Bi-component hydrogel of perylene-3,4,9,10-tetracarboxylic potassium salt and L-tyrosine†
Partha
Bairi
,
Bappaditya
Roy
and
Arun K.
Nandi
*
Polymer Science Unit, Indian Association for the cultivation of Science, Jadavpur, Kolkata-700 032, India. E-mail: psuakn@iacs.res.in
First published on 2nd November 2011
Abstract
Potassium perylene-3,4,9,10-tetracarboxylate (P) and L-tyrosine (T) produces supramolecular PT complexes viz.PT11, PT12, PT13 and PT14 where the numbers indicate respective molar ratios. PT11 does not produce a hydrogel but the other three produce hydrogels with a minimum gelation concentration of 1.30% (w/v). All PT gels exhibit a fibrillar morphology and the gel melting temperature follows the order PT14 > PT13 > PT12. The frequency independent G′ (G′ > G′′) for PT12, PT13 and PT14 systems characterize them as a gel and the critical yield stress values are 6.0, 3.18 and 0.76 Pa for PT14, PT13 and PT12, respectively. The presence of H-bonding and electrostatic interactions in PT complexes is evident from the FTIR spectra. NMR spectra suggest π–π stacking in both gels and in the PT11 complex, however, there is less π-stacking in the latter. The red shift of the absorption peaks of PT gels compared to that of P is attributed to the J-aggregate formation. The emission peaks also show red shifts and the emission intensity increases significantly. The average life time value decreases in the gels. With increasing temperature, PL-intensity gradually decreases due to the de-stacking of the J-aggregates but the emission peak position shows a step like increase due to batch-wise slipping of PT motifs from J-aggregates.
Introduction
Small molecular bi-component gelators (SMBGs) have gathered an enormous research interest during the past few years from material scientists for their interesting tunable properties.1,2 The SMBGs show tunability of their morphologies, as well as their mechanical and optical properties, by changing the composition of the components2c and also by changing one of the components of the gel.2g In our earlier studies we chose melamine as a central molecule and changed the complimentary components from riboflavin to galic acid, lumichrome, different positional isomers of hydroxyl benzoic acidetc. To generalize the formation mechanism and behaviour of the bi-component gels we chose perylene-3,4,9,10-tetracarboxylic acid (PA) as another central molecule, because of its planar structure and eight H-bonding sites capable of strong supramolecular complex formation and π-stacking. But this compound has difficulty in forming a hydrogel because of its insolubility in water. However, its potassium salt (P) has good solubility in water providing an opportunity to make a supramolecular complex with another suitable complimentary component. P is an important extended π-conjugated molecule and it exhibits good photoluminescence (PL) properties.3 Its derivatives are used in biolabelling4 and in the fabrication of solar cell devices.5,6 Among the twenty essential amino acidsL-tyrosine (T) is an important member with a polar side group7 and it occurs in proteins that are part of the signal transduction processes.8 It also plays an important role in photosynthesis as an electron donor during the reduction of oxidized chlorophylls.9 So it would be interesting to study the supramolecular complexes of a P and T produced hydrogel.In a recent report, a carboxylic acid, e.g. tetra-proline modified calix-[4]-arene, forms complexes with amino acids (e.g.arginine, histidine, and lysine) producing a hydrogel.10 It is well established that the donor–acceptor pair, coronene tetracarboxylate tetra-potassium salt, and dodecyl functionalized methyl viologen derivative11a,b produces a bi-component hydrogel by a charge transfer process. It is also known that the hydrogel of N-(fluorenylmethoxycarbonyl) amino acid—NPC 15199 and Fmoc-lysine is formed in the presence of sodium carbonate mainly due to H-bonding and π-stacking.12 Encouraged from these results we have studied L-tyrosine and perylene-3,4,9,10-tetracarboxylic potassium salt (Scheme 1) as a new two component hydrogel system where H-bonding and π-stacking might be responsible for hydro-gelation. Additionally, L-tyrosine provides an electrostatic interaction13 strengthening the supramolecular complex to self-organise into fibrils producing a gel. The perylene moiety is fluorescent in nature hence the derived hydrogel also shows strong green fluorescence under UV light. Also the stacking pattern of the complex during the gel formation induces the hydrophobic core formation causing ∼3 times enhancement of PL-intensity than that of pure P. With an increase of temperature the PL-intensity gradually decreases in all the PT gels but the emission peak position shows a step like increase.
Results
Hydrogel formation
A mixture of potassium perylene-3,4,9,10-tetracarboxylate (P) and L-tyrosine (T) in different molar ratios (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, 1:3, 1:4) are dissolved in water (1.5% (w/v)) in a closed vial at 100 °C as L-tyrosine (T) has some difficulty in being fully soluble at the concentration range used here. The homogeneous solution is gradually transformed into a reddish-yellow coloured gel after it is cooled to 30 °C (Fig. 1). The gel formation is confirmed by the cessation of the flow in the test tube tilting method and the minimum gelation concentration (MGC) is found to be 1.30% (w/v) for all the compositions (1:2, 1:3, 1:4) of P and T. The gels are designated as PT14, PT13 and PT12, where the numbers associated with PT indicate the molar ratios of P and T. It has been observed that P and T in the 1:1 molar ratio (PT11) does not produce a gel under similar conditions. Instead of L-tyrosine we have also tested the gelation possibility of P with alanine, phenylalanine, leucine, isoleucine, cysteine and methionine, but they do not produce a gel under similar experimental conditions. The gel melting temperatures are measured by a dropping ball method using a temperature controlled oil bath and have the values 63.8 ± 0.4, 62.0 ± 0.7 and 61.3 ± 0.4 °C for PT14, PT13 and PT12 systems, respectively. The gradual decrease of gel melting point with decreasing T concentration may be attributed to the weaker aggregate formation required to produce the fibrils forming the gel network.
:2, 1:3, 1:4) are dissolved in water (1.5% (w/v)) in a closed vial at 100 °C as L-tyrosine (T) has some difficulty in being fully soluble at the concentration range used here. The homogeneous solution is gradually transformed into a reddish-yellow coloured gel after it is cooled to 30 °C (Fig. 1). The gel formation is confirmed by the cessation of the flow in the test tube tilting method and the minimum gelation concentration (MGC) is found to be 1.30% (w/v) for all the compositions (1:2, 1:3, 1:4) of P and T. The gels are designated as PT14, PT13 and PT12, where the numbers associated with PT indicate the molar ratios of P and T. It has been observed that P and T in the 1:1 molar ratio (PT11) does not produce a gel under similar conditions. Instead of L-tyrosine we have also tested the gelation possibility of P with alanine, phenylalanine, leucine, isoleucine, cysteine and methionine, but they do not produce a gel under similar experimental conditions. The gel melting temperatures are measured by a dropping ball method using a temperature controlled oil bath and have the values 63.8 ± 0.4, 62.0 ± 0.7 and 61.3 ± 0.4 °C for PT14, PT13 and PT12 systems, respectively. The gradual decrease of gel melting point with decreasing T concentration may be attributed to the weaker aggregate formation required to produce the fibrils forming the gel network.
 | ||
| Fig. 1 Photograph of the PT14 gel before and after UV light irradiation. | ||
Morphology
The well-organized fibrilar network morphology in the PT14 hydrogel is birefringent in nature as evident from the polarized optical micrograph (POM) made under a perfectly crossed polarizer (Fig. 2a), indicating a high degree of molecular anisotropy in the molecular packing of the PT14 complex. The FESEM image of the dried PT14 hydrogel (Fig. 2b), indicates the fibrillar network morphology of the xerogel. In ESI† Fig. 1 the FESEM micrographs of the other compositions are presented and similar fibrilar morphology is observed in both PT12, PT13 hydrogels and in the PT11 supramolecular complex. The fibrilar diameters are 0.94 ± 0.06, 1.27 ± 0.55, 0.41 ± 0.08 and 0.81 ± 0.25 μm for PT11, PT12, PT13 and PT14 gels, respectively. It is surprising that though PT11 has fibrilar morphology of similar dimensions to the other systems, it does not produce gel and there are also similar reports where the self assembled fibers are produced but do not produce gel.13 It is probable, that the density of the fibers is insufficient to make the surface force effective for entrapping solvent molecules. The same morphology of all the systems points out the similar pattern of molecular arrangements for different molar compositions of P and T. | ||
| Fig. 2 (a) POM image of PT14 hydrogel and (b) FESEM image of PT14 xerogel at 1.5% (w/v). | ||
Structure
The FTIR spectra of P, T and PT14 xerogel are presented in Fig. 3 and those of PT11, PT12 and PT13 are presented in ESI† Fig. 2. From the figures it is apparent that for pure P the peaks at 1556 and 1413 cm−1 correspond to the stretching vibration of carboxylate ions and they are shifted to 1566 and 1416 cm−1 respectively.14a This shift to higher energy may be attributed to the hydrogen bonding of COO− with the –OH group of T. On the other hand, T has a peak at 1591 cm−1 for the zwitterionic carboxylate ion14b which remains unchanged at 1591 cm−1 in PT14, indicating carboxylate ion of T is not involved in H-bond formation and the electrostatic interaction prevails in the supramolecular complex as it is present in pure T. These results prove the presence of both H-bonding and ionic interactions in the PT14 supramolecular complex. The 1598 cm−1 peak of P is due to the aromatic ring stretching vibration which gradually shifts to 1591 cm−1 with increasing T concentration (ESI† Fig. 2). It might be due to the easier vibration of the aromatic ring for the delocalization of ring electrons on π-stacking of the complex and also for the overlapping of the peaks of P and T at this region. The 3206 cm−1 peak is due to the –NH3+ ion of T and it remains unchanged in all the complexes,14c further supporting that the ionic interaction in T remains unchanged even for the complex formation with P. | ||
| Fig. 3 FTIR spectrum of PT14 xerogel with those of pure P and T. | ||
The chemical shifts of the aromatic –CH protons of P, T and PT14 systems are shown in the 1H NMR spectra (Fig. 4) and for other systems they are presented in ESI† Fig. 3. The upfield shifts of aromatic –CH protons during the gel formation are also compared in Table 1 as it is an index of π-stacking of the supramolecular complexes in the gel.2e,15 In all cases the aromatic protons of P and T show an upfield shift due to the π-stacking but in the case of the PT11 complex, the upfield shift is lower than that of the other systems. The extent of π-stacking depends on the electron density of the π-clouds.2g,16 The H-bonding of T causes a deactivation of the P ring and the larger the number of T units attached, the higher the deactivation of the P ring becomes, causing a lowering of the electron density. This facilitates effective π-stacking for complexes with higher concentrations of T and it is the lowest in the PT11 system.
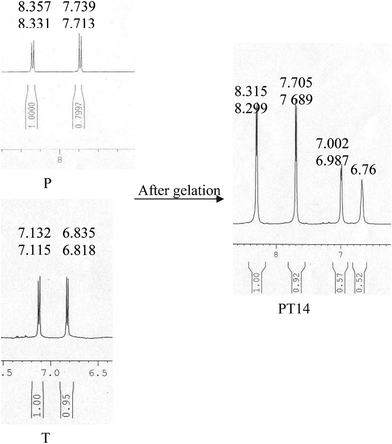 | ||
| Fig. 4 Comparison of the 1H NMR spectra of P and T (aromatic CH protons) with that of the PT14 hydrogel. | ||
| Compositions | Peak Position (δ in ppm) | |||||
|---|---|---|---|---|---|---|
| Pure state | Gel state | Δδ (ppm) | ||||
| P | T | P | T | P | T | |
| PT14 | 8.357 | 7.132 | 8.315 | 7.002 | 0.042 | 0.130 |
| 8.331 | 7.115 | 8.299 | 6.987 | 0.032 | 0.180 | |
| 7.739 | 6.835 | 7.705 | 6.76 | 0.034 | 0.073 | |
| 7.713 | 6.818 | 7.689 | 0.024 | |||
| PT13 | 8.357 | 7.132 | 8.313 | 6.998 | 0.044 | 0.134 |
| 8.331 | 7.115 | 8.300 | 6.981 | 0.031 | 0.134 | |
| 7.739 | 6.835 | 7.707 | 6.684 | 0.032 | 0.151 | |
| 7.713 | 6.818 | 7.691 | 6.668 | 0.022 | 0.150 | |
| PT12 | 8.357 | 7.132 | 8.310 | 7.003 | 0.047 | 0.129 |
| 8.331 | 7.115 | 8.294 | 6.987 | 0.082 | 0.128 | |
| 7.739 | 6.835 | 7.704 | 6.678 | 0.035 | 0.157 | |
| 7.713 | 6.818 | 7.689 | 6.668 | 0.024 | 0.150 | |
| PT11 | 8.357 | 7.132 | 8.349 | 7.049 | 0.008 | 0.083 |
| 8.331 | 7.115 | 8.322 | 7.022 | 0.009 | 0.093 | |
| 7.739 | 6.835 | 7.736 | 6.729 | 0.003 | 0.106 | |
| 7.713 | 6.818 | 7.710 | 6.701 | 0.003 | 0.117 | |
The molecular packing is delineated from the WAXS patterns of the dried hydrogels (Fig. 5) where the diffraction pattern of PT14 is compared with those of pure P and T. The WAXS patterns of PT11, PT12 and PT13 are presented in ESI† Fig. 4 and it is apparent that a different WAXS pattern is observed in PT systems from those of the components but they are almost the same with each other. The sharp peaks of the dried PT samples indicate that crystallites of the PT complex constitute the fibrils producing the network. The WAXS patterns of the PT gels and PT11 complex are almost the same indicating the nature of molecular packing is the same. In all cases, a new peak at θ = 25.3° of PT complex appears for the inter-planar stacking distance (3.5 Å) of the π-stacked complex.2g,17
 | ||
| Fig. 5 WAXS patterns of pure P, T and PT14 xerogel. | ||
Rheology
The gel formation and also the strength of the hydrogels are discussed here under the results of rheological experiments. The frequency independent storage modulus (G′) (Fig. 6a) with a significantly higher value of G′ than that of G′′ (loss modulus) supports their gel nature.18 Both the G′ and G′′ values follow the order PT14 > PT13 > PT12 hydrogel systems. It is interesting to note that in the PT12 hydrogel system the G′ and G′′ show a crossover point at higher frequency region (431 Hz) implying the weaker gel structure than those of PT13 and PT14 systems. On the application of stress to the hydrogel systems under the metal geometry the hydrogels break down at different oscillator stresses (Fig. 6b) depending upon the strength of the gels. The critical yield stress values are 6.0, 3.18 and 0.76 Pa for PT14, PT13 and PT12, respectively. As both G′ and critical yield stress follow the same order (PT14 > PT13 > PT12), it may be concluded that the gel strength is the highest for PT14 system probably due to good packing of the system. | ||
| Fig. 6 (a) Storage modulus (G′) and loss modulus (G′′) vs. angular frequency plot at a concentration of 1.5% (w/v) of PT12, PT13 and PT14 hydrogels at 30 °C. (b) G′ and G′′ vs. oscillator stress plot at a constant frequency of 1 Hz of the three different hydrogels at a concentration of 1.5% (w/v) at 30 °C. | ||
It is to be noted here that in Fig. 6b, there is an appreciable fluctuation of loss modulus data in the linear viscoelastic region (LVR) for the PT14 and PT13 systems. No definite reason for this oscillating behaviour can be afforded, however it might possibly occur due to the applied oscillator stress, which may not be isotropically distributed through these strong gels; the more localized fibrils dissipate more stress, squeezing some associated solvent molecules which further get reversibly absorbed due to the surface tensional forces maintaining the network structure. This reversible and transient loss and gain of solvent molecules by the gel network during the experiment causes an oscillatory behaviour in the G′′ vs. stress plot. However, the effect is less pronounced in the case of the storage modulus because while storing the energy the fibrils transfer it to the entrapped solvent molecules. The fluctuation of G′′ with applied stress is lower in the weak (soft) PT12 gel, as the applied stress becomes dissipated more isotropically due to the presence of well dispersed fibrils.
Photophysical properties
It is now necessary to understand the variation of optical properties due to gelation in order to get an insight into the finer details on the stacking of PT complexes producing a fibrillar network structure. In Fig. 7, the normalized UV-vis spectra of P, PT11, PT12, PT13 and PT14 sols are presented. The absorbance spectrum of pure P has two sharp peaks at 468 nm and 438 nm and two weak shoulders at 415 nm and 388 nm corresponding to the 0–0, 0–1, 0–2 and 0–3 electronic transition,19 respectively. In the PT14, PT13, and PT12 sols, the 438 nm peak shows a red shift to 442 nm and the 464 nm peak also shows a red shift by 4–5 nm, but in case of PT11 complex, the 438 nm and 464 nm peaks remain unchanged. The red shifts of the absorption peaks are attributed to the formation of PT complexes where the perylene moieties are stacked side by side which results in the lowering of the band gap compared to that of pure P.![Normalized UV-vis spectra of different PT complexes and pure P in a water medium at 0.05% (w/v) [inset: Job’s plot with respect to mole fraction of T (probed at 464 nm)].](/image/article/2012/RA/c1ra00506e/c1ra00506e-f7.gif) | ||
| Fig. 7 Normalized UV-vis spectra of different PT complexes and pure P in a water medium at 0.05% (w/v) [inset: Job’s plot with respect to mole fraction of T (probed at 464 nm)]. | ||
Aggregation of small molecules by a π-stacking process may be classified as J or H-aggregates. In the former, side by side stacking occurs while in the latter, face to face aggregation occurs,20–22 the former usually yields a red shift and the latter exhibits a blue shift in the absorption peak. Hence the red shift observed in the present systems is indicative of the J-aggregate formation as presented in Scheme 2. In the Job’s plot of absorbance vs. mole fraction of T (inset: Fig. 7) we have found the higher absorbance value at the PT14 composition, suggesting this complex is the most favourable to form a stable hydrogel.
 | ||
| Scheme 1 Chemical structure of perylene tetracarboxylate ion (P) and L-tyrosine (T). | ||
P has a good photoluminescence (PL) property, so it is important to know how this property changes with gelation. The normalized PL spectra of P and PT gels by exciting with a light of 440 nm are presented in Fig. 8; showing a PL peak at 511 nm and two weak shoulders at 550 nm and 590 nm. The PT14, PT13 and PT12 gels exhibit two sharp emission peaks but PT11 sol exhibits one sharp peak at 515 nm and two weak shoulders at around 550 nm and 590 nm, respectively. The 511 nm peak of P shows a red shift at 516 for PT13 and PT14 and at 518 nm for PT12 gel. In the PT12 gel the shoulder of 550 nm transforms into a peak at 563 nm but in PT13 and PT14, the shoulders merge giving a sharp peak at 596 and 593 nm, respectively. Thus there is a red shift in the emission peaks suggesting that excitons are stabilised during J-aggregate formation.23 The occurrence of two emission peaks may be attributed to the presence of two different excited states in the gel state arising from the J-aggregate formation.
 | ||
| Fig. 8 Normalized photoluminescence spectra of pure P, different PT hydrogels and PT11 complex at 1.5% (w/v) concentration, excited at 440 nm. | ||
In all the PT12, PT13 and PT14 gels, the PL-intensity is higher than that of P by 3.8, 2.5, and 3.5 times, respectively. The enhancement of the PL-intensity may be attributed to the hydrophobic core formation due to the π-stacking process2a–c and also due to J-aggregate formation.24 But in case of the PT11 system, the PL-intensity is not at all increased, suggesting no appreciable hydrophobic core formation. The lifetime values of all the systems are measured from the decay curves (ESI† Fig. 5), and the average lifetime values together with component decay times are presented in Table 2. It is evident from the table that the average lifetime values have decreased from that of pure P for both the PT11 complex and in the PT12, PT13 and PT14 gels, however, the decrease is smaller in case of the PT11 complex. It is interesting to note that in spite of the decrease in the lifetimes, the PL-intensity increases in all the systems suggesting that the hydrophobic core formation is the major cause by decreasing the decay paths with the solvent molecules (water). Thus the very small changes in the absorbance spectra and high fluorescence of the gels suggest that chromophores are in isolated environments with not much π–π interaction occurring between the aromatic cores (supported by the sharp peaks in the NMR spectra), describing the J-type packing. So, from the red shift in UV-vis absorption peak, the PL-intensity enhancement and the lower average lifetime value than that of pure P, it can be concluded that J-aggregate formation occurs during the stacking process of the PT complexes.20–24
| Systems | τ 1 (ns) | Relative amplitudes (a1) | τ 2 (ns) | Relative amplitudes (a2) | Av. Lifetime (ns) |
|---|---|---|---|---|---|
| P | 6.18 | −1.90 | 7.23 | 2.15 | 15.14 |
| PT11 | 3.89 | −0.35 | 5.31 | 0.51 | 8.44 |
| PT12 | 2.41 | 7.05×10−3 | 5.32 | 0.16 | 5.21 |
| PT13 | 2.22 | 1.03×10−2 | 4.86 | 0.16 | 4.71 |
| PT14 | 2.86 | 3.75×10−3 | 5.70 | 0.16 | 5.64 |
Both the UV-vis and the PL spectral data indicate that in the PT11 system the aggregation of the complex is weak and this is also supported by the very low upfield shift of aromatic protons (NMR spectra) of the PT11 complex. The weak π-stacking of the complex is the probable cause for the PT11 system for not forming the gel. This indicates that the relative concentration of T with respect to P has a significant role for the gel formation in the PT system.
The temperature dependency of the PL emission is presented in Fig. 9a for the PT14 gel (ESI† Fig. 6 and 7 for the PT12 and PT13 gel). It is clear that the PL-intensity gradually decreases suggesting that the aggregation decreases with rising temperature and both the emission peaks at 515 and 594 nm show a red shift (Fig. 9b). The gradual decrease of PL-intensity with an increase of temperature may be related to the de-stacking which gradually decreases the hydrophobic cores. On the gradual increase of temperature, the emission peak position shows a step like increase on which the horizontal sections occur as a result of the constancy of the excited energy state for a particular range of temperature. This result suggests the disaggregation of J-aggregates appears to be occurring in batches.
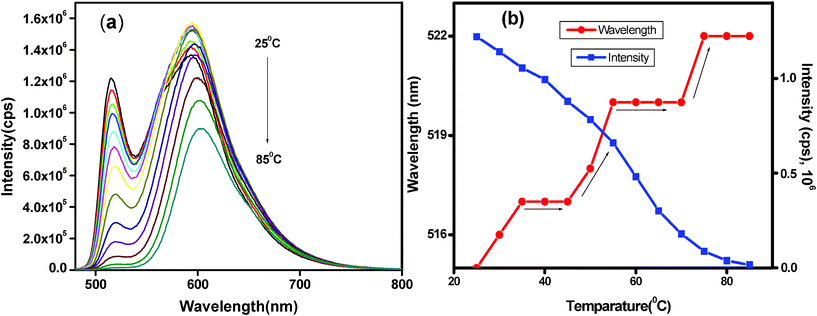 | ||
| Fig. 9 (a) Temperature dependent PL spectra of PT14 hydrogel at 1.5% (w/v) excited at 440 nm during heating (25–85 °C) and (b) corresponding shift of λmax and intensity with respect to temperature. | ||
Discussion
It is important to discuss here how the hydrogel is formed in the case of T with P but not in the case of L-phenylalanine with P. Here the polar –OH group present in the phenyl ring in T plays an important role for the formation of the gel. This group is H-bonded with the COO−group of P and forms a supramolecular complex (Scheme 2) which further extends laterally through electrostatic interaction and then longitudinally through π–π interaction. These π-stacked motifs organize into fibrils which produce the network, entrapping a large amount of water producing the hydrogel. The lack of an –OH group in L-phenylalanine is the main reason for its inability for gel formation. The other amino acids (alanine, leucine, isoleucine, cysteine, methionine) do not produce gels with P due to the absence of the aromatic ring that provides the π-stacking interaction. | ||
| Scheme 2 Schematic model of PT14 supramolecular complex forming gels by H-bonding, electrostatic interaction and π-stacking processes. | ||
The formation of the J-aggregated supramolecular architecture is not expected here because P is a flat molecule and its face to face overlapping (i.e. formation of H-aggregate) of the PT complex is easier than side by side overlapping of the complex as it occurs here. A probable reason is the presence of an asymmetric carbon atom present in the amino acid T. The non-superimposability of this part of the PT complex prevents it from H-aggregate formation, but due to the strong supramolecular force of the perylene moiety, the aggregation of the PT complex occurs in the mode of side by side stacking of the complexes. The relative strength and stability of the PT gels depend on the extent of the π-stacking process and it is best in the case of PT14 due to the complexation in all four arms of P by T, giving four additional aromatic moieties with P. However, in the PT13 system, one arm of P remains vacant and in the PT12 system, two arms of P are empty. This results in progressively reduced π-stacking, causing the strength and stability of the gel to follow the order PT14 > PT13 > PT12.
It is now necessary to discuss the mode of de-stacking of the PT14 complex with rising temperature. It is evident from Fig. 9b that there is a step like increase of emission peak (three stages) but a continuous decrease of PL-intensity with increasing temperature. One probable reason is the slipping of the complex motifs of the J-aggregates to occur in batches; an extraction of a batch of motifs keeps the aggregate stable for 10–15 °C until the next slipping event starts with a rise in temperature. The energy level of excitons consequently decreases lowering the emission band gap, hence showing a red shift. This batchwise slipping of the motifs occurs due to the strong π-stacking force of the PT complexes and the slipping occurs from the defect regions produced during the packing of the complexes. At the second stage of slipping the gel melts by destroying the fibrils but at the third stage the increase of emission peak position suggests that there are still some small J-aggregates in the form of microgels and it completely disaggregates at 75 °C. The same phenomenon is occurring in the PT13 and PT12 hydrogels with a larger number of steps and smaller step size. This is probably due to the reduced stability of the gel arising from weaker π-stacking.
In contrast to the step like increase of the emission peak, the PL-intensity decreases continuously with rising temperature for all cases. A probable explanation may lie in the quenching of both the slipped motifs and the remaining J-aggregate of the PT complex. With a rise of temperature, the slipped motif further disaggregates into the PT complex and finally back to the components by the destruction of H-bonds. In this step the remaining J-aggregate remains stable, but the smaller motifs completely break into P and T, decreasing the hydrophobic cores, thus increasing the overall quenching by the solvent molecules. The resultant manifestation of the overall process is the cause of a continuous decrease of PL-intensity in the PT gels with rising temperature.
Experimental section
Materials
Perylene-3,4,9,10-tetracarboxylic dianhydride was purchased from Aldrich Chemical.Co., USA and L-tyrosine (T) from Sisco Research Laboratories PVT. LTD. (Mumbai). They were used without further purification. Potassium hydroxide was purchased from Merck Specialities Private Limited (Mumbai).Perylene-3,4,9,10-tetracarboxylic dianhydride was hydrolyzed with KOH/ethanol to prepare water soluble perylene-3,4,9,10-tetracarboxylic acid potassium salt (P).
Microscopy
The polarized optical microscopy (POM) images of the gels were studied by taking a thin layer of the gel on a glass slide and taking observations through an optical microscope under a perfectly crossed polarizer (Biomed, Leitz, Germany) fitted with a digital camera. The morphology of the PT gel was investigated by field emission scanning electron microscopy (FESEM). A small portion of the gels, produced at a different molar ratio, was taken in a glass cover slip, dried in air at 30 °C and finally under vacuum at 30 °C. They were observed through an FESEM instrument (JEOL, JSM 6700F) operating at 5 kV after a platinum coating.Measurement of gel–sol transition temperature (Tgel)
The gel–sol transition temperature (Tgel) was determined using the following “falling steel ball method”.25 For the measurement, a gelator solution with different weight ratios of P and T (1:2, 1:3, and 1:3) was filled into a screw cap vial (inner diameter: 11.8 mm, filling height: 16 mm), sealed and kept at room temperature for 6 h. A steel ball (diameter: 2.1 mm, weight: 45 mg) was put on the top of the gel, and the vial was heated slowly in a temperature controlled water bath (heating rate: 1 °C min−1). Tgel was defined as the temperature at which the steel ball reached the bottom of the vial. An average of four such measurements was taken as Tgel. The temperature was measured in a reference vial filled with 1 mL of pure water.
Spectroscopy
The UV-vis spectra of the samples were recorded with a Hewlett-Packard UV-vis spectrophotometer (model 8453) using a cuvette of 0.1 cm path length. The PT hydrogel samples were prepared in sealed cuvettes and fluorescence studies were carried out in a Horiba Jobin Yvon Fluoromax 3 instrument. Each gel sample in a quartz cell of 1 cm path length was excited at 440 nm wavelength and the emission scans were recorded from 460 to 800 nm using a slit width of 2 nm with a 1 nm wavelength increment having an integration time of 0.1 s. Fluorescence lifetime values were measured by using a time-correlated single photon counting fluorometer (Fluorecule, Horiba Jobin Yvon). The system was excited with 440 nm nano LED of Horiba Jobin Yvon having λmax at 368 nm with pulse duration < 200 ps. All the samples were prepared for room temperature measurement (30 °C) in double distilled water. Average fluorescence lifetimes (τf) for the exponential iterative fitting were calculated from the decay times (τi) and the relative amplitudes (ai) using the following relation| <τf> = a1τ1 + a2τ2 + a3τ3 | (1) |
The FTIR spectra of pure P, T and PT xerogels were recorded using KBr pellets of the xerogels in a FTIR-8400S instrument (Shimadzu). 1H NMR spectra of pure P and PT gels in D2O (1.5% w/v) were recorded via a Bruker DPX 300 instrument at a frequency of 500 MHz. The concentration of P is kept the same in all the samples.
Diffraction study
The wide-angle X-ray scattering (WAXS) experiments of all the xerogels of PT and pure P were performed using a Bruker AXS diffractometer (model D8 Advance) using a Lynx Eye detector. The instrument was operated at a 40 kV voltage and at a 40 mA current. Samples were placed on glass slides and were scanned in the range of 2θ = 4−50° at the scan rate of 0.5 s step−1 with a step width of 0.02°.Rheology
To understand the mechanical properties of the PT gel, a rheological experiment was performed with an advanced rheometer (AR 2000, TA Instrument, USA) using cone plate geometry on a Peltier plate. The diameter of the plate was 40 nm and the angle was 4° with a plate gap of 121 μm. Both frequency sweep and temperature ramp experiments were performed.Conclusions
PT complexes produce hydrogels for PT12, PT13 and PT14 compositions but the PT11 complex does not form a gel due to significantly reduced amount of T which causes lower π-stacking than found in the former systems. All of the PT gels and PT11 complexes produce fibrillar network morphologies and the gel melting temperature follows the order PT14 > PT13 > PT12. H-bonding and electrostatic interactions are responsible for the PT complex formation and NMR spectra suggest the presence of an appreciable amount of π–π stacking in the gels. The frequency independent G′ and G′′ characterize the systems as gels and both the G′ and G′′ values follow the order PT14 > PT13 > PT12 gels. The critical yield stress values for the gels are 6.0, 3.18 and 0.76 Pa for PT14, PT13 and PT12, respectively, suggesting the strength and stability of the gels increase with an increase in T concentration. The red shift of the absorption peaks of the PT gels compared with that of P is attributed to the J-aggregate formation. The emission peaks also show a red shift due to the stabilization of excitons during J-aggregate formation and emission intensity increases significantly in the gels due to both hydrophobic core and J-aggregate formation. The PL-intensity gradually decreases in all the PT gels due to de-stacking of J-aggregates with an increase of temperature but the emission peak position shows an interesting step like increase. The batch wise slipping of PT motifs from the J-aggregates has been attributed to the step like increase of the emission peak.Acknowledgements
We gratefully acknowledge CSIR, New Delhi (Grant No. 01(2224)/08 EMR-11) and DST, New Delhi (grant no. SR/P1/PC/26/2009) for financial support. P. B. and B. R. acknowledge CSIR, for providing the fellowship.References
- (a) S. Yagai, M. Higashi, T. Karatsu and A. Kitamura, Chem. Mater., 2004, 16, 3582 CrossRef CAS; (b) S. Yagai, H. Aonuma, Y. S. Kubota, T. Karatsu, A. Kitamura, S. Mahesh and A. Ajayaghosh, Chem.–Eur. J., 2010, 16, 8652 CrossRef CAS; (c) S. Mahesh, R. Thirumalai, S. Yagai, A. Kitamura and A. Ajayaghosh, Chem. Commun., 2009, 5984 RSC; (d) Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208 RSC; (e) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS; (f) A. R. Hirst and D. K. Smith, Chem.–Eur. J., 2005, 11, 5496 CrossRef CAS; (g) S. Yagai, M. Higashi, T. Karatsu and A. Kitamura, Chem. Mater., 2004, 16, 3582 CrossRef CAS; (h) M. George, G. P. Funkhouser and R. G. Weiss, Langmuir, 2008, 24, 3537 CrossRef CAS.
- (a) S. Manna, A. Saha and A. K. Nandi, Chem. Commun., 2006, 4285 RSC; (b) A. Saha, S. Manna and A. K. Nandi, Langmuir, 2007, 23, 13126 CrossRef CAS; (c) A. Saha, S. Manna and A. K. Nandi, Chem. Commun., 2008, 3732 RSC; (d) A. Saha, B. Roy, A. Garai and A. K. Nandi, Langmuir, 2009, 25, 8457 CrossRef CAS; (e) B. Roy, A. Saha, A. Esterrani and A. K. Nandi, Soft Matter, 2010, 6, 3337 RSC; (f) P. Bairi, B. Roy and A. K. Nandi, J. Phys. Chem. B, 2010, 114, 11454 CrossRef CAS; (g) B. Roy, P. Bairi, A. Saha and A. K. Nandi, Soft Matter, 2011, 7, 8067 RSC.
- (a) E. Krieg, E. Shirman, H. Weissman, E. Shimoni, S. G. Wolf, I. Pinkas and B. Rybtchinsk, J. Am. Chem. Soc., 2009, 131, 14365 CrossRef CAS; (b) S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Roger, F. Wurthner, N. Sakai and S. Matile, Science, 2006, 313, 84 CrossRef CAS; (c) F. Wu1rthner, B. Hanke, M. Lysetska, G. Lambright and G. S. Harms, Org. Lett., 2005, 7, 967 CrossRef CAS; (d) X. Zhang, Z. Chen and Frank Wurthner, J. Am. Chem. Soc., 2007, 129, 4886 CrossRef CAS; (e) A.P. H. J. Schenning, J. V. Herrikhuyzen, P. Jonkheijm, Z. Chen, F. Wurthner and E. W. Meijer, J. Am. Chem. Soc., 2002, 124, 10252 CrossRef CAS.
- (a) K. Peneva, G. Mihov, A. Herrmann, N. Zarrabi, M. Börsch, T. M. Duncan and K. Müllen, J. Am. Chem. Soc., 2008, 130, 5398 CrossRef CAS; (b) Y. Zhaoa, X. Zhang, D. Li, D. Liu, W. Jiang, C. Han and Z. Shi, Luminescence, 2009, 24, 140 CrossRef CAS.
- J. V. Herrikhuyzen, A. Syamakumari, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2004, 126, 10021 CrossRef.
- F. Wurthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C-C. You, P. Jonkheijm, J. v. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611 CrossRef.
- B. Hernandez, F. Pfluger, A. Adenier, S. G. Kruglik and M. Ghom, J. Phys. Chem. B, 2010, 114, 15319 CrossRef CAS.
- K. H. Dittmann, C. Mayer and H. P. Rodemann, Int. J. Cancer, 2001, 96, 1 Search PubMed.
- J. Wachtveitl, J. W. Farchaus, P. Mathis and D. Oesterhelt, Biochemistry, 1993, 32, 10894 Search PubMed.
- J. Zhang, D. S. Guo, L. H. Wang, Z. Wang and Y. Liu, Soft Matter, 2011, 7, 1756 RSC.
- (a) K. V. Rao, K. Jayaramulu, T. K. Maji and S. J. George, Angew. Chem., Int. Ed., 2010, 49, 4218 CrossRef CAS; (b) K. V. Rao, K. K. R. Datta, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2011, 50, 1179 CrossRef CAS.
- Z. Yang, H. Gu, Y. Zhang, L. Wanga and B. Xu, Chem. Commun., 2004, 208 RSC.
- (a) H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4267 Search PubMed; (b) K. Liu, C. Wang, Z. Li and X. Zhang, Angew. Chem., Int. Ed., 2011, 50, 1 CrossRef; (c) J. L. Lpez, C. Atienza, W. Seitz, D. M. Guldi and N. Martn, Angew. Chem., Int. Ed., 2010, 49, 9876 Search PubMed.
- (a) J. M. Berg, J. L. Tymoczko, L. StryerBiochemistry 5-th ed. W .H. Freeman and Co Search PubMed; (b) D. Jang, H. Y. Lee, M. Park, S. R. Nam and J.-I. Hong, Chem. Eur. J., 2010, 16, 4836 CAS; (c) M. Barthes, A. F. Vik, A. Spire, H. N. Bordallo and J. Eckert, J. Phys. Chem. A, 2002, 106, 5230 CrossRef CAS.
- (a) R. N. Mitra, D. Das, S. Roy and P. K. Das, J. Phys. Chem. B, 2007, 111, 14107 CrossRef CAS; (b) T. Yajima, G. Maccarrone, M. Takani, A. Contino, G. Arena, R. Takamido, M. Hanaki, Y. Funahashi, A. Odani and O. Yamauchi, Chem.–Eur. J., 2003, 9, 3341 CrossRef CAS.
- (a) S. E. Wheeler and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10854 CrossRef CAS; (b) A. Saha, B. Roy, A. Esterrani and A. K. Nandi, Org. Biomol. Chem., 2011, 9, 770 RSC.
- F. Li, Y. Zhu, B. You, D. Zhao, Q. Ruan, Y. Zeng and C. Ding, Adv. Funct. Mater., 2010, 20, 669 CrossRef CAS.
- (a) C. Daniel, C. Dammer and J. M. Guenet, Polymer, 1994, 35, 4243 CrossRef CAS; (b) R.G. Weiss, P. TerechMolecular Gels: Materials with Self Assembled Fibrillar Network; ed.; Springer; Dordrecht, 2006 Search PubMed; (c) A. Garai and A. K. Nandi, J. Nanosci. Nanotechnol., 2008, 8, 1842 CrossRef CAS; (d) A. Garai and A. K. Nandi, J. Polym. Sci., Part B: Polym. Phys., 2008, 46, 28 CrossRef CAS.
- X. Rena, W. Yub, Z. Zhangb, N. Xiab, G. Fub, X. Lua and W. Wang, Colloids Surf., A, 2011, 375, 156 Search PubMed.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Wurthner, Angew. Chem., Int. Ed., 2008, 47, 3367 CrossRef CAS.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS.
- M. Sikorski, E. Sikorska, A. Koziolowa, R. G. Moreno, J. L. Bourdelande, R. P. Steer and F. Wilkinson, J. Photochem. Photobiol., B, 2001, 60, 114 CrossRef CAS.
- (a) N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528 CrossRef CAS; (b) P. Xue, R. Lu, X. Yang, L. Zhao, D. Xu, Y. Liu, H. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2009, 15, 9824 CrossRef CAS; (c) H. Wang, D. Pang, H. Xin, M. Li, P. Zhang and W. Tian, Liq. Cryst., 2006, 33, 439 CrossRef CAS.
- (a) C. Bao, R. Lu, M. Jin, P. Xue, C. Tan, G. Liu and Y. Zha, Org. Biomol. Chem., 2005, 3, 2508 RSC; (b) B. K. An, D. S Lee, J. S. Lee, Y. S. Park, H. S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232 CrossRef CAS.
- J. W. Chung, B. K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00506e |
| This journal is © The Royal Society of Chemistry 2012 |