An efficient hybrid design to prepare highly dense imprinted layer-coated silica particles for selective uptake of trace metsulfuron-methyl from complicated matrices
Ying
Xie
ab,
Danjun
Chen
a,
Jiawei
Zhao
a,
Yan
Peng
a,
Nan
Jiang
a,
Xuemin
Zhou
a,
Shuhu
Du
*a and
Zhongping
Zhang
*c
aSchool of Pharmacy, Nanjing Medical University, Nanjing, 210029, China. E-mail: shuhudu@njmu.edu.cn; Fax: +86-025-86862762; Tel: +86-025-86862762
bThe Third Affiliated Hospital of Xuzhou Medical College, Xuzhou, 221009, China
cInstitute of Intelligent Machines, Chinese Academy of Sciences, Hefei, 230031, China. E-mail: zpzhang@iim.ac.cn
First published on 2nd November 2011
Abstract
This work presents an approach called “hybrid design” which combines “grafting from” and “grafting through” methods, in order to prepare highly dense imprinted layer-coated silica particles for improved binding capacity to metsulfuron-methyl (MSM). The imprinted conditions including the kind of solvent and molar ratio of template to functional monomer were carefully optimized by quantum mechanics method. The obtained imprinted materials were evaluated by transmission electron microscopy and rebinding experiments, exhibiting well-controlled shell thickness (up to ∼30 nm) and high binding affinity to MSM. Moreover, the rebinding amount of MSM-imprinted particles to MSM was nearly 1.6- and 1.4-folds that of normal surface-imprinted particles, respectively. When the MSM-imprinted particles were used as dispersive solid-phase extraction materials, the recoveries (RSDs) of MSM determined by high-performance liquid chromatography (HPLC) were 93.4% (1.5%), 86.8% (3.2%) and 88.4% (2.7%) in the spiked water, soil and wheat samples, respectively. Therefore, the method will provide new opportunities for enrichment and detection of trace MSM from complicated matrices.
1 Introduction
Sulfonylureas, such as metsulfuron-methyl (MSM), are environmentally deleterious substances that have been of societal security concern, because they are widely used for a variety of crops to control weeds.1 Compared with other herbicides, sulfonylureas have been used with a lower dose of 10–40 g h m−2,2 and are more rapidly degraded in soil.3 So the concentration of these herbicides is usually found very low (ppt range) in soil, water or corn samples. These greatly increase the difficulty of their detection in chromatographic analysis. Presently, the determination of sulfonylurea herbicides (SHs) mainly rely on high performance liquid chromatography (HPLC),4liquid chromatography-mass spectrometry (LC-MS),5capillary electrophoresis (CE),6,7gas chromatography (GC)8 and immunoassay9 method, which are usually of low efficiency owing to the lack of specific adsorptive materials. Moreover, the complicated matrices can seriously interfere with the enrichment and detection of SH residues to reduce the reliability and reproducibility of SH analysis. Thus, the exploration in enrichment and detection for trace SHs from environment or food has attracted considerable research interest in recent years. And, the stable antibody-like materials with specific binding properties have been developed. For example, molecular imprinting polymers (MIPs) with specific recognition sites may make effective enrichment of trace substances possible.Although molecular imprinting for the recognition of SHs has already been reported,10–12 the most widely used method is bulk polymerization. The obtained MIPs exhibit some disadvantages including incomplete template removal, slow mass transfer and irregular material shapes.13–15 To overcome the above drawbacks, the surface-imprinting method has recently been put forward.16–24 In the imprinting process, the surface of inorganic materials are generally functionalized with polymer chains either by the “grafting from” and “grafting to” or “grafting through” methods. As the “grafting from” method, the initiator groups were immobilized at the surface of supports. The imprinting polymerization could be carried out at the surface of initiator-modified support particles suspended in a mixture of the monomers and solvent. A high grafting density of polymer chains can be achieved because the steric barrier to incoming polymers imposed by the in situ grafted chains does not limit the access of smaller monomer molecules to the active initiation sites.25–30 However, as the initiator is bound to the surface of the substrates only by one end, and the second part of the initiator can diffuse into solution and start a polymerization reaction there, not all polymers which had formed are attached to the substrate, leading to a low graft ratio and low yield of imprinted sites.31 In the manner of “grafting to”, often end-functionalized polymers are first synthesized and then react with appropriate surface sites. Another straightforward technique, which was similar to the “grafting to”, is the “grafting through” approach. But the link of polymer chains or networks to the surface is a random process in the “grafting through” manner.32–34 Once the surface becomes significantly covered, additional polymer molecules, which are trying to reach the surface, have to diffuse against the concentration gradient built up by the already deposited polymer chains. This leads to a strong kinetic and steric hindrance for the attachment of additional polymer chains to the surface and impedes further imprinted layer growth. Therefore, the formation of surface-bound polymer monolayers by such a “grafting through” method is intrinsically limited to low graft density and low imprinted layer thickness.35
Compared with traditional MIPs, surface-imprinting polymers have several advantages, such as the complete removal of templates, good accessibility to the target species, and low mass-transfer resistance. However, the surface-imprinting materials have limited (or few) imprinted sites, resulting in a low rebinding capacity.
In this paper, an efficient hybrid design which combines “grafting from” and “grafting through” methods was firstly developed to prepare highly dense imprinted layer-coated silica particles for improved binding capacity to MSM. Simultaneously, a quantum mechanics method was used to predict the suitable polymerization solvent and the molar ratio of template to functional monomer, and to identify functional monomers capable of interacting with MSM. The volume ratio of two silylated reagents was carefully optimized. The obtained MSM-imprinted particles showed higher binding capacity, compared to normal surface-imprinted particles.
2 Experimental
2.1 Chemicals
3-Aminopropyltriethoxysilane (APTS), 3-(methacryloxy)propyltrimethoxysilane (MPTS), trimethylolpropane trimethacrylate (TRIM) and 4,4′-azobis(4-cyanopentanoic acid) (ACPA) were purchased from Sigma-Aldrich (Steinheim, Germany). Tetraethylosilicate (TEOS), ammonia, azo(bis)-isobutyronitrile (AIBN), methacrylic acid (MAA), thionyl chloride and acetic acid were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Analytical grade toluene, acetonitrile, dichloromethane, chloroform, methanol and ethanol were purchased from Nanjing Chemical reagent Co., Ltd. (Nanjing, China). AIBN was purified by recrystallization from ethanol before use. All the other chemicals were used as received. Metsulfuron-methyl (MSM), chlorsulfuron (CS), bensulfuron-methyl (BSM), tribenuron-methyl (TBM) and thifensulfuron-methyl (TSM) were purchased from Sigma-Aldrich (Steinheim, Germany). The methanol of HPLC grade was purchased from Merck (Darmstadt, Germany). Commercial C18 was from YMC Co. Ltd. (Shimogyo-ku, Japan). HPLC water was doubly distilled. Tap water samples were collected from a suburb of Nanjing in China. Soil samples were from a suburb of Nanjing in China. Wheat samples were from a farm of Xuzhou in China.The individual stock (10 mmol L−1) solutions were prepared in chloroform, and the standard solutions of lower concentration were prepared by the serial dilution of stock solutions. All stock and standard solutions were stored at −18 °C in a refrigerator and re-prepared every month.
2.2 Apparatus and analytical methods
The instruments used in this study were as follows: UV-2450 UV-vis spectrophotometer (Shimadzu, Kyoto, Japan), Bruker Vector 27 FT-IR spectrometer (Bruker, Ettlingen, Germany), JEM-1010 transmission electron microscope (JEOL, Tokyo, Japan), thermogravimetric analyzer Linseis L81 (Linseis, Selb, Germany) and a Shimadzu HPLC system (Shimadzu, Kyoto, Japan) equipped with LC-20AT pump, SPD-M20A detector, CTO-20A column oven and Shimadzu shim-pack C18 column (5 μm, 250 mm × 4.6 mm I.D.). The column temperature was 30 °C. The mobile phase was methanol/water (65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35, v/v), the pH of which was adjusted to 3.0 with acetic acid, at a flow rate of 1.0 mL min−1. MSM was monitored at 230 nm. The injection volume was 20 μL. Sample solutions were filtered through a nylon 0.22 μm filter before use.
:35, v/v), the pH of which was adjusted to 3.0 with acetic acid, at a flow rate of 1.0 mL min−1. MSM was monitored at 230 nm. The injection volume was 20 μL. Sample solutions were filtered through a nylon 0.22 μm filter before use.
2.3 Chlorination of 4,4′-azobis(4-cyanopentanoic acid) with thionyl chloride
ACPA (1.40 g) was dispersed in dichloromethane (150 mL), followed by the addition of thionyl chloride (2.0 mL) within 20 min. The reaction mixture was stirred for 12 h while in an ice bath under high-purity nitrogen. After reaction, the solvent was evaporated and 4,4′-azobis(4-cyanopentanoyl chloride) (ACPC) was finally obtained. The product was washed with dichlormethane to remove unreacted thionyl chloride, and then dried at 4 °C under vacuum overnight.2.4 Synthesis and chemical modification of silica particles
Subsequently, the amino end groups of APTS monolayer were further acryloylated by ACPC. Typically, ACPC (2.66 g) was dispersed in SiO2@(MPTS+APTS) toluene solution (50 mL), and anhydrous potassium carbonate was added into this reaction system as a catalyst. The mixture was vigorously stirred for 12 h while remaining in ice bath under high-purity nitrogen. The product was separated by centrifuge and washed with toluene, water and ethanol, in that order, and dried under vacuum at room temperature. Finally, SiO2@[MPTS+(APTS-ACPC)] was obtained.
The SiO2@MPTS was prepared by immobilized MPTS monolayer at the surface of silica particles. Typically, dried silica particles (0.3 g) and MPTS (6.0 mL) were added into toluene to make the mixture solution (50 mL). The mixture was refluxed for 12 h under high-purity nitrogen. The resultant SiO2@MPTS was separated by centrifuge and washed with toluene.
2.5 Molecular modeling studies
Quantum mechanics (QM) was employed to select the optimal solvent and molar ratio of template to functional monomer. The solvent effects on the electronic structures of the studied systems were evaluated through polarized continuum model (PCM) (dielectric constant ε = 35.69, 2.37, 4.71 and 7.43 for acetonitrile, toluene, chloroform and tetrahydrofuran, respectively). This model had been successfully employed to predict the surface morphology and molecular recognition properties of MIPs.37–39 The full geometry optimization of template, monomer and their complexes with different ratios were carried out with density functional theory (DFT) at B3LYP/3-21G level. Then, the corresponding energetic calculations were performed at the B3LYP/6-31G(d,p) level. The binding energy (ΔEbind) of MSM with the monomer in solvent was calculated according to the following formula:| ΔEbind = Ecomplex – Etemplate – ΣEmonomer | (1) |
2.6 Preparation of MSM-imprinted particles
Prior to polymerization, the prearranged solution was prepared by MSM (0.625 mmol) and MAA (2.5 mmol) dissolved in 25 mL of mixture solvent with toluene/acetonitrile (4:1, v/v) and placed in the dark for 12 h. Subsequently, 0.1 g of SiO2@(APTS-ACPC), SiO2@MPTS and SiO2@[MPTS+(APTS-ACPC)] particles were dispersed in 25 mL of mixture solvent with toluene/acetonitrile (4:1, v/v) by ultrasonic vibration, respectively. The obtained three mixture solutions were mixed with the prearranged solution, TRIM (2.5 mmol) and different amounts of AIBN (i.e., 0 mmol, 0.10 mmol and 0.06 mmol), respectively. And the mixing solutions were purged with high-purity nitrogen while cooled in ice bath. The polymerization was first done at 50 °C for 6 h, then at 60 °C for 24 h. The products were further aged at 85 °C for 6 h obtaining a high cross-linking density,20 and separated by centrifuge and rinsed with methanol, respectively. To remove template molecules, the imprinted particles were ultrasonically cleaned by methanol/acetic acid (9:1, v/v) and dried under vacuum at room temperature. The obtained MSM-imprinted particles were termed SiO2@MSM-MIP1, SiO2@MSM-MIP2 and SiO2@MSM-MIP3.
The non-imprinted particles (SiO2@NIP1, SiO2@NIP2 and SiO2@NIP3) were also prepared using an identical procedure but without the addition of a template.
2.7 Binding properties of MSM-imprinted particles
Twenty milligrams of MSM-imprinted particles (SiO2@MSM-MIP1, SiO2@MSM-MIP2 and SiO2@MSM-MIP3) and non-imprinted particles (SiO2@NIP1, SiO2@NIP2 and SiO2@NIP3) were suspended in chloroform (5.0 mL) with various MSM concentrations (0.3–10 mmol L−1), and the samples were shaken on a reciprocating shaking-table at room temperature, respectively. The particles in the solution were removed through a 0.22 μm microporous membrane after reaching adsorption equilibrium, and the equilibrium concentrations of MSM-imprinted and non-imprinted particles were determined with UV-vis spectrophotometer. The equilibrium adsorption amounts of MSM-imprinted and non-imprinted particles to MSM were calculated according to eqn (2), the binding isotherm was figured. The specific recognition property of MSM-imprinted particles was evaluated by imprinting factor (α), which was calculated according to eqn (3), and the binding constants of binding sites of MSM-imprinted particles were calculated according to the Langmuir eqn (4). | (2) |
 | (3) |
 | (4) |
Meanwhile, the binding kinetics was tested by monitoring the temporal evolution of MSM concentration in the solutions. 20 mg of MSM-imprinted particles (SiO2@MSM-MIP1, SiO2@MSM-MIP2 and SiO2@MSM-MIP3) and non-imprinted particles (SiO2@NIP1, SiO2@NIP2 and SiO2@NIP3) were suspended in chloroform (5 mL) with MSM (2 mmol L−1) solution, shaken on a reciprocating shaking-table at room temperature, respectively. The mixtures (including MSM-imprinted/non-imprinted particles and chloroform with MSM solutions) were taken suction after 5, 10, 15, 20, 30, 45, 60 and 120 min, then filtered through 0.22 μm microporous membranes. And the concentrations of MSM in the filtrate were determined with a UV-vis spectrophotometer. The dynamic binding curve was plotted.
The selectivity of SiO2@MSM-MIP3 was measured using the structure analogues CS, BSM, TSM and TBM. 20 mg of SiO2@MSM-MIP3 particles and SiO2@NIP3 particles were suspended in chloroform (5.0 mL) with MSM, CS, BSM, TSM and TBM individual solutions at the 2 mmol L−1 level, respectively. The equilibrium concentrations of SiO2@MSM-MIP3 and SiO2@NIP3 were determined with UV-vis spectrophotometer, and the equilibrium adsorption amounts of SiO2@MSM-MIP3 and SiO2@NIP3 were also calculated according to eqn (2). The selectivity factor (β) was defined as expressed in eqn (5).
 | (5) |
2.8 Detection of MSM from the spiked samples
The wheat and soil were taken and crushed into homogenates. The homogenates (20 g) of wheat and soil were mixed 20 mL of MSM standard solution (0.015 μmol L−1), incubated for 10 h and filtered, respectively. The filtrate was evaporated to dryness. Then, the residue was re-dissolved in chloroform (5.0 mL) to form the spiked samples. Water samples (20 mL) were spiked at a concentration of 0.015 μmol L−1 with MSM. Subsequently, the mixture was filtered, evaporated to dryness under vacuum and then the residue was re-dissolved in chloroform (5.0 mL). So, the concentration of MSM in the three spiked samples was about 0.060 μmol L−1.
SiO2@MSM-MIP3, SiO2@NIP3 and commercial C18 (20 mg) were dispersed in the spiked solutions (5.0 mL) and incubated for 60 min at room temperature. The particles were collected through a 0.22 μm microporous membrane, washed with chloroform (3.0 mL) to reduce nonspecific adsorption, and then eluted with 5.0 mL of methanol/acetic acid (9:1, v/v). The particles in the desorption solution were separated through 0.22 μm microporous membrane. The filtrate was dried by nitrogen and re-dissolved in methanol (0.5 mL), and then analyzed with HPLC by the procedures and conditions as mentioned in Section 2.2.
3 Results and discussion
3.1 Preparation and characterization of modified silica particles
To obtain MSM-imprinted particles with high binding capacity, two silylated reagents (APTS and MPTS) were simultaneously used in this work. The amount of APTS and MPTS modified at the surface of silica particles is a key factor for imprinting performance. At low graft density, few recognition sites caused by the low degree of cross-linking decrease rebinding capacity. With the increasing of graft density, the high degree of cross-linking may enhance the difficulties of the accessibility for template molecules in polymerization. Therefore, a suitable amount of grafting yield is critical for maximum adsorption capacity. With the fixed amount of silica particles (0.3 g) and the total volume of APTS and MPTS (6.0 mL), the volume ratio of MPTS to APTS from 1.0:2.0 to 3.0:1.0 was optimized using rebinding capacities. As shown in Table 1, at the region of low values of volume ratio, the rebinding capacities increased as the value of the volume ratio was increased. When the ratio was 2.0:1.0, the rebinding capacity of SiO2@MSM-MIP to MSM reached to the maximum at 117.0 μmol g−1. Further increasing the volume ratio led to the decrease of rebinding capacity. This is likely because the dense vinyl groups decreased the steric space and the polymer chains could not easily access the reactive sites to copolymerize the with cross-linker. Thus, a ratio of 2.0:1.0 of MPTS to APTS was chosen to obtain SiO2@MSM-MIP with the maximum adsorption capacity.
| Volume ratio of MPTS to APTS | Rebinding capacity (μmol g−1) | RSD (%) (n = 5) |
|---|---|---|
| 1.0:2.0 |
29.14 | 3.3 |
| 1.0:1.0 |
56.69 | 2.3 |
| 1.7:1.0 |
101.7 | 1.7 |
| 2.0:1.0 |
117.0 | 1.5 |
| 2.3:1.0 |
100.6 | 2.1 |
| 3.0:1.0 |
45.02 | 2.7 |
Fig. 1 showed the infrared spectra of SiO2@(APTS-ACPC) (a), SiO2@MPTS (b), SiO2@[MPTS+(APTS-ACPC)] (c) and pure silica (d). Compared with the infrared data of pure silica, the SiO2@(APTS-ACPC) displayed a characteristic peak at 1715 cm−1 (due to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in amide group). In Fig. 1(b), the CO band centered at 1735 cm−1 (typical stretching vibration of CO in ester group). The characteristic peaks of SiO2@[MPTS+(APTS-ACPC)] appeared at 1710 cm−1 and 1737 cm−1 (attributed to the stretching vibration of CO in amide group and ester group, respectively). These characteristic peaks found in SiO2@(APTS-ACPC), SiO2@MPTS and SiO2@[MPTS+(APTS-ACPC)] verified the successful grafting of the silane coupling agents or azo-initiators to the surface of particles.
O in amide group). In Fig. 1(b), the CO band centered at 1735 cm−1 (typical stretching vibration of CO in ester group). The characteristic peaks of SiO2@[MPTS+(APTS-ACPC)] appeared at 1710 cm−1 and 1737 cm−1 (attributed to the stretching vibration of CO in amide group and ester group, respectively). These characteristic peaks found in SiO2@(APTS-ACPC), SiO2@MPTS and SiO2@[MPTS+(APTS-ACPC)] verified the successful grafting of the silane coupling agents or azo-initiators to the surface of particles.
![FTIR spectra of SiO2@(APTS-ACPC) (a), SiO2@MPTS (b), SiO2@[MPTS+(APTS-ACPC)] (c) and pure silica (d).](/image/article/2012/RA/c1ra00438g/c1ra00438g-f1.gif) | ||
| Fig. 1 FTIR spectra of SiO2@(APTS-ACPC) (a), SiO2@MPTS (b), SiO2@[MPTS+(APTS-ACPC)] (c) and pure silica (d). | ||
3.2 Molecular modeling of template–monomer interaction
Currently, a typical imprinting protocol, such as the selection of functional monomer and polymerization solvent, is tedious and time-consuming. Now, we have developed a method, i.e., quantum mechanics (QM) for selecting the most suitable solvent and optimizing the optimal molar ratio of template to functional monomer.In order to understand the electric structure of MSM molecule, the Mulliken charges are investigated at B3LYP/6-31G(d,p) level and the results are displayed in Fig. 2a. It can be seen that there were several potential binding sites on the MSM molecule, which can interact with the MAA molecule through hydrogen bonding or electrostatic interactions. For example, the three nitrogens of the triazine ring and the four oxygens with negative partial charges in the MSM molecule were likely to become the hydrogen bond acceptors for the hydrogen atoms on the hydroxyl group of the MAA molecule (Fig. 2b). Because of the higher negative charges on the nitrogen atoms of the triazine ring than the oxygen atoms, the nitrogen atoms were firstly selected as the most preferable binding site for MAA.
 | ||
| Fig. 2 The charges obtained by molecular modeling of atoms on the MSM molecule (a), MAA molecule (b) and the complex formed between the MSM molecule and four molecules of MAA (c). Nitrogen is represented in blue, carbon in gray, oxygen in red, hydrogen in white and sulphur in yellow. | ||
To select the optimal solvent, the binding energies (ΔEbind) of MSM with the monomer in various solvents, including acetonitrile, toluene, chloroform and tetrahydrofuran, were compared. As shown in Table 2, the interaction energies between MSM and MAA in different solvents were ordered as follows: ΔEbind (acetonitrile) > ΔEbind (toluene) > ΔEbind (chloroform) > ΔEbind (tetrahydrofuran). It indicated that the MSM–MAA complexes in acetonitrile are more stable than those in toluene, chloroform and tetrahydrofuran. Furthermore, because the boiling point (b.p.) of acetonitrile, chloroform and tetrahydrofuran are 81 °C, 61 °C and 65 °C, respectively, which were lower than that of the aged temperature (85 °C) of the polymers, only toluene (b.p. 110 °C) can be used as the solvent. However, the MSM molecule was difficult to dissolve in toluene. So, a mixture of toluene/acetonitrile (4:1, v/v) was chosen as the polymerization solvent.
| Solvent | E template (a.u.) | E monomer (a.u.) | E complex (a.u.)a | ΔEbind (kcal mol−1) |
|---|---|---|---|---|
|
a The Ecomplex was the total energy of template-monomer complex with the molar ratio of 1:3.
|
||||
| Acetonitrile | −1656.74226 | −304.78948 | −2571.23402 | −77.39 |
| Toluene | −1656.72606 | −304.79397 | −2571.21701 | −68.42 |
| Chloroform | −1656.73351 | −304.79550 | −2571.22466 | −65.66 |
| THF | −1656.73688 | −304.79616 | −2571.22821 | −64.53 |
In addition, the influence of molar ratio between MSM and MAA are also studied by QM method. The MSM-MAA complexes with different molar radios were investigated in toluene. The detailed geometric parameters and the interaction energies were given in Fig. 2c and Table 3, respectively. It can be seen that MSM–MAA complexes with molar radios of 1:1, 1:2, 1:3 and 1:4 are formed through intermolecular hydrogen bonding interactions. The average hydrogen bonding lengths (angle) are 1.57 Å (174.9°), 1.57 Å (172.8°), 1.60 Å (167.4°) and 1.64 Å (167.2°), respectively. With the addition of MAA molecules, there are more hydrogen bonds between MSM and MAA, resulting in the increased ΔEbind. Further additions of MAA will not lead to a greater level of complexation. So, the optimal molar ratio of MSM to MAA was 1:4.
| MSM:MAA | Intermolecular distance (Å) | Intermolecular angle (°) | Binding energy (kcal mol−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r N1⋯H2 | r O5⋯H1 | r O2⋯H3 | r O1⋯H4 | r O3⋯H5 | Average | ∠N1⋯H2-O4 | ∠O5⋯H1-N2 | ∠O2⋯H3-O6 | ∠O1⋯H4-O7 | ∠O3⋯H5-O8 | Average | ΔEbind | |
| 1:1 |
1.54 | 1.59 | 1.57 | 176.4 | 173.3 | 174.9 | −77.39 | ||||||
| 1:2 |
1.55 | 1.59 | 1.56 | 1.57 | 176.2 | 172.1 | 168.8 | 172.8 | −68.42 | ||||
| 1:3 |
1.56 | 1.57 | 1.57 | 1.68 | 1.60 | 176.0 | 172.6 | 169.1 | 151.0 | 167.4 | −65.66 | ||
| 1:4 |
1.58 | 1.57 | 1.60 | 1.80 | 1.64 | 1.64 | 176.1 | 171.4 | 168.4 | 157.8 | 166.6 | 167.2 | −64.53 |
3.3 Preparation of MSM-imprinted particles
Recently, several methods have been adopted to increase the thickness of the MIPs layer coated on silica particles to improve the rebinding capacity of imprinted particles to template molecules, for instance, by tuning the amount of surface end vinyl groups36 and by controlling the ratio of acrylamide-APTS-silica to the polymerization precursors.20 But the critical scale of the shell with the most imprinted sites was only ∼25 nm. Thus, in this work, we attempted a novel and straightforward strategy for the preparation of MSM-imprinted particles through combining “grafting from” and “grafting through” methods.Here, the organic modifying layer on the silica support is very important. For SiO2@(APTS-ACPC), APTS-ACPC with an azo group (initiator) was introduced on the surface of silica particles. When polymerization occurred at the silica particle’s surface, only one end of the initiator was bound to the surface, the second part dispersed into solution and started a polymerization reaction and formed some nonbonded polymers (as indicated with swallow tail arrow in the inset TEM image of Fig. 3A). Thus, the formation of surface-bound polymers by the “grafting from” technique was intrinsically limited to low graft ratio and thin shell thickness (Fig. 3A-a).
 | ||
| Fig. 3 Outline of fixation of different functional monomer at silica particles and the coating MIP layer at silica particles by the “grafting from” method (A), the “grafting through” method (B) and the combination of “grafting from” and “grafting through” methods (C). SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c). | ||
For SiO2@MPTS, MPTS with an active end group was modified onto the surface of silica particles, ensuring that the imprinting polymerization selectively occurred at the particle's surface. With the growth of surface polymer chains, additional polymers had difficultly reaching the silica particle’s surface due to steric crowding (as indicated with curved arrow in Fig. 3B). This impeded further the MIPs layer growth.36 So, the surface-imprinted layer formed by the “grafting through” technique contained a limited number of recognition sites and its thickness was thin (Fig. 3B-b).
For SiO2@[MPTS+(APTS-ACPC)], APTS-ACPC and MPTS were simultaneously modified on the surface of silica particles. And the copolymers at the silica particle’s surface included two parts. One part (polymer chains or netwoks) which had mainly formed by the “grafting through” method, entered in the space of the substrate's surface, as indicated with hollowed arrow in Fig. 3C. The other formed in the manner of “grafting from” distributed in a far space from the surface, as indicated with solid black arrow in Fig. 3C. Compared with SiO2@MPTS, the vinyl groups on the SiO2@[MPTS+(APTS-ACPC)] surface were not so crowded that most of the polymer chains were attached to the surface. The nonbonded polymers in solution could enter reactive sites at the silica particle’s surface (as indicated with curved arrow in Fig. 3C), crosslinked with excessive vinyl groups. Therefore, both the graft density and graft ratio of the polymers were improved. As a result, the imprinted layer of SiO2@MSM-MIP3 contained so many recognition sites and was thick (Fig. 3C-c). The mechanism mentioned above is only qualitative. Detailed quantitative analysis including the determination or characterization of shell thickness, weight loss, binding capacity and binding affinity of the three imprinted materials can be seen in Section 3.4 and 3.5.
3.4 Characterization of MSM-imprinted particles
The obtained SiO2@MSM-MIP1, SiO2@MSM-MIP2 and SiO2@MSM-MIP3 were characterized by transmission electron microscopy (TEM) and thermogravimetric analysis (TGA), respectively.By TEM observation (Fig. 4), the thicknesses of imprinted layer at the surface of SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c) were about 18 nm, 20 nm and 30 nm, respectively.
 | ||
| Fig. 4 TEM images of SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c). | ||
Additionally, Fig. 5 displayed the TGA curves of SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c). TGA curves of SiO2@MSM-MIP1 (a) and SiO2@MSM-MIP3 (c) approximately plateaued at the range 100–150 °C, showing weight losses of 5.7 and 5.5%, respectively, due to the thermal decomposition of the remaining azo groups and the evaporation of the adsorbed water.41 In a successive heating from 150 to 800 °C, the steep decrease was attributed to the further thermal decomposition of the imprinted layer. And the weights (wt%) of imprinted layers of SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c) were about 7.4%, 9.2% and 12.8%, respectively. The above data showed that the thicker the imprinted layer is, the higher the weight of the imprinted layer is, possibly resulting in the higher binding capacity.
 | ||
| Fig. 5 Thermogravimetic weight loss curves of SiO2@MSM-MIP1 (a), SiO2@MSM-MIP2 (b) and SiO2@MSM-MIP3 (c). | ||
3.5 Binding properties of MSM-imprinted particles
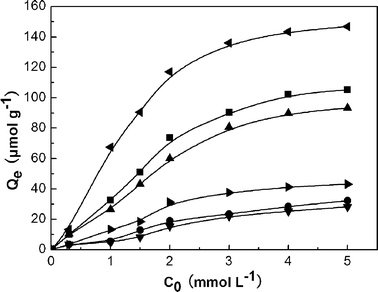 | ||
| Fig. 6 Rebinding capacity curves of SiO2@MSM-MIP1 (▲), SiO2@NIP1 (▼), SiO2@MSM-MIP2 (■), SiO2@NIP2 (●), SiO2@MSM-MIP3 (◀) and SiO2@NIP3 (▶) to MSM. The amounts of rebinding MSM were measured by suspending 20 mg of materials in 5 mL of solution with different MSM concentrations. The adsorption time was 60 min. | ||
| Particles | Q max (μmol g−1) (RSD%) | α a | K e (L mmol−1)b | K s (μmol g−1)b | r2 |
|---|---|---|---|---|---|
| a The imprinted factor (α) was calculated from maximum adsorption amounts of SiO2@MSM-MIP (QMIP) and that of SiO2@NIP (QNIP): α = QMIP/QNIP. b The equilibrium binding constant (Ke), saturation constant (Ks) of higher affinity binding sites were calculated by Langmuir equation: 1/Qe = 1/KeKsCe + 1/Ks, having a slope of (KeKs)−1 and intercept of Ks−1. | |||||
| SiO2@MSM-MIP1 | 93.22 (1.5) | 3.31 | 4.057 | 13.23 | 0.99 |
| SiO2@NIP1 | 28.20 (3.1) | 1.567 | 3.569 | 0.97 | |
| SiO2@MSM-MIP2 | 105.1 (1.2) | 3.26 | 4.435 | 13.50 | 0.99 |
| SiO2@NIP2 | 32.23 (2.3) | 1.752 | 3.855 | 0.99 | |
| SiO2@MSM-MIP3 | 146.7 (1.0) | 3.41 | 6.338 | 19.72 | 0.99 |
| SiO2@NIP3 | 43.05 (2.9) | 2.021 | 4.647 | 0.98 |
To obtain further knowledge of MSM binding on MSM-imprinted polymers, the Langmuir equation was employed to evaluate the binding affinity of the imprinted polymers.42 A plot of Qe−1versus Ce−1 gave a straight line having a slope of (KeKs)−1 and intercept of Ks−1. From the slope and intercept of the plot, Ke and Ks of higher affinity binding sites were calculated. Meanwhile, Ke and Ks values are summarized in Table 4. From the data of Table 4, it is evident that the imprinted polymers have a higher binding affinity to MSM than non-imprinted polymers, and MSM interacts more strongly with SiO2@MSM-MIP3 compared to SiO2@MSM-MIP1 and SiO2@MSM-MIP2.
The above results indicate that there were more recognition sites in SiO2@MSM-MIP3 than that in SiO2@MSM-MIP1 and SiO2@ MSM-MIP2. This was because the imprinted layer of SiO2@MSM-MIP3 was the thickest among three kinds of MSM-imprinted particles (as shown in Fig. 4). In other words, the thicker the imprinted layer is, the more imprinted sites there are, resulting in the higher binding affinity and binding capacity.
 | ||
| Fig. 7 Adsorption time curves of SiO2@MSM-MIP1 (▲), SiO2@NIP1 (▼), SiO2@MSM-MIP2 (■), SiO2@NIP2 (●), SiO2@MSM-MIP3 (◀) and SiO2@NIP3 (▶) to MSM. The amounts of rebinding MSM were measured by suspending 20 mg of materials in 5 mL of 2 mmol L−1MSM solution at different adsorption time. | ||
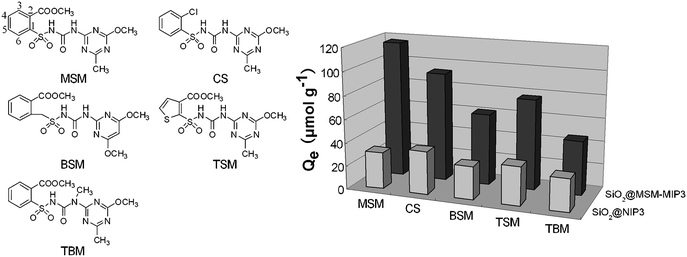 | ||
| Fig. 8 Binding selectivity of SiO2@MSM-MIP3 and SiO2@NIP3. The measurements were carried out by suspending samples (20 mg) in chloroform (5 mL) with MSM, CS, BSM, TSM and TBM individual solutions at the 2 mmol L−1 level. Before the measurement, the samples were incubated for 60 min at room temperature on a reciprocating shaking-table. | ||
| Compounds | Q MIP (μmol g−1) (RSD%) | Q NIP (μmol g−1) (RSD%) |
|---|---|---|
| a The individual solution concentration of MSM, CS, BSM, TSM, and TBM used for binding capacities was 2 mmol L−1. | ||
| MSM | 117.0 (1.5) | 31.24 (3.2) |
| CS | 93.10 (2.0) | 36.41 (2.9) |
| BSM | 61.24 (2.4) | 28.40 (3.3) |
| TSM | 77.11 (1.9) | 32.85 (3.1) |
| TBM | 46.17 (2.8) | 27.73 (3.4) |
In contrast, the rebinding capacities of SiO2@NIP3 to target analytes were much lower than those of SiO2@MSM-MIP3, and SiO2@NIP3 did not exhibit the obvious difference in the rebinding capacities to MSM, CS, BSM, TSM and TBM. Thus, the small binding capacities of SiO2@NIP3 to target compounds mainly depended on the physical adsorption. These comparisons further demonstrate that the MSM-imprinted layer at the surface of silica particles have high molecular selectivity for the target species.
3.6 Selective enrichment of MSM from the spiked samples
To assess the application of SiO2@MSM-MIP3 to selective enrichment of MSM, the wheat, soil and water samples spiked with MSM (0.060 μmol L−1) were analyzed. Simultaneously, SiO2@NIP3 and commercial C18 were also investigated for comparison. As can be clearly seen from Fig. 9d, no MSM was detected in the three spiked samples due to low concentration. After the dispersive solid-phase extraction by SiO2@MSM-MIP3, the peak of MSM sharply increased (Fig. 9a) and the enriched concentration of MSM was high enough to be quantitatively analyzed. Under the same conditions, SiO2@NIP3 and commercial C18 did not show a selectively enriched ability for MSM due to no specific binding sites existing on them, so only a small quantity of MSM was detected after extraction by SiO2@NIP3 (Fig. 9b) and commercial C18 (Fig. 9c). Most of the MSM was washed during the chloroform cleaning up process, thus, higher sensitivity can be achieved by using SiO2@MSM-MIP3 as the extraction sorbent. Furthermore, recoveries of MSM in the spiked water, soil and wheat samples were 93.4%, 86.8%, 88.4%; the RSDs were 1.5%, 3.2%, 2.7%, respectively. The results clearly demonstrate that the SiO2@MSM-MIP3 can be effectively applied in the enrichment and detection of trace MSM in environment and foodstuff. | ||
| Fig. 9
HPLC
chromatograms of spiked water, soil and wheat samples. (a) extracted with SiO2@MSM-MIP3; (b) extracted with SiO2@NIP3; (c) extracted with commercial C18; (d) spiked solution containing 0.060 μmol L−1MSM. Experimental conditions: 5.0 mL of spiked solutions; 20 mg of SiO2@MSM-MIP3; wash solution, 3.0 mL of chloroform; eluent solution, 5.0 mL of methanol/acetic acid (9:1, v/v); re-dissolve solution, 5.0 mL of methanol. HPLC conditions: mobile phase, methanol/water (65:35, v/v), the pH of which was adjusted to 3.0 with acetic acid; flow rate, 1.0 mL min−1; column temperature, 30 °C; UV detection, at 230 nm. | ||
Conclusions
In summary, an effective method was developed for coating silica particles with a molecularly imprinted polymervia a hybrid strategy. As a model, the organic MPTS+(APTS-ACPC) monolayer at the surface of silica particles can not only direct the selective occurrence of imprinting polymerization at the surface of supports, but also drive template molecules into the formed polymer shells during the imprinting process. The resultant SiO2@MSM-MIP3 particles show some attractive characteristics, such as uniform morphology, controlled shell thickness (up to ∼30 nm) and higher binding capacity or binding affinity. It has clearly been shown that the combination of “grafting from” and “grafting through” methods can more significantly improve the rebinding capacity of the imprinted materials to MSM (up to 146.7 μmol g−1), indicating the success of the hybrid imprinting technique used in this study. In addition, the analytical method based on SiO2@MSM-MIP3 was successfully applied to MSM analysis in spiked water, soil and wheat samples with higher selectivity and sensitivity. The approach reported herein could be potentially exploited in detecting the other environmentally deleterious chemicals.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No.21075066 and 20875048), and the National 863 High Technology Project of China (No. 2008AA10Z421).References
- J. F. Liu, J. B. Chao, G. B. Jiang, Y. Q. Cai and J. M. Liu, J. Chromatogr., A, 2003, 995, 21–28 CrossRef CAS.
- K. V. Penmetsa, R. B. Leidy and D. Shea, J. Chromatogr., A, 1997, 790, 225–234 CrossRef CAS.
- A. D. Corcia, C. Crescenzi, R. Samperi and L. Scappaticcio, Anal. Chem., 1997, 69, 2819–2826 CrossRef CAS.
- E. Ayano, H. Kanazawa, M. Ando and T. Nishimura, Anal. Chim. Acta, 2004, 507, 211–218 CrossRef CAS.
- S. Polati, M. Bottaro, P. Frascarolo, F. Gosetti, V. Gianotti and M. C. Gennaro, Anal. Chim. Acta, 2006, 579, 146–151 CrossRef CAS.
- A. J. Krynitsky, J. AOCA Int, 1997, 80, 392 Search PubMed.
- G. Dinelli, A. Vicari and V. Brandolini, J. Chromatogr., A, 1995, 700, 201–207 CrossRef CAS.
- P. Klaffenbach and P. T. Holland, J. Agric. Food Chem., 1993, 41, 396 CrossRef CAS.
- S. A. Eremin, I. A. Ryabova, J. N. Yakovleva, E. V. Yazynina, A. V. Zherdev and B. B. Dzantiev, Anal. Chim. Acta, 2002, 468, 229–236 CrossRef CAS.
- Q. Z. Zhu, K. Haupt, D. Knopp and R. Niessner, Anal. Chim. Acta, 2002, 468, 217–227 CrossRef CAS.
- J. Bastide, J. P. Cambon, F. Breton, S. A. Piletsky and R. Rouillon, Anal. Chim. Acta, 2005, 542, 97–103 CrossRef CAS.
- K. J. Tang, S. W. Chen, X. H. Gu, H. J. Wang, J. Dai and J. Tang, Anal. Chim. Acta, 2008, 614, 112–118 CrossRef CAS.
- K. Haupt, Anal. Chem., 2003, 75, 376A–383A CAS.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- E. Yilmaz, K. Haupt and K. Mosbach, Angew. Chem., Int. Ed., 2000, 39, 2115–2118 CrossRef CAS.
- C. Sulitzky, B. Rückert, A. J. Hall, F. Lanza, K. Unger and B. Sellergren, Macromolecules, 2002, 35, 79–91 CrossRef CAS.
- F. G. Tamayo, M. M. Titirici, A. M. Esteban and B. Sellergren, Anal. Chim. Acta, 2005, 542, 38–46 CrossRef CAS.
- M. Quaglia, E. D. Lorenzi, C. Sulitzky, G. Massoloni and B. Sellergren, Analyst, 2001, 126, 1495–1498 RSC.
- M. Quaglia, E. D. Lorenzi, C. Sulitzky, G. Caccialanza and B. Sellergren, Electrophoresis, 2003, 24, 952–957 CrossRef CAS.
- D. M. Gao, Z. P. Zhang, M. H. Wu, C. G. Xie, G. J. Guan and D. P. Wang, J. Am. Chem. Soc., 2007, 129, 7859–7866 CrossRef CAS.
- X. G. Hu, J. L. Pan, Y. L. Hu, Y. Huo and G. K. Li, J. Chromatogr., A, 2008, 1188, 97–107 CrossRef CAS.
- L. Schweitz, Anal. Chem., 2002, 74, 1192–1196 CAS.
- J. J. Ou, X. Li, S. Feng, J. Dong, X. L. Dong and L. Kong, Anal. Chem., 2007, 79, 639–646 CrossRef CAS.
- Y. Peng, Y. Xie, J. Luo, L. Nie, Y. Chen, L. N. Chen and S. H. Du, Anal. Chim. Acta, 2010, 674, 190–200 CrossRef CAS.
- M. Ejaz, S. Yamamoto, K. Ohno, Y. Tsujii and T. Fukuda, Macromolecules, 1998, 31, 5934–5936 CrossRef CAS.
- X. Huang and M. J. Wirth, Macromolecules, 1999, 32, 1694–1696 CrossRef CAS.
- K. Matyjaszewski, P. J. Miller, N. Shukla, B. Immaraporn, A. Gelman and B. B. Luokola, Macromolecules, 1999, 32, 8716–8724 CrossRef CAS.
- J. B. Kim, M. L. Bruening and G. L. Baker, J. Am. Chem. Soc., 2000, 122, 7616–7617 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- W. Feng, J. Brash and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2931–2942 CrossRef CAS.
- O. Prucker and J. Rühe, Macromolecules, 1998, 31, 592–601 CrossRef.
- S. Mona and P. Sébastien, Nat. Chem., 2010, 2, 811–820 CrossRef CAS.
- N. B. Zhitenev, A. Sidorenko, D. M. Tennant and R. A. Cirelli, Nat. Nanotechnol., 2007, 2, 237–242 CrossRef CAS.
- C. D. Grande, M.C. Tria1, M.J. Felipe, F. Zuluaga and R. Advincula, Eur. Phys. J. E, 2011, 34, 15–24 CrossRef.
- Y. Lyatskaya and A. C. Balazs, Macromolecules, 1998, 31, 6676–6680 CrossRef CAS.
- Q. Lu, X. M. Chen, L. Nie, J. Luo, H. J. Jiang, L. N. Chen and S. H. Du, Talanta, 2010, 81, 959–966 CrossRef CAS.
- B. Sellergren and K. J. Shea, J. Chromatogr., A, 1993, 635, 31–49 CrossRef CAS.
- L. Wu, K. Zhu, M. Zhao and Y. Li, Anal. Chim. Acta, 2005, 549, 39–44 CrossRef CAS.
- R. H. Schmidt, A. S. Belmont and K. Haupt, Anal. Chim. Acta, 2005, 542, 118–124 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, J. R. Cheeseman, Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Czaun, L. Hevesi, M. Takafuji and H. Ihara, Macromolecules, 2009, 42, 4539–4546 CrossRef CAS.
- J. J. Hsu, I. B. Huang, C. C. Hwang and M. C. Wu, Indian J. Chem. Technol., 2011, 18, 7–12 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |