Colloidal calcium nanoparticles: digestive ripening in the presence of a capping agent and coalescence of particles under an electron beam†
Udishnu
Sanyal
a,
Ranjan
Datta
b and
Balaji R.
Jagirdar
*a
aDepartment of Inorganic & Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: jagirdar@ipc.iisc.ernet.in; Fax: +91-80-2360 1552; Tel: +91-80-2293 2825
bInternational Centre for Materials Science, Jawaharlal Nehru Centre for Advanced Scientific Research, Jakkur P.O., Bangalore 560064, India
First published on 2nd November 2011
Abstract
The nanochemistry of calcium remains unexplored, which is largely due to the inaccessibility of calcium nanoparticles in an easy to handle form by conventional methods of synthesis as well as its highly reactive and pyrophoric nature. The synthesis of colloidal Ca nanoparticles by the solvated metal atom dispersion (SMAD) method is described. The as-prepared Ca–THF nanoparticles, which are polydisperse, undergo digestive ripening in the presence of a capping agent, hexadecyl amine (HDA) to afford highly monodisperse colloids consisting of 2–3 nm sized Ca–HDA nanoparticles. These are quite stable towards precipitation for long periods of time, thereby providing access to the study of the nanochemistry of Ca. Particles synthesized in this manner were characterized by UV-visible spectroscopy, high resolution electron microscopy, and powder X-ray diffraction methods. Under an electron beam, two adjacent Ca nanoparticles undergo coalescence to form a larger particle.
Introduction
Nanostructuring of most metals in the size regime of 2–10 nm, where the quantum confinement effects predominate,1 could be realized; however, the same is rather challenging in the case of alkaline earth metals. This is primarily due to their high reduction potential, which makes it difficult to use the conventional chemical reduction methods and very high oxophilicity, especially in the size regime of 2–10 nm that they pose difficulties in handling such materials. Efforts on the synthesis of alkaline earth metal nanoparticles have been largely dominated by magnesium from the standpoint of hydrogen storage.2,3 The nanochemistry of the other alkaline earths, wherein the metal is in its elemental form, remains unexplored. This is primarily due to the inaccessibility of these materials in an easy to handle form by conventional methods of synthesis as well as their highly reactive and pyrophoric nature. Studies on calcium have either been limited to matrix isolation spectroscopy of Ca, Ca2, and Cax4–8 or theoretical aspects of hydrogen storage on calcium decorated graphene.9,10 In recent years, there has been increased interest on developing systems with high gravimetric storage of hydrogen. First principles plane wave calculations by Ciraci and co-workers predict a hydrogen storage capacity of 8.4 wt% on calcium atoms adsorbed on graphene.11 Herein, we report the synthesis of highly monodisperse colloidal calcium nanoparticles by the solvated metal atom dispersion (SMAD) method. These colloids are quite stable towards precipitation of particles, thereby providing easy handling of these highly reactive materials.Results and discussion
A synthetic protocol of broad applicability, solvated metal atom dispersion (SMAD) process, involving the growth of clusters from atoms in low temperature matrices,12 in combination with a process termed as digestive ripening was used earlier by us for the synthesis of hexadecyl amine (HDA) capped 2–3 nm Mg nanoparticles.13 The synthesis procedure involves co-condensation of metal atoms and a stabilizing organic solvent on the walls of a reactor maintained at 77 K. Warming the matrix results in a slurry of metal atoms weakly solvated by the organic solvent molecules. The metal atoms interact with one another resulting in the growth of the particles. Halting of the growth is achieved by the addition of an organic ligand/surfactant. This, the as-prepared colloid consisting of polydisperse nanoparticles, could be transformed into a colloid of highly monodisperse nanoparticlesvia the digestive ripening process. This process involves heating the as-prepared colloid at or near the boiling point of the solvent;14,15 it takes place even at room temperature as demonstrated by us13 and by Klabunde and co-workers.16 Although not a fully understood process, digestive ripening renders the breakdown of larger particles and the growth of smaller ones until the sample becomes homogeneous and a dynamic equilibrium is reached. The strengths of the SMAD process, in combination with the digestive ripening, include easy scale-up, high reproducibility, avoidance of tedious purification procedures, and more importantly a colloid consisting of highly monodisperse nanoparticles. Motivated by the advantages that the SMAD method offers, we attempted to prepare colloids of hitherto unknown calcium nanoparticles.We prepared calcium nanoparticles of 8 nm in diameter by the SMAD method. In a typical experiment, Ca atoms and THF were co-condensed on the walls of a SMAD reactor at 77 K over a period of 3 h. A dark blue colored matrix, which we believe is a mixture of Ca atoms, calcium dimers and oligomers was obtained. Once the metal vaporization was complete, the matrix was slowly warmed to room temperature under an argon atmosphere. During the warm up procedure, the dark blue matrix gave a dark blue colloid, which finally turned to an orange colored colloid within 15 min. The colloid was stirred for 1 h before it was siphoned into a Schlenk tube under Ar for further characterization. The colloid was stable towards precipitation of particles for 2–3 h after which a small amount of precipitate was noted. The precipitate could be redispersed easily upon sonication (90 W, 15 min). Fig. 1 shows different stages of the SMAD experiment.
 | ||
| Fig. 1 Different stages of the preparation of colloids of Ca nanoparticles by the SMAD method. | ||
The UV-visible spectrum of the calcium colloid recorded under an Ar atmosphere showed a broad band at 460 nm (Fig. 2a). The absorption spectra of atomic Ca as well as Ca2, which is purely a van der Waals species, both stabilized by cryogenic inert gas matrix, were studied extensively in the 1970s.4–8 Atomic calcium is characterized by a strong absorption due to the 4s 4p ← 4s transition (1P1 ← 1S0) around 410 nm in an argon matrix; in the gas phase, this band appears as an asymmetric doublet at 422.7 nm and another band at 456 nm, which has been assigned to the forbidden 1D1 ← 1S0 atomic transition.6 The calcium dimer Ca2 exhibits an interesting 12-membered band centered at 648 nm with a spacing of 113 cm−1 between each member; this band has been assigned to the 1Σ+u (1S + 1P) ← 1Σ+g (1S + 1S) transition. In addition to this band, two weaker bands at 500 nm and 374 nm were also noted for Ca2. The higher aggregate Cax showed an absorption band at 484 nm; the value of x, however, could not be deciphered. Theoretical calculations on calcium nanoparticles with a diameter of 10 nm have been predicted to exhibit a surface plasmon resonance band at 480 nm.17 Based on this, we assign the band at 460 nm (Fig. 2a) to the SPR of the Ca(0) nanoparticles. The blue shift in our case compared to the calculated one could be ascribed to the presence of smaller sized particles, whereas, the broad nature of the band could be attributed to the polydispersity of the sample. Exposure of this sample to air rendered the orange colloid, colorless instantaneously. The UV-visible spectrum of this sample was featureless.
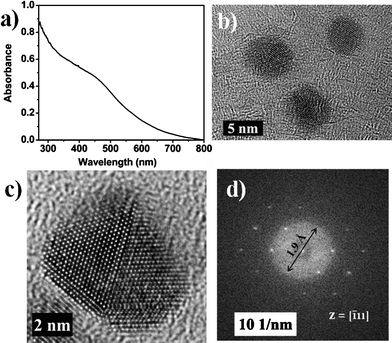 | ||
| Fig. 2 Ca–THF colloid: (a) UV-visible spectrum; (b) HRTEM image; (c) atomic resolution TEM image; (d) FFT pattern. | ||
Calcium nanoparticles were precipitated by centrifugation in an inert atmosphere glove box. The brown calcium nanopowder could be redispersed in THF by sonication. This powder was found to be highly pyrophoric and must be stored under a dry atmosphere of an inert gas. The shape, size, and the crystal structure of the Ca nanoparticles were established using an aberration corrected FEI TITAN3™ 80–300 kV microscope operating at 80 kV. The TEM bright field image of the Ca–THF sample revealed the presence of well separated Ca nanoparticles. The particles were quite polydisperse in nature (2–10 nm) and almost all of them were found to be nearly spherical (Fig. 2b) except for a few that possessed irregular shape. The average particle size was estimated to be 8.3 ± 1.0 nm. The polycrystalline nature of the sample was quite apparent from the atomic resolution TEM image (Fig. 2c). The lattice fringes measured to be 1.9 Å, could be assigned to the <220> plane of pure fcc phase of Ca(0). The FFT pattern (Fig. 2d) generated from Fig. 2b further lends support for the presence of pure Ca(0). A small amount of CaO was also found in this sample. Partial oxidation takes place while mounting the TEM sample in the instrument, which incidentally has to be done in air within a period of a minute and cannot be avoided. The powder XRD pattern of the Ca–THF nanopowder loaded in a 0.5 mm diameter quartz capillary inside the glove box and flame sealed under Ar was found to be broad and featureless. The powder sample was found to be pyrophoric in nature and readily gives Ca(OH)2 upon exposure to air/moisture, which was apparent from the brown sample turning colorless. The powder XRD pattern of the hydrolyzed sample was comprised of peaks assignable to Ca(OH)2 (JCPDS-841276) (see ESI†).
A colloid of Ca nanoparticles that is stable towards precipitation of particles and towards oxidation/hydrolysis would be extremely useful. Such a colloid could be handled easily for a variety of purposes. We noted that Ca nanoparticles with THF as the sole stabilizer cannot prevent agglomeration of particles with time and thus the colloid was not stable towards precipitation of particles. Complete precipitation of nanoparticles took place in ca. 6–7 h. This could be circumvented by employing a ligand with a long alkyl chain, hexadecyl amine (HDA), as the capping agent. We took HDA at the bottom of the SMAD reactor and carried out the experiment with THF as the dispersing solvent. Here, a blue colored matrix, which upon melting and warm up to room temperature followed by vigorous stirring gave a clear and homogeneous orange colored colloid that was stable towards precipitation of particles for a period as long as one week.
The UV-visible spectrum (Fig. 3a) of the Ca–HDA–THF colloid recorded under Ar showed a well defined, relatively sharp band compared to that of Ca–THF nanoparticles at 325 nm. Exposure of this orange colored colloid to air rendered it colorless accompanied by the disappearance of the 325 nm band (Fig. 3a).
 | ||
| Fig. 3 Ca–HDA–THF nanoparticles: (a) UV-visible spectra; (b) HRTEM image; (c) atomic resolution TEM image; (d) FFT pattern; (e) powder XRD pattern. | ||
The high resolution TEM micrograph (Fig. 3b) of Ca–HDA–THF colloid revealed the presence of highly monodisperse, well separated, spherical 2–3 nm sized particles suggesting that digestive ripening had taken place when the colloid was stirred with HDA at room temperature. The lattice fringes of 2.0 Å obtained from the HRTEM image evidenced the presence of the <220> plane of FCC calcium. The atomic resolution image of a ∼2 nm particle (Fig. 3c) clearly shows the single crystalline nature of the sample. The corresponding FFT pattern (Fig. 3d) further corroborates the d-spacing of pure Ca(0) (JCPDS 230430). The powder XRD pattern of Ca–HDA–THF (Fig. 3e) exhibits peaks corresponding to the FCC phase of Ca(0). In addition, we also noted peaks for CaH2 (JCPDS 731391). The origin of CaH2 could be from the C–H bond activation within HDA or THF. Although Mendoza and Tacke reported the formation of (C4H3O)CaHvia C–H bond activation within furan, the hydride species has not been well characterized.18 The powder XRD of the sample showed peaks for both Ca(0) (FCC) and CaH2 (orthorhombic), we found no hydride particles in the TEM studies. This suggests that CaH2 is present in the bulk state together with Ca nanoparticles.
During the imaging of the particles, coalescence of a pair of calcium nanoparticles, that are adjacent to one another, takes place to form a larger particle under the electron beam. Fig. 4 shows a series of TEM images taken at approximately one minute intervals. These images clearly depict the evolution of a large particle from smaller particles. We monitored the morphological changes in the particles in areas 1 (comprised of 4 particles) and 2 (comprised of 2 particles). In the first image (Fig. 4a), two small particles in very close proximity were already conjoined, i.e., coalescence of the nanoparticles commences immediately upon exposure to the electron beam. Hexadecyl amine (HDA) has been used as the surfactant to stabilize the particles against aggregation. We propose the loss of HDA in the area of conjugation of the two particles when they are exposed to the electron beam. van Huis et al. reported the removal of surfactant prior to the coalescence of two PbSe nanoparticles. It was found that coalescence of particles occurred at a significantly faster rate as well as at much lower temperature by using a low boiling point surfactant.19 In the present case, two such conjoined particles could be seen in area 1. These two twins further come closer to form a larger particle (Fig. 4b–e), the diameter of which is ∼10 nm, approximately four times that of the individual small particles of diameter 2.5 nm (the average diameter). On the other hand, in area 2, an already conjoined twin (Fig. 4a) evolved into a nearly spherical particle (Fig. 4b–e). No phase changes were noted as a result of this transformation. During the process of coalescence, the shape and size of the particles change continuously. Recrystallization takes place as a result of migration of surface atoms in order to minimize the total surface free energy. This has been demonstrated in the case of coalescence of Pt nanocrystals by others.20 Atoms on the convex region of the surface possess higher chemical potentials compared to the concave region. Hence there will always be an atomic migration towards a neck region i.e., the joining point of the two particles,19,21 which results in a morphological evolution, which in our case is spherical. Even though no apparent phase changes were noted as a result of this process, we could observe the recrystallization of the conjoined particles after each coalescence event.
 | ||
| Fig. 4 TEM snapshots of coalescence of calcium nanoparticles. | ||
Detailed TEM analysis of our sample revealed that only calcium nanoparticles that are within 20 Å or less from one another coalesce. The coalescence of two particles is very fast so we found it difficult to take TEM images of two particles in close proximity as separate entities. Before the image could be captured, the two particles started undergoing coalescence. Thus, the requirement of a distance of 20 Å (length of the hexadecyl amine molecule) or less between two conjoining particles is a rough estimate. We believe that this is the critical inter-particle distance for two particles to conjoin in our case. Additionally, the hexadecyl amine chains between two conjoining particles have to be well packed. If the chains are end to end, the closest distance between the two particles would be greater than 20 Å, consequently, conjoining will not take place. In order to get an insight into the coalescence process, we are studying the effect of the chain length of the capping agent as well as employing surfactants bearing different functional groups. Studies toward this direction are in progress in our laboratories. The coalescence process is shown in Scheme 1.
 | ||
| Scheme 1 Schematic representation of the coalescence of two calcium nanoparticles under the electron beam. | ||
Conclusions
In conclusion, we have developed a protocol to synthesize colloids of calcium nanoparticles by the solvated metal atom dispersion method. Taking advantage of the digestive ripening process, highly monodisperse Ca–HDA–THF nanoparticles were obtained in gram scale quantities. These colloids are quite stable towards precipitation of particles under Ar, thereby providing easy handling of these highly reactive materials. Under the electron beam, two small particles that are adjacent to one another undergo coalescence to give a larger particle, which has been demonstrated by way of snapshots of the conjoining process. Overall, the SMAD method in combination with the digestive ripening process provides new possibilities for accessing materials in an easy to handle form that are hitherto not accessible by any other means and yields gram scale quantities of highly monodisperse colloids. In turn, research towards the development of such materials for hydrogen storage applications will get a greater boost.Experimental section
Materials
Calcium shots were purchased from Sigma Aldrich. Hexadecyl amine (HDA) was dried and degassed for 12 h at 100 °C. Tetrahydrofuran (THF) was dried over sodium-benzophenone. The solvents were degassed by several freeze-pump-thaw cycles. All glassware were dried in a hot air oven around 130 °C and evacuated when hot, just before use to ensure the removal of trace quantity of moisture.Instrumentation
The TEM images were acquired using a FEI TITAN3™ 80–300 kV instrument operating at 80 kV. Atomic resolution images of Ca nanoparticles were obtained using the FEI TITAN3™ 80–300 kV aberration corrected TEM with a negative spherical aberration coefficient (Cs) of ∼–16 μm and a positive defocus of about +9 nm. All the TEM samples were prepared inside a N2 filled glovebox by placing a 2 μL drop of the sample on a holy carbon grid. The powder X-ray diffraction measurements were carried out on samples placed in 0.7 mm diameter capillaries flame sealed under a N2 atmosphere using Bruker ADVANCE X-ray diffractometer using Cu Kα radiation. A Perkin Elmer Lambda 750 UV-visible spectrometer was used to record the UV-visible spectra of the Ca colloids.Preparation of the Ca–THF colloids by the SMAD method
The SMAD setup is described in detail in ref. 12. The tungsten crucible coated with alumina cement was connected between two water-cooled copper electrodes and a 3000 mL reactor vessel was connected to a reactor head that is equipped with copper electrodes. The entire setup was evacuated to 1–3 × 10−3 torr. The crucible was heated in steps and at each step the pressure was allowed to come down to 2–3 × 10−3 torr. The crucible was kept under gentle heating overnight. This curing process ensures the removal of moisture and other volatile impurities from the crucible. In a typical experiment, about 170 mg of Ca-metal was loaded in a tungsten crucible and the crucible was resistively heated by applying an appropriate voltage between the two water-cooled copper electrodes. The reactor walls were maintained at 77 K using a liquid nitrogen bath and precoated with 20 mL of THF before metal vaporization. The voltage was increased stepwise until metal vaporization began, which was apparent by the appearance of a blue color on the white matrix. The voltage was maintained at this point for 3 h and about 80–90 mL of THF was co-condensed on the walls of the reactor. Once the metal vaporization was completed, the liquid nitrogen dewar was removed and the matrix was allowed to warm up to room temperature slowly under an argon atmosphere. During the warming process, the blue colored matrix turned dark blue, which persisted during the melting and formed a dark blue colored colloid, which finally became orange within 15 min upon stirring. There was no further color change even after stirring for a long time. The colloid was finally siphoned to a Schlenk tube under Ar. The overall yield in this control experiment is ∼60%.This experiment was scaled up to a gram scale but at the expense of the particle size and the size distribution. This was necessary to complete the experiment in a reasonable period of time by increasing the rate of metal evaporation.
Preparation of the Ca–HDA–THF colloids by the SMAD method
To prepare Ca–HDA–THF colloids, about 1.5 g of HDA was taken at the bottom of the reactor and rest of the experiment was carried out in a similar manner to that of Ca–THF experiment. The molar ratio of Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HDA was maintained at ∼1:3. During the evaporation of the metal the color of the matrix was blue and upon warming of the matrix, followed by stirring of the resulting colloids for an hour, complete dissolution of HDA and ripening of the colloid took place. The color of the colloids also changed from dark blue to orange. The colloids were siphoned into a Schlenk tube under Ar. This orange colored, transparent, HDA capped Ca-colloid was found to be very stable towards precipitation of particles for nearly a week. The typical evaporation rate to realize stable Ca colloids is around 30–35 mg h−1.
:HDA was maintained at ∼1:3. During the evaporation of the metal the color of the matrix was blue and upon warming of the matrix, followed by stirring of the resulting colloids for an hour, complete dissolution of HDA and ripening of the colloid took place. The color of the colloids also changed from dark blue to orange. The colloids were siphoned into a Schlenk tube under Ar. This orange colored, transparent, HDA capped Ca-colloid was found to be very stable towards precipitation of particles for nearly a week. The typical evaporation rate to realize stable Ca colloids is around 30–35 mg h−1.
This experiment was scaled up to a gram scale. In order to complete the experiment in a reasonable period of time, the rate of metal evaporation was increased, however, it did not affect either the particle size or the size distribution.
Isolation of HDA capped Ca nanopowders
Ca–HDA–THF colloids were diluted with a large excess of degassed THF and centrifuged at 3000 rpm inside a glovebox for 2–3 h. The brownish black precipitate that resulted was washed several times with THF to remove the excess HDA. Finally, the powders were dried and stored inside the glovebox. These powders were found to be extremely pyrophoric in nature.Acknowledgements
We are grateful for the financial support from the Council of Scientific & Industrial Research, India. We thank Prof. C. N. R. Rao for allowing us to access the TEM facilities at JNCASR, Bangalore. US thanks CSIR, India for a fellowship.References
- G. Schmid, Chem. Rev., 1992, 92, 1709 CrossRef CAS.
- L. Schlapbach and A. Züttel, Nature(London). 2001, 414, 353 Search PubMed.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS.
- J. C. Miller, R. L. Mowery, E. R. Krausz, S. M. Jacobs, H. W. Kim, P. N. Schatz and L. Andrews, J. Chem. Phys., 1981, 74, 6349 CrossRef CAS.
- J. C. Miller, B. S. Ault and L. Andrews, J. Chem. Phys., 1977, 67, 2478 CrossRef CAS.
- J. E. Francis and S. E. Webber, J. Chem. Phys., 1972, 56, 5879 CrossRef CAS.
- L. Andrews, W. W. Dulley and L. Brewer, J. Mol. Spectrosc., 1978, 70, 41 CrossRef CAS.
- K. J. Klabunde and A. Whetten, J. Am. Chem. Soc., 1986, 108, 6529 CrossRef CAS.
- H. Lee, J. Ihm, M. L. Cohen and S. G. Louie, Nano Lett., 2010, 10, 793 CrossRef CAS.
- H. Lee, J. Ihm, M. L. Cohen and S. G. Louie, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115412 CrossRef.
- C. Ataca, E. Aktürk and S. Ciraci, Phys. Rev. B, 2009, 79(041406) Search PubMed.
- K. J. Klabunde, P. L. Timms, P. S. Skell and S. Ittel, Inorg. Synth., 1979, 19, 59 CAS.
- S. B. Kalidindi and B. R. Jagirdar, Inorg. Chem., 2009, 48, 4524 CrossRef CAS.
- X. M. Lin, C. M. Sorensen and K. J. Klabunde, J. Nanopart. Res., 2000, 2, 157 CrossRef CAS.
- K. J. Klabunde, C. M. Sorensen, S. I. Stoeva, B. L. V. Prasad, A. B. Smetana and X. M. Lin, In Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control, Part II Methodologies; C. Corrain, G. Schmid and N. Toshima, Ed.; Elsevier Science: Amsterdam, 2008; Chapter 11, po 233-252 Search PubMed.
- S. Cingarapu, Z. Yang, C. M. Sorensen and K. J. Klabunde, Inorg. Chem., 2011, 50, 5000 CrossRef CAS.
- J. A. Creighton and D. G. Eadon, J. Chem. Soc., Faraday Trans., 1991, 87, 3881 RSC.
- O. Mendoza and M. Tacke, J. Organomet. Chem., 2006, 691, 1110 CrossRef CAS.
- M. A. van Huis, L. T. Kunneman, K. Overgaag, Q. Xu, G. Pandraud, H. W. Zandbergen and D. Vanmaekelbergh, Nano Lett., 2008, 8, 3959 CrossRef CAS.
- H. Zheng, R. K. Smith, Y. –W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309 CrossRef CAS.
- T. H. Lim, D. McCarthy, S. C. Hendy, K. J. Stevens, S. A. Brown and R. D. Tilley, ACS Nano, 2009, 3, 3809 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Photographs of the colloids, UV-visible spectrum, TEM images and powder XRD patterns of the samples. See DOI: 10.1039/c1ra00468a/ |
| This journal is © The Royal Society of Chemistry 2012 |