ag ions solution to complex the Ag(I) ions with the available carboxyl groups of PAA for subsequent formation of AgNPs through sodium borohydride reduction. The AgNP-immobilized nanofibrous mats were thoroughly characterized using scanning electron microscopy (SEM), transmission electron microscopy (TEM), selected area electron diffraction (SAED), energy dispersive spectroscopy (EDS), X-ray diffraction (XRD), Fourier transform infrared (FTIR) spectroscopy, and thermogravimetric analysis (TGA).Typical SEM images and fiber diameter histograms of AgNP-immobilized PAA/PVA nanofibrous mats fabricated with different AgNO3 concentrations are presented in Fig. 1. It shows that the hybrid nanofibers exhibited a porous nanofibrous structure with a smooth surface regardnless of the AgNO3 concentrations, similar to that of the PAA/PVA nanofiber without AgNPs immobilized.38 Moreover, the mean diameter of AgNP-immobilized nanofibers at any AgNO3 concentration was apparently larger than that of the PAA/PVA nanofibers without AgNPs (170 ± 27 nm). This suggests that the AgNPs have been successfully immobilized into the nanofibers, thus resulting in the diameter increase of nanofibers. It is interesting that the mean diameter of hybrid nanofibers increased first and then decreased with the increase of AgNO3 concentrations. When the AgNO3 concentration was lower than 0.05 M, the mean diameter of the nanofibers regularly increased from 502 ± 83 nm for 0.025 M AgNO3 solution to 584 ± 106 nm for 0.05 M AgNO3 (Fig. 1b and 1d). However, further increasing the concentration of AgNO3 solution resulted in a mean diameter decrease of hybrid nanofibers inversely. The mean diameter of AgNP-immobilized nanofibers synthesized with the AgNO3 concentrations of 0.1 M and 0.2 M was 314 ± 50 nm and 282 ± 76 nm (Fig. 1f and 1h), respectively. We thought that the Ag content and particle size of AgNPs immobilized into the nanofibers might be responsible for the mean diameter variations of hybrid nanofibers.
 | ||
| Fig. 1 SEM images and diameter histograms of AgNP-immobilized nanofibers synthesized with 0.025 M (a, b), 0.05 M (c, d), 0.1 M (e, f) and 0.2 M (g, h) AgNO3 solution concentrations. | ||
The formation of AgNPs in nanofibrous mats was further confirmed by the cross-sectional TEM images of AgNP-immobilized nanofibers as shown in Fig. 2. Fig. 2a shows a typical cross-sectional TEM image of AgNP-containing polymer nanofibers synthesized with 0.2 M AgNO3 solution. It showed that individual AgNPs with a relatively uniform distribution along the cross-section of fibers was clearly observed in a magnified TEM image (Fig. 2b), which was quite different from the uniform distribution of zero-valent iron NPs (1.6 nm) in the PAA/PVA nanofibers observed in our previous studies.37,38 We noted that the AgNPs were mainly assembled at the outer layer of the PAA/PVA nanofibers. This could be due to the higher molecular weight of the Ag(I) ions compared to that of Fe(III) ions used to complex with the PAA/PVA nanofibers, thus limiting Ag(I) ions entering into interior of nanofibers. The mean size of AgNPs synthesized with 0.2 M AgNO3 solution was estimated to be 5.8 ± 2.4 nm (Fig. 2c). A high-resolution TEM image (Fig. 2d) of the individual AgNPs shows that the AgNP was crystalline, as lattices of Ag crystals were clearly observed. The selected area electron diffraction (SAED) of AgNPs demonstrated the face-centred-cubic (fcc) crystal structures (Fig. 2e), in good agreement with the literature.25EDS analysis on the hybrid nanofibers (Fig. 3) evidenced the dominance of the element Ag, further demonstrating that the AgNPs were successfully synthesized and immobilized in the PAA/PVA nanofibers. The elemental carbon and oxygen might be from the polymer PAA and PVA, even probably from the silver oxide. Sodium was detected, likely as a residue from the sodium borohydride used for Ag(I) ion reduction. Platinum was from the Pt film sputter-coated onto the nanofibrous mats before measurement.
 | ||
| Fig. 2 Cross-sectional TEM image (a), a magnified TEM image (b) and the high-resolution TEM image (d) of AgNP-immobilized nanofibers synthesized at the 0.2 M AgNO3 solution. (c) and (e) show the particle size distribution histogram and selected area electron diffraction of the immobilized AgNPs. | ||
 | ||
| Fig. 3 EDS spectrum of Ag-immobilized PAA/PVA nanofibrous mats. | ||
In order to further determine the crystal nature of AgNPs, the crystallinity of AgNPs immobilized into the PAA/PVA nanofibers was also analyzed using X-ray diffraction (XRD) (Fig. 4). A typical XRD pattern of as-prepared AgNPs shows broad Bragg reflections at 2θ = 38.1°, 44.3°, 64.3°, 77.5°, and 81.5°, which were respectively corresponding to the (111), (200), (220), (311), and (222) reflections of fcc structure of metallic silver, in agreement with the literature.14,44,45 These data were also in good agreement with data in the SAED of AgNPs as shown in Fig. 2d, demonstrating the crystalline structure of AgNPs. Moreover, no diffraction peaks corresponding to silver oxide were observed, which confirmed that only metallic Ag was formed during the NaBH4 reduction process, and thus evidenced that the elemental oxygen in the EDS spectrum (Fig. 3) was totally ascribed to the carboxyl groups of PAA and hydroxyl groups of PVA.
 | ||
| Fig. 4 X-Ray diffraction pattern of AgNP immobilized into the PAA/PVA nanofibers. | ||
Fig. 5 displays a significant difference in the FTIR spectra of PAA/PVA nanofibrous mats before and after immobilization of AgNPs. A strong absorption peak at 1710 cm−1 assigned to the free carboxylic group (COO-asymmetric stretching) of PAA in the PAA/PVA nanofibrous mats before Ag loading disappeared and shifted to 1560 cm−1 (attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric vibrations in the carboxylate after the immobilization of AgNPs), which is similar to that of PAA/PVA nanofibrous mats after the immobilization of zero-valent iron nanoparticles reported in our previous study.37 Changes at 1410 cm−1 and 1330 cm−1 indicate the interaction between AgNPs and the carboxyl groups of PAA. Moreover, a very broad absorption band in the region 3690 cm−1–2980 cm−1 for both samples indicates the presence of a tiny amount of water molecules because the polymer PAA and PVA can absorb moisture in air. Moreover, after the introduction of AgNPs into the PAA/PVA nanofibers, the spectral features of the nanofibers seem to be broader.
O asymmetric vibrations in the carboxylate after the immobilization of AgNPs), which is similar to that of PAA/PVA nanofibrous mats after the immobilization of zero-valent iron nanoparticles reported in our previous study.37 Changes at 1410 cm−1 and 1330 cm−1 indicate the interaction between AgNPs and the carboxyl groups of PAA. Moreover, a very broad absorption band in the region 3690 cm−1–2980 cm−1 for both samples indicates the presence of a tiny amount of water molecules because the polymer PAA and PVA can absorb moisture in air. Moreover, after the introduction of AgNPs into the PAA/PVA nanofibers, the spectral features of the nanofibers seem to be broader.
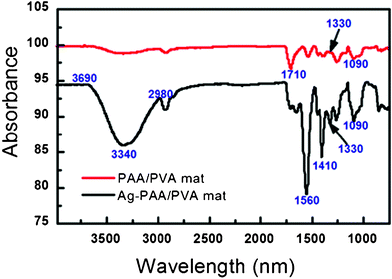 | ||
| Fig. 5 FTIR spectra of PAA/PVA nanofibrous mats before and after the immobilization of AgNPs. | ||
It is well known that the unique physicochemical properties offered by nanoparticles arise from their large specific surface area, a property inversely related to the particle size. Therefore, the particle size manipulation of AgNPs immobilized in the nanofibers is vital to their properties for various applications. Herein, we further examined the size distribution of AgNPs in the hybrid nanofibers fabricated with AgNO3 solutions of different concentrations. Fig. S1 (ESI†) shows the TEM images and size distribution histogram of AgNPs immobilized in the hybrid nanofibers synthesized with 0.025 M, 0.05 M, 0.1 M AgNO3 solution, respectively. It is clear that all the AgNPs, formed at various silver concentrations, are mainly located along the cross-section of nanofibers, just like what we observed in the typical TEM image of AgNPs in Fig. 2a. However, the mean particle size of AgNPs in the nanofibers did not correspondingly increase with the increase of AgNO3 solution concentration, but increased first and then decreased sharply. When the AgNO3 solution concentration was 0.025 M, the mean size of AgNPs was 6.3 ± 2.6 nm, obviously smaller than that of AgNPs synthesized at 0.05 M AgNO3 solution (20.4 ± 7.1 nm). However, further increasing the concentration of AgNO3 solution (0.1 M and 0.2 M) resulted in a lower mean size of AgNPs immobilized in the nanofibers. The mean size of AgNPs synthesized with 0.1 M and 0.2 M AgNO3 solution was 10.8 ± 3.47 nm and 5.8 ± 2.4 nm. We thought that at lower silver cation concentrations (≤ 0.5 M), particles grow until they reach a size with a surface area that can be stabilized by the polymer,24 thus resulting in the obvious increase of AgNPs sizes and the mean diameters of AgNP-immobilized nanofibers (Fig. 1b and 1d). Whereas, at higher silver cation concentrations, large numbers of silver ions complexed with carboxyl groups of PAA at the same time, which accelerated the nucleation of particles, resulting in the smaller sized AgNPs being stabilized in the limited steric spaces in the nanofibers. Therefore, the AgNPs synthesized at higher AgNO3 solution concentration should be relatively smaller, supporting the argument that the mean diameter of hybrid nanofibers decreases when the AgNO3 solution concentration is above 0.05 M (Fig. 1f and 1h). In general, these data suggest that the particle size of AgNPs can be tuned by varying the AgNO3 solution concentrations.
The Ag loading in the nanofibrous mats prepared at various AgNO3 solution concentrations was examined by TGA (Fig. S2, ESI†) and the data are listed in Table 1. It showed that no significant Ag loading differences were observed with the increase of AgNO3 solution concentration. When the concentration of AgNO3 solution was 0.2 M, the Ag content in the nanofibrous mats was 26.6%, only 3.4% higher than that of the Ag content in the hybrid nanofibrous mats prepared with 0.025 M AgNO3 solution. It means that the Ag(I) ions were mainly introduced via ion exchange rather than adsorption, thus the Ag content in the nanofibrous mats was independent of the AgNO3 solution concentration used to complex with the carboxyl groups of PAA, consistent with the literature data.25 In addition, the results from the TGA analysis further demonstrated that the mean diameter variation of AgNP-immobilized nanofibers was mainly due to the size changes of AgNPs in the nanofibers.
| AgNO3 concentration (M) | Ag loading percentage (%) |
|---|---|
| 0.025 | 23.2 |
| 0.05 | 24.4 |
| 0.1 | 25.8 |
| 0.2 | 26.6 |
Catalytic reduction of 4-nitrophenol
To investigate the potential catalytic properties of the AgNP-immobilized PAA/PVA nanofibrous mat, we selected a reaction to transform 4-nitrophenol to 4-aminophenol as a model. The reaction was monitored using UV-vis spectroscopy. We showed that the AgNP-immobilized nanofibrous mats were able to effectively catalyze the reaction to transform 4-nitrophenol to 4-aminophenol in the presence of NaBH4 (Fig. 6a and 6c). After the addition of nanofibrous mats containing AgNPs, the intensity of characteristic absorption peak of 4-nitrophenol at 400 nm decreases significantly with time and a new peak appears at 290 nm (Fig. 6a), which is ascribed to the product 4-aminophenol.46 Within the time frame of 30 min, the catalytic reduction efficiency of AgNP-immobilized nanofibrous mats could approach 100% (Fig. S3, ESI†). The result is consistent with the observation in Fig. 6c. The yellow color of 4-nitrophenol solution gradually faded within 15 min and ultimately goes colorless. In contrast, when PAA/PVA nanofibrous mats without AgNPs were exposed to the same 4-nitrophenol aqueous solution, no significant color change was observed within the reaction time (Fig. 6d). A slight decrease in the intensity of the absorption peak at 400 nm, which resulted in 6.8% decoloration efficiency (Fig. S3, curve b, ESI†), is due to the physical adsorption of the 4-nitrophenol onto the surface of PAA/PVA nanofibrous mats. This suggested that the superior transformation of 4-nitrophenol to 4-aminophenol was solely related to the excellent catalytic property of the immobilized AgNPs. In addition, AgNPs immobilized in the nanofibers are quite stable and do not escape from the fibrous mats. Inductively coupled plasma atomic emission spectroscopy studies showed that no Ag is released during the whole catalytic reaction process. | ||
| Fig. 6 UV-vis spectra of a solution of 4-nitrophenol aqueous solution catalyzed with AgNP-immobilized PAA/PVA nanofibrous mats (a) and pure PAA/PVA nanofibrous mats (b) at different time intervals, and photos of the 4-nitrophenol aqueous solution treated with AgNP-immobilized nanofibrous mats (c) and PAA/PVA nanofibrous mats without AgNPs (d) at 0, 3, 5, 10, 15, 20, 30 min, respectively. (AgNP-immobilized nanofibrous mats synthesized with 0.2 M AgNO3 solution.) | ||
The AgNP-immobilized PAA/PVA nanofibrous mat could be reusable and recyclable. The excellent catalytic activity of the AgNP-immobilized nanofibrous mats was thoroughly confirmed by plotting the remaining fraction of 4-nitrophenol as a function of exposure time (Fig. 7). After catalyzing the 4-nitrophenol solution reduction for the first time, the hybrid nanofibrous mats without any treatments still performed well for the second, third and even for the forth catalytic experiments. The ultimate catalytic reduction efficiency of 4-nitrophenol could be approximate to 100% at 30 min. However, we note that when the nanofibrous mats containing AgNPs was reused for the forth time, the catalytic reaction time was prolonged to 60 min for completely transforming 4-nitrophenol to 4-aminophenol, which indicated that the catalytic rate of the AgNP-immobilized nanofibrous mats would slow down after several repeated uses.
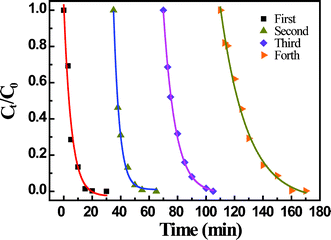 | ||
| Fig. 7 Remaining fraction of 4-nitrophenol as a function of time after treatment with the same AgNP-immobilized nanofibrous mats for the first, second, third and forth time. (AgNP-immobilized nanofibrous mats synthesized with 0.2 M AgNO3 solution) | ||
To illustrate the effect of AgNP size on the catalytic activity of the nanofibrous mats, we further investigated the catalytic reduction capacity of hybrid nanofibrous mats synthesized with different AgNO3 solution concentrations. The differences in the catalytic efficiency of hybrid nanofibrous mats were demonstrated by plotting the remaining fraction of 4-nitrophenol concentration as a function of the exposure time (Fig. 8). Within the given time frame of 60 min, the 4-nitrophenol could be effectively transformed to 4-aminophenol by the hybrid nanofibrous mats. The residual 4-nitrophenol fraction decreased with the exposure time, and the ultimate remaining fraction of 4-nitrophenol in the solution were close to 0 for all the cases. The 4-nitrophenol catalytic reduction rate followed the order of 0.2 M > 0.025 M > 0.1 ≥ 0.05 M.
 | ||
| Fig. 8 Remaining fraction of 4-nitrophenol solution as a function of time after treatment with AgNP-immobilized nanofibrous mats synthesized with different AgNO3 solution concentrations. | ||
As for the catalytic activity difference using AgNPs synthesized with different AgNO3 solution concentrations, we think that the catalytic reduction capacity of AgNP-immobilized nanofibrous mats to 4-nitrophenol could be mainly dependent on the size of AgNPs immobilized into the nanofibers. With smaller sizes of AgNPs, larger surface areas were obtained, thus resulting in higher reactivity toward the reagent. In this scenario, the 4-nitrophenol catalytic reduction rate of AgNPs followed the order of 5.8 nm (0.2 M AgNO3 solution) > 6.3 nm (0.025 M AgNO3 solution) > 10.8 nm (0.1 M AgNO3 solution) ≥ 20.4 nm (0.05 M AgNO3 solution). It should be noted that although the size of AgNPs synthesized with 0.05 M AgNO3 solution was much bigger (20.4 nm) than that of AgNPs synthesized with 0.1 M AgNO3 solution (10.8 nm), the catalytic activity of hybrid nanofibrous mats with 20.4 nm AgNPs was comparable to that of hybrid nanofibrous mats with 10.8 nm AgNPs. We think that in the case of hybrid nanofibrous mats with 20.4 nm AgNPs, the space between the adjacent AgNPs was larger than that of 10.8 nm AgNPs immobilized in the nanofibrous mats, which was beneficial for the diffusion of 4-nitrophenol molecular into the interior of the fibers, resulting in a increased catalytic reaction rate to some extend. In this case, both the size effect of AgNPs and permeability of nanofiber should be responsible for the catalytic activity of AgNP-immobilized nanofibrous mats synthesized with 0.1 M and 0.05 M AgNO3 solution, respectively.
Conclusion
In summary, we developed a facile approach to simultaneously synthesize and immobilize AgNPs within the PAA/PVA nanofibers using electrospun PAA/PVA nanofibers as nanoreactors. The AgNP-immobilized nanofibrous mats exhibited a porous structure with a smooth and uniform surface. TEM analysis showed that spherical AgNPs with a fcc crystaline structrue were uniformly distributed along the cross-section of nanofibers. The AgNP-immobilized nanofibrous mats were demonstrated to be able to catalyze the reaction of transforming 4-nitrophenol to 4-aminophenol with an efficiency approaching 100% at 30 min. Furthermore, the AgNP-immobilized PAA/PVA nanofibrous mats can be easily recycled and reused for at least 4 times with similar catalytic performance. In addition, the effect of AgNO3 solution concentration on the immobilized AgNPs size, the Ag content in the hybrid nanofibrous mat, and the catalytic activity are systematically investigated. The results show that the Ag loading in the nanofibrous mats seems independent of the AgNO3 solution, while the size of AgNPs, the spacial distribution of AgNPs and the catalytic activity of AgNP-immobilized nanofibrous mats can be conveniently manipulated by varying the concentration of AgNO3 solution. This methodology is generally applicable to polymer nanofibers with other specific functional group (e.g., amino groups) and other metal ions or semiconductor precursor can be introduced to fabricate a wide variety of functional hybrid nanofibers for catalysis, energy, sensing, photonic and biomedical applications.Acknowledgements
This research is financially supported by 973 project of China (No. 2009CB526402) and the National Natural Science Foundation of China (No. 50873079).References
- P. Alivisatos, Nat. Biotechnol., 2004, 22, 47–52 CrossRef.
- J. Dai and M. L. Bruening, Nano Lett., 2002, 2, 497–501 CrossRef CAS.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer and M. Ballauff, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- X. Shi, K. Sun and J. R. J. Baker, J. Phys. Chem. C, 2008, 112, 8251–8258 CrossRef CAS.
- M. Schrinner, S. Proch, Y. Mei, R. Kempe, N. Miyajima and M. Ballauff, Adv. Mater., 2008, 20, 1928–1933 CrossRef CAS.
- K.-S. Lee and M. A. EI-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS.
- M. D. Malinsky, K. L. Kelly, G. C. Schatz and R. P. V. Duyne, J. Am. Chem. Soc., 2001, 123, 1471–1482 CrossRef CAS.
- R. J. Tseng, C. Tsai, L. Ma, J. Ouyang, C. S. Ozkan and Y. Yang, Nat. Nanotechnol., 2006, 1, 72–77 CrossRef CAS.
- L. Balogh, D. R. Swanson, D. A. Tomalia, G. L. Hagnauer and A. T. McManus, Nano Lett., 2001, 1, 18–21 CrossRef CAS.
- Z. Shi, K. G. Neoh and E. T. Kang, Langmuir, 2004, 20, 6847–6852 CrossRef CAS.
- Q. Wang, H. Yu, L. Zhong, J. Liu and J. Shen, Chem. Mater., 2006, 18, 1988–1994 CrossRef CAS.
- Y. Lu, Y. Mei, M. Drechsler and M. Ballauff, Angew. Chem., Int. Ed., 2006, 45, 813–816 CrossRef CAS.
- Y. Lu, Y. Mei, M. Schrinner, M. Ballauff, M. W. Moller and J. Breu, J. Phys. Chem. C, 2007, 111, 7676–7681 CrossRef CAS.
- W. Lesniak, A. U. Bielinska, K. Sun, K. W. Janczak, X. Shi, J. R. J. Baker and L. P. Balogh, Nano Lett., 2005, 5, 2123–2130 CrossRef CAS.
- A. D. McFarland and P. V. Duyne, Nano Lett., 2003, 3, 1057–1062 CrossRef CAS.
- K. J. Lee, P. D. Nallathamby, L. M. Browning, C. J. Osgood and X.-H. N. Xu, ACS Nano, 2007, 1, 133–143 CrossRef CAS.
- Y. Li, Y. Wu and B. S. Ong, J. Am. Chem. Soc., 2005, 127, 3266–3267 CrossRef CAS.
- T. Dadosh, Mater. Lett., 2009, 63, 2236–2238 CrossRef CAS.
- Y. M. Mohan, K. Vimala, V. Thomas, K. Varaprasad, B. Sreedhar, S. K. Bajpai and K. M. Raju, J. Colloid Interface Sci., 2010, 342, 73–82 CrossRef.
- R. A. Salkar, P. Jeevanandam, S. T. Aruna, Y. Koltypin and A. Gedanken, J. Mater. Chem., 1999, 9, 1333–1335 RSC.
- D. Spadaro, E. Barletta, F. Barreca, G. Curro and F. Neri, Appl. Surf. Sci., 2010, 256, 3812–3816 CrossRef CAS.
- N. Toshima, Y. Shiraishi and T. Teranishi, J. Mol. Catal. A: Chem., 2001, 177, 139–147 CrossRef CAS.
- T. C. Wang, M. F. Rubner and R. E. Cohen, Langmuir, 2002, 18, 3370–3375 CrossRef CAS.
- X. Zan and Z. Su, Langmuir, 2009, 25, 12355–12360 CrossRef CAS.
- B. Yin, H. Ma, S. Wang and S. Chen, J. Phys. Chem. B, 2003, 107, 8898–8904 CrossRef CAS.
- D. H. Reneker and I. Chun, Nanotechnology, 1996, 7, 216–223 CrossRef CAS.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384–1391 CrossRef CAS.
- K. Yoon, B. S. Hsiao and B. Chu, J. Mater. Chem., 2008, 18, 5326–5334 RSC.
- W. He, Z. Ma, T. Yong, W. E. Teo and S. Ramakrishna, Biomaterials, 2005, 26, 7606–7615 CrossRef CAS.
- X. Zhu, W. Cui, X. Li and Y. Jin, Biomacromolecules, 2008, 9, 1795–1801 CrossRef CAS.
- H. Kong and J. Jang, Langmuir, 2008, 24, 2051–2056 CrossRef CAS.
- W. K. Son, J. H. Youk and W. H. Park, Carbohydr. Polym., 2006, 65, 430–434 CrossRef CAS.
- X. Wang, C. Drew, S.-H. Lee, K. J. Senecal, J. Kumar and L. A. Samuelson, Nano Lett., 2002, 2, 1273–1275 CrossRef CAS.
- Y. Hong, D. Li, J. Zheng and G. Zou, Nanotechnology, 2006, 17, 1986–1993 CrossRef CAS.
- J. A. Lee, K. C. Krogman, M. Ma, R. M. Hill, P. T. Hammond and G. C. Rutledge, Adv. Mater., 2009, 21, 1252–1256 CrossRef CAS.
- S. Xiao, M. Shen, R. Guo, Q. Huang, S. Wang and X. Shi, J. Mater. Chem., 2010, 20, 5700–5708 RSC.
- S. Xiao, M. Shen, R. Guo, S. Wang and X. Shi, J. Phys. Chem. C, 2009, 113, 18062–18068 CrossRef CAS.
- Q. B. Yang, D. M. Li, Y. L. Hong, Z. Y. Li, C. Wang, S. L. Qiu and Y. Wei, Synth. Met., 2003, 137, 973–974 CrossRef CAS.
- A. C. Patel, S. Li, C. Wang, W. Zhang and Y. Wei, Chem. Mater., 2007, 19, 1231–1238 CrossRef.
- F. Liu, R. Guo, M. Shen, S. Wang and X. Shi, Macromol. Mater. Eng., 2009, 294, 666–672 CrossRef CAS.
- J. Yue and R. E. Cohen, Supramol. Sci., 1994, 1, 117–122 CrossRef CAS.
- R. T. Clay and R. E. Cohen, Supramol. Sci., 1995, 2, 183–191 CrossRef CAS.
- D. Cheng, X. Zhou, H. Xia and H. S. O. Chan, Chem. Mater., 2005, 17, 3578–3581 CrossRef CAS.
- G.-H. Jiang, L. Wang, T. Chen, H.-J. Yu and J.-J. Wang, J. Mater. Sci., 2005, 40, 1681–1683 CrossRef CAS.
- A. A. Antipov, G. B. Sukhorukov, Y. A. Fedutik, J. Hartmann, M. Giersig and H. Mohwald, Langmuir, 2002, 18, 6687–6693 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional cross-sectional TEM images of ZVI NP-immobilized nanofibrous mats synthesized with 0.025 M, 0.05 M and 0.1 M AgNO3 solution concentrations, respectively, TGA analysis, and the evaluation of catalytic reduction efficiency of nanofibrous mats with and without AgNPs. See DOI: 10.1039/c1ra00127b/ |
| This journal is © The Royal Society of Chemistry 2012 |