On the organic/inorganic interface between mesoporous SBA-16 silica and its structural directing polymer: a combined FT-IR and solid state NMR study†
Fabio
Carniato
a,
Geo
Paul
a,
Chiara
Bisio
*ab,
Stefano
Caldarelli
c and
Leonardo
Marchese
a
aDipartimento di Scienze e Tecnologie Avanzate and Nano-SISTEMI Interdisciplinary Centre Università del Piemonte Orientale “A Avogadro”, V. le Teresa Michel 11, 15121, Alessandria, Italy. E-mail: chiara.bisio@mfn.unipmn.it; Fax: (+)39 0131360250; Tel: (+39) 0131360216
bISTM-CRN Istituto di Scienze e Tecnologie Molecolari, via G. Venezian 21, Milano, Italy
cAix Marseille Université, ISm2 UMR 6263, Campus de Saint Jérôme, Service 511 F-13013, Marseille, France
First published on 12th December 2011
Abstract
In the last years large attention was devoted to the synthesis and characterization of SBA-16 material focusing the interest on the formation mechanisms of copolymer micelles which drive the organization of the final siliceous mesostructure. However, there are relatively few reports on the block copolymer-silica self-assembly interaction phenomena which mainly occur at solid state and which help to understand how the copolymer is organized in the final solid sample. The physico-chemical properties at the interface between silica framework and triblock EO106PO70EO106 co-polymer in a SBA-16 material were investigated. In particular, the combination of infrared spectroscopy with solid state NMR allowed to obtain complementary information on how the surfactant co-polymer interacts with the SBA-16 surface silanols in the presence or absence of physisorbed water and to follow the evolution of the structural organization of the co-polymer, which depends on the hydration degree of the SBA-16 sample.
1. Introduction
Mesoporous silica materials have been widely studied because of their important role as molecular sieves, supports for catalysts1–4 and for their use in optical applications5–8 or for immobilization of biomolecules.9–12 Many synthetic methodologies have been investigated in order to tailor the pore size of mesoporous silica to specific uses and to maximize the stability of the final product.13 A large variety of surfactants, from molecules containing a hydrophilic head group and a long hydrophobic tail group to copolymers containing both hydrophilic and hydrophobic regions, have been used to drive the formation of inorganic mesostructures from solubilized precursors.14–16In the present work, mesoporous SBA-16 silica characterized by a Im3hm body-centred cubic structure with pores approximately of 65 Å in diameter connected through 25 Å wide windows and surface areas in excess of 1000 m2 g−1 was specifically studied. This material can be prepared using various synthetic procedures,15,17–20 which can drastically influence its final textural and morphological properties. If a ternary triblock copolymer/water/butanol system is used, and the synthesis is carried out at a lower acid concentration than in the traditional copolymer/water/HCl system, it is possible to enhance the structural order over the final product.17 The synthesis is driven by a self-assembling of the polymer in micelles, which minimizes the contact of its hydrophobic parts with the water solution and leads to the formation of the silica inorganic structure by hydrolysis of tetraethoxysilane (TEOS) at the water/polymer interface.
In the last years, a considerable number of papers have appeared focusing on the synthesis and characterization of SBA-16, including the mechanism of mesostructure formation.21–23 However, there are relatively few reports on the block copolymer–silica SBA-16 interface interactions in which it is stated that the interactions between the hydrophilic part of the surfactant, used as a structure directing agent, and the silica phase play a key-role in driving the formation of mesoporous structure in aqueous media.15 Nevertheless, a systematic study focused to the comprehension of the interactions of both hydrophilic and hydrophobic moieties of surfactant with the inorganic part of the solid can provide additional information on the formation of SBA-16 phase. Due to the complexity of this study, a multidisciplinary approach is required to collect information on the interactions existing at the interface between the organic surfactants and the silica phase.
In this context, we describe a combined study of Fourier transform infrared (FT-IR) and solid-state nuclear magnetic resonance (SS-NMR) spectroscopy, to assess the interactions occurring between the triblock copolymer EO106PO70EO106 and the SBA-16 siliceous framework. Particular emphasis is given to investigate the as-synthesised SBA-16 at different hydration levels, in order to clarify the role of water on the interactions occurring between the organic and inorganic phases.
2. Experimental details
Material synthesis
The synthesis of silica SBA-16 was made following a ternary system based on a previously described method reported in literature.17 A solution of 2.5 g of Pluronic F127 (EO106PO70EO106, by Sigma Aldrich) in a mixture of 120 g of distilled water, 5 g of concentrated HCl (37%) and 7.5 g of butanol was first prepared. After one hour stirring at 45 °C, 12 g of TEOS (98%, Sigma Aldrich) were added and the mixture stirred for a further 24 h. The step was followed by a hydrothermal treatment of 24 h at 100 °C under static conditions in a PTFE bottle. Drying of the as synthesized sample was performed at room temperature overnight (sample hereafter named as-SBA-16). A part of the dry sample was calcined under air flow by increasing the temperature 1.5 °C min−1 up to 550 °C and maintaining it at this temperature for 6 h (sample hereafter named calc-SBA-16).Characterization techniques
X-ray diffraction (XRD) patterns were obtained on a ARL XTRA48 diffractometer using Cu-Kα radiation (lambda = 1.54062 Å). The X-ray profiles were recorded at room temperature between 0.5 and 10° 2Theta with a step size of 0.02° and a rate of 1° min−1.High-resolution transmission electron micrographs were obtained with a JEOL 3010-UHR HRTEM operating at 300 kV. Samples were ultrasonically dispersed in isopropanol and a drop of the suspension was deposited on a copper grid covered with a lacey carbon film.
N2 physisorption measurements were carried out at −196 °C in the relative pressure range from 1×10−6 to 1 P/P0 by using a Quantachrome Autosorb1MP/TCD instrument. Prior to the analysis the samples were outgassed at 100 °C for 3 h (residual pressure lower than 10−6 Torr). Apparent surface areas were determined by using Brunauer-Emmett-Teller equation, in the relative pressure range from 0.01 to 0.1 P/P0. Pore size distributions were obtained by applying the NLDFT method (N2silica kernel based on a cylindrical pore model applied to the desorption branch).
The thermogravimetric analysis (TGA) was performed on a Q500 balance (TA Instrument) in N2 flow (20 mL min−1) from 50 to 800 °C (heating rate of 5 °C min−1).
Variable temperature IR analysis were performed on a FTIR Nicolet 5700 spectrometer (Thermo Optics) using a resolution of 4 cm−1. The samples were pressed in the form of self-supporting wafers and placed into a IR cell equipped with KBr windows permanently attached to high vacuum line (residual pressure: 1.0×10−6 Torr, 1 Torr = 133.33 Pa). The experimental setup allowed all temperature treatment experiments to be carried out in situ and in flowing conditions. Spectra were collected by heating the samples from room temperature to 600 °C (10 °C min−1) under nitrogen flow (20 mL min−1).
Prior to diethyl ether adsorption the sample was treated in vacuum at 300 °C for 3 h and then cooled down to RT. The ether adsorption was carried out at rt, by adsorbing increasing pressure of probe molecule (from 0.03 to 10 mbar).
All NMR spectra were acquired on a Bruker Avance III 500 spectrometer and a wide bore 11.7 Tesla magnet with operational frequencies for 1H, 29Si and 13C of 500.13, 99.35 and 125.77 MHz, respectively. A suite of one-dimensional (1D) and two-dimensional (2D) NMR were used, including 1D 1H MAS NMR, 29Si MAS NMR, 13C CPMAS NMR and 2D 1H–13C and 1H–29Si frequency switched Lee-Goldburg (CPFSLG) heteronuclear correlation (HETCOR) NMR.24 A 4 mm triple resonance probe with MAS was employed in all the experiments. The samples were packed on a Zirconia rotor and spun at a MAS rate between 5 and 15 kHz. The magnitudes of radio frequency fields were 100 and 42 kHz and the relaxation delays between accumulations were between 5 and 120 s for 1H and 29Si MAS NMR, respectively. For the 13C{1H} CPMAS experiments, the magnetic fields υrfH of 55 and 28 kHz were used for initial excitation and decoupling, respectively. During the CP period the 1H RF field υrfH was ramped using 100 increments, whereas the 13C RF field υrfC was maintained at a constant level. During the acquisition, the protons are decoupled from the carbons by using a TPPM decoupling scheme. A moderate ramped RF field υrfH of 62 kHz was used for spin locking, while the carbon RF field υrfC was matched to obtain optimal signal and the CP contact time of 2 ms was used. For the CPFSLG HETCOR experiment, the sample was packed in a ZrO2 HRMAS rotor and the volume was restricted to the middle of the rotor. A protonυrf of 100 kHz was used for FSLG and TPPM decoupling. These experiments were recorded with 512-1k scans, 32–64 rows, MAS rate of 12.5–13.84 kHz and a CP contact time of 2 ms. All chemical shifts were reported using δ scale and were externally referenced to TMS at 0 ppm.
3. Results and discussion
3.1 Physico-chemical properties of as-synthesized and calcined SBA-16
SBA-16 material, synthesized by using as structure directing agent (SDA) a poly(alkyleneoxide) triblock copolymer, EO106PO70EO106 (Pluronic F127), (hereafter named as-SBA-16) was firstly characterized, along with the calcined sample (hereafter named calc-SBA-16), by X-Ray diffraction, TEM microscopy and porosimetric analysis in order to investigate the structural and textural features of the material.The XRD pattern of as-SBA-16 (Fig. 1, (a)) showed a strong reflection at 0.83° (2Theta) with a large d-spacing of 10.6 nm, indexed as (110). After calcination at 550 °C in air for 6 h, the X-ray profile of the calc-SBA-16 became better resolved. Beside the (110) reflection, which became more intense, two weak signals at 1.5° and 1.72° 2θ have been observed, corresponding to (200) and (211) reflections of the SBA-16 cubic structure (Im3hm space group) (Fig. 1, (b)).
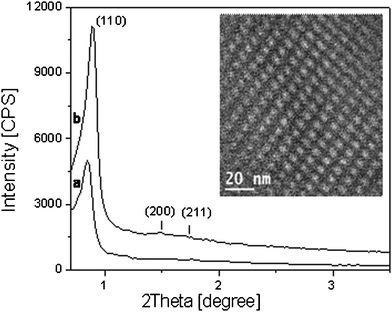 | ||
| Fig. 1 Powder X-ray diffraction patterns of as-synthesized (a) and calcined SBA-16 sample (b); TEM micrograph of calc-SBA-16 is reported in the panel. | ||
The mesoporous structure of the SBA-16 sample was also investigated by using high resolution transmission electron microscopy (HRTEM). A micrograph of the arrangement of SBA-16 pores array is shown in the inset of Fig. 1. A well-ordered cubic structure of the pores with ca. 5 nm in diameter was detected in agreement with the pore size distribution analysis (vide infra).
The textural properties of the as-synthesized and calcined SBA-16 have been also studied by nitrogen adsorption/desorption analysis and NLDFT calculations (Fig. 2). The isotherms of both samples were a mixture of type IV and I shape, with a clear hysteresis loop at high relative pressures indicating the presence of mesoporosity in the sample.15 The pore size of the material remained essentially unchanged upon calcination and a diameter distribution around 50 Å was obtained for both samples, while a distribution of smaller micropores and mesopores (10–35 Å) appeared only in the calcined sample (Fig. 2 B). In addition, the surface area of the as-SBA-16 drastically changed upon calcination, going from 300 m2 g−1 to 1020 m2 g−1 (Fig. 2 A). Additional textural details are reported in the Table 1.
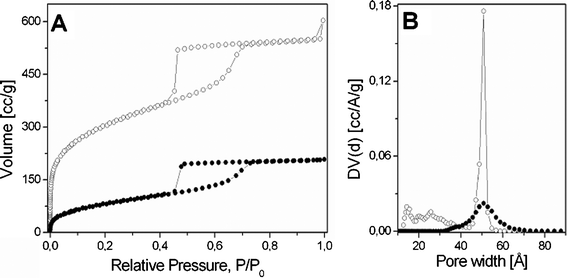 | ||
| Fig. 2 N2 adsorption-desorption isotherms at 77 K and relative pressures (P/P0) from 1×10−6 to 1 of as-synthesized (–•–) and calcined SBA-16 (–○–) (A); Pore size distribution, determined by NLDFT method, for the as-synthesized (–•–) and calcined SBA-16 (–○–) (B). | ||
| Sample | SBET m2 g−1 | Pores volume cm3 g−1 | Pores Diameter Å |
|---|---|---|---|
| as-SBA-16 | 300 | 0.31 | 51 |
| calc-SBA-16 | 1020 | 0.82 | 51 |
These results can be explained by assuming that, upon calcination, the pores are liberated from the polymer and made available to nitrogen adsorption, hence the dramatic increase of both surface area and mesopores volume observed along with the presence of microporosity.
The amount of copolymer present in the as-SBA-16 channels was estimated by thermogravimetric analysis (TGA). TGA profile, collected under oxygen flow, of as-SBA-16 was compared to that of calc-SBA-16 in order to assign the weight losses typical of the copolymer. The thermal profile of both samples highlighted a weight loss around 100 °C, which can be assigned to the presence of physisorbed water. Moreover, the water amount was significantly different in the two solids, going from 12 wt% for the calc-SBA-16 to 3 wt% for the as-SBA-16 silica.
In addition, the TGA profile of the as-SBA-16 evidenced a second important weight loss of ca. 35 wt% in the range 150–500 °C with a maximum centered at 180 °C (see derivative curves in the bottom panel of Fig. 3) which can be ascribed to the complete degradation of the PluronicF127 polymer.
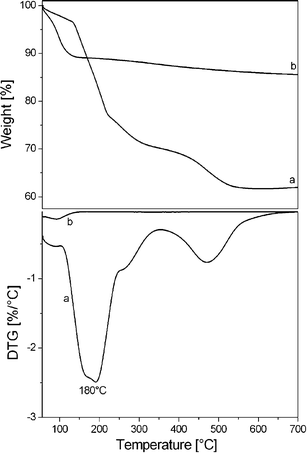 | ||
| Fig. 3 TGA measurements (10 °C min−1) under O2 flow for as-synthesized (a) and calcined SBA-16 (b); the derivative curves (DTG), are shown in the bottom panel. | ||
3.2 Monitoring the host–guest interactions in as-synthesized SBA-16: a combined FT-IR and SS-NMR study.
A detailed investigation of the interactions occuring at the interface between the silica framework and the Pluronic F127 in the as-SBA-16 was carried out by IR spectroscopy and mono- and bi-dimensional MAS and CP MAS solid state NMR experiments.The thermal decomposition of F-127 in as-SBA-16 sample was followed by variable temperature FT-IR, under N2 flow, recorded from room temperature (rt) to 600 °C (Fig. 4).
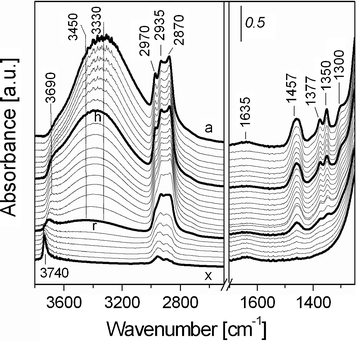 | ||
| Fig. 4 Infrared spectra at variable temperature of as-synthesized SBA-16 from (a) room temperature under nitrogen flow (20 mL min−1) to b) 50 °C, c) 75 °C, d) 100 °C, e) 125 °C, f) 150 °C, g) 175 °C, h) 200 °C, i) 225 °C, j) 250 °C, k) 275 °C, l) 300 °C, m) 325 °C, n) 350 °C, o) 375 °C, p) 400 °C, q) 425 °C, r) 450 °C, s) 475 °C, t) 500 °C, u) 525 °C, v) 550 °C, w) 575 °C and x) 600 °C. | ||
The spectrum collected at room temperature (Fig. 4, a) showed an intense broad band in the 3600–3100 cm−1 with a weaker broad component at ca. 3690 cm−1, together with narrower bands at 2970, 2935 and 2870 cm−1. The low frequency side in the spectrum of as-SBA-16 is characterized by the presence of bands at 1635 (weak and broad), 1457, 1377, 1350 and 1300 cm−1.
The broad band at high frequencies is due to the O–H stretching mode of the surface Si–OH species involved in hydrogen bonds with water (the bending mode falling at 1635 cm−1) and with moieties of the SDA polymer. The OH stretching mode of adsorbed water falls also in this region.
The absorption at 2970 cm−1 is assigned to the asymmetric stretching mode of CH3 group of the PPO unit, whereas the two bands at 2935 and 2870 cm−1 are due to stretching of CH and CH2 species of F127 block co-polymer. The band at 1457 cm−1 (broad) is due to bending modes of CH2 groups directly bound to oxygen atoms (of both PPO and PEO units) and of CH3 species of PPO, and the band at 1377 cm−1 can be related either to the bending mode (wagging) of CH2 species directly bound to the oxygen or to a bending mode of CH3 of PPO. Finally, the band at 1350 cm−1 is assigned to a bending mode of CH2 species bound to oxygen and the weak band at 1300 cm−1 associated to a bending vibration of CH groups.25
The treatment at 100 °C (Fig. 4, curve d) allowed to completely eliminate the adsorbed water, as witnessed by the diminishing of the band at 3600–3100 cm−1 range and of the peak at 1635 cm−1 (data in agreement with TGA analysis). Upon water removal, the band at 3600–3100 cm−1 became more asymmetric with two components at ca. 3450 and 3330 cm−1: it is proposed that these bands are associated to the interactions of surface silanols with ether oxygen atoms of the copolymer. This assignment is based on an adsorption experiment of a model molecule, diethyl ether, on calc-SBA-16 (vide infra).
Moreover, broad absorption at ca. 3500 cm−1, which is masked by the components at 3450 and 3330 cm−1, can be assigned to H-bonded vicinal silanol groups.26
By heating from 200 to 450 °C (Fig. 4, curves h–r), significant modifications of the PluronicF127 IR bands were observed. In particular, the bands at 2970, 1377 and 1350 cm−1 decreased in intensity together with the broad band in the high frequency range (3600–3100 cm−1). This behavior indicated that at 200 °C, Pluronic F127 starts to gradually decompose, as witnessed by the decrease of bands due to the stretching and bending modes of CH3 and CH2 groups and to the strong diminishing of the components at 3450 and 3330 cm−1. In this range of temperatures the bands at 3690 cm−1, which is due to surface silanols interacting with polymer alkyl groups, become progressively more evident.
After thermal treatment from 450 to 600 °C (Fig. 4, curves r–x), the bands of the alkyl groups of the Pluronic F127 almost completely disappear witnessing that the co-polymer practically fully decomposed in this range of temperatures. In parallel, a new sharp OH stretching band appeared at 3740 cm−1, which is assigned to the stretching vibration of isolated Si–OH groups, previously interacting with the polymer. The IR profiles, here presented, suggested the presence of significant interactions between surface SiOH groups and the Pluronic F127, probably occurring through both PEO hydrophilic groups and alkyl groups of the polymer, as confirmed by SS-NMR data below described.
Diethyl ether (Et2O) was used as model molecule adsorbed on calcined SBA-16 to evaluate the nature of the interactions of the hydrophilic PEO groups with the silica surface. The confirmation of hydrogen bonds formation with surface silanols has been obtained by using FT-IR spectroscopy (Fig. 5).
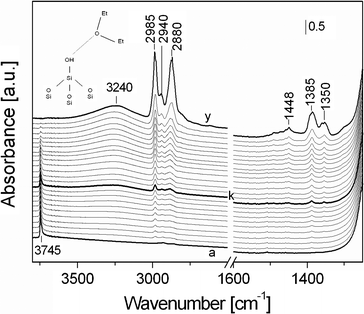 | ||
| Fig. 5 FTIR spectra of diethylether adsorbed at rt on calcined SBA-16. Spectrum a) was collected in vacuum after treating the sample at 300 °C for 3 h, spectra b–y) were registered upon adsorption of increasing pressure of Et2O, from 0.03 to 10 mbar (spectrum y). | ||
The spectrum of calc-SBA-16 upon thermal treatment (Fig. 5, a) showed a band at 3745 cm−1 due to the presence of isolated silanol species on the silica surface.
The Et2O adsorption results in the progressive formation of bands related to stretching and bending modes of the model molecule. In particular, bands at 2985, 2940 and 2880 cm−1 with a shoulder at 2850 cm−1 are clearly visible upon adsorption of low pressure of Et2O and can be assigned to the asymmetric stretching modes of –CH3 and –CH2groups, the asymmetric modes of –CH2groups and to the symmetric stretching vibrations of both –CH2 and CH3 species, respectively (the latter vibrations being overlapped are not distinguishable).
In addition, bands at 1448 (weak), 1385 and 1350 cm−1 are also present. The band at 1448 cm−1 is due to bending modes of CH2 and CH3 species, the peak at 1385 cm−1 is associated to either the bending mode (wagging) of CH2 species or a bending mode of CH3. The CH2 groups bound to the etheric oxygen have in addition a bending mode at 1350 cm−1.
For Et2O pressure higher than 0.5 mbar (Fig. 5, curve k), a broad band with maximum at ca. 3240 cm−1 is formed, with a parallel reduction of the intensity of the band at 3745 cm−1. This behavior is associated to the interaction of Et2O molecules with SiOH species, occurring through H-bonding. These interactions are particularly evident for high Et2O pressure (10 mbar, Fig. 5, curve y), where a complete erosion of the band at 3745 cm−1 is observed due to the fact that all silanols (both on the internal and external surface, see Fig. S1†) are bound to ether molecules by H-bonds of medium strength (shift for the stretching mode of surface OH groups of ca. 500 cm−1).27
The shift observed in the case of Et2O interaction is higher than those found for Pluronic F127, where two bands at 3450 and 3330 cm−1 were found (shift of 300–400 cm−1 range, see above Fig. 4), thus suggesting that Et2O has a stronger interaction than the polymer with the silica surface. This may depend on the fact that the oxygen atoms of the bulk Pluronic polymer could be sterically less accessible than those of the diethyl ether molecules.
Additional information of the interactions between Pluronic F127 and the silica were also obtained by solid-state NMR spectroscopy. Fig. 6 shows the CPMAS spectra of 13C (top) and 29Si MAS one (bottom) for as-SBA-16 in comparison to the dehydrated sample (named dehyd-SBA-16). The dehydration of the sample was carried out at 80 °C in vacuum conditions for 8 h. This dehydrated sample was analyzed to investigate if and how the presence of water molecules influences the interactions between the copolymer and the silica framework.
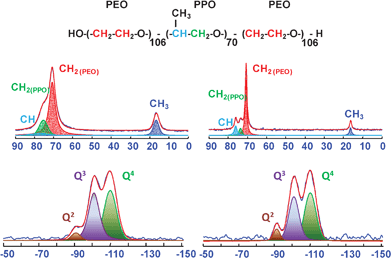 | ||
| Fig. 6 13C CPMAS (top) and 29Si MAS NMR spectra (bottom) of the as-SBA-16 (right) and dehyd-SBA-16 (left). | ||
In both cases, the carbon spectra bear close resemblance to previously reported ones of SBA-16 related materials.15,28 Thus, the resonances of the PPO block (CH3 at 16.3 ppm, CHO and CH2O at 76 and 73.5 ppm, respectively) and of the PEO one (CH2O at 70.6 ppm), can be distinguished in the as-SBA-16 (Fig. 6, right), while the CHO and CH2O peaks of PPO merge in the dehydrated sample (Fig. 6, left). This difference is probably due from a generalized increasing in the linewidth of all signals in going from as-SBA-16 to dehyd-SBA-16. Similar differences in linewidth have been observed in the literature in going from SBA-16 (broad) to SBA-16/THF films (narrower).29 The reason for this lineshape variation can be found in a change in the molecular mobility of F127 in the presence or in the absence of water. However, indistinguishable line widths of PPO and PEO block pairs in as-SBA-16 and dehyd-SBA-16 reflects a water controlled molecular environment in these materials and absence of microphase separations. 13C carbon signals have been deconvoluted and confirmed the expected intensity ratios, justified by the more abundant fraction of PEO (of about a factor of 6) in the F127 polymer, considering that the CP transfer function is likely to possess similar values for the CH2O groups in PPO and PEO moieties. This approximation is based on inspection of the 1H spectrum, which does not reveal severe changes in mobility between the PEO and the PPO block A quantitative 13C MAS NMR was also performed and confirmed the validity of this assumption (see Fig. S2†).
The distribution of surface silanol groups in both as-SBA-16 and dehyd-SBA-16 was evaluated by 29Si MAS NMR and 1H MAS NMR. The 29Si MAS spectrum of the as-SBA-16 sample exhibited three resolved signals at ca. −90, −101 and −111 ppm, corresponding to Q2, Q3 and Q4 units (Qn = Si(OSi)n(OH)4-n), respectively (Fig. 6, bottom). The collected data indicated that the dehydration step does not alter the distribution of silicon population, as no significant differences in the two 29Si spectra are observed.
The 1H MAS spectra (Fig. 7) reinforce the indications above described, showing peaks of both the F127 protons and the silanols, with additional information about water. The signal of this latter is the most evident difference between the spectra of as-SBA-16 (where it appears as a broad feature at 4.4 ppm) and dehyd-SBA-16 (where it is reduced beyond detection). With the exception of hydrogen bonded SiOH peak, which relocates from a broad feature at 6.1 ppm to a narrower resonance at 5.6 ppm upon dehydration, all remaining peaks broaden in the dehyd-SBA-16 spectrum, consistently with the observations concerning the 13C MAS signals. The presence of sharper peaks in the as-SBA-16 spectrum allows to distinguish two very close signals, at 3.7 and 3.6 ppm. A similar splitting has been observed previously in the case of silica films analogues of SBA materials28 but the spectral quality is not sufficient to positively assign the two peaks to either the different CH2O protons of the copolymer constituent or to a single CH2O signal shifting due to structural variations along the sample. The two signals merge in the dehydrated sample. A finer line-shape analysis of the methyl signal, around 0.9–1.1 ppm, reveals a composite resonance, made of a minimum of two components (1.1 ppm/57 Hz and 0.9 ppm/205 Hz for the as-SBA-16 and 1.1 ppm/235 Hz and 0.8 ppm/800 Hz for dehyd-SBA-16). The variations in chemical shift and linewidth suggest that the methyl groups are probing different regions of the sample characterized by specific mobilities or relaxation and magnetic susceptibilities. It is thus likely that the F127 is organized in a heterogeneous phase distribution, consisting of regions with different segmental mobility.
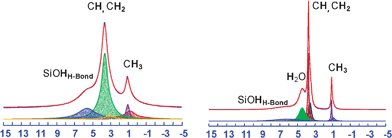 | ||
| Fig. 7 1H MAS NMR spectra of the as-SBA-16 (right) and dehyd-SBA-16 (left) at a MAS rate of 15 kHz. | ||
Key experiments to detect possible specific contacts and proximities between the F127 template and the silica are heteronuclear correlations (HETCOR). Based on hydrogen to silicon or carbon dipolar couplings, these experiments are sensitive concurrently to the atomic distances of interest and to the polymer segmental mobilities. Fig. 8 shows the 1H–29Si CPFSLG-HETCOR experiments performed on both the as synthesized and dehydrated sample. The most noticeable features in the as-SBA-16 spectrum are clear correlations of the Q3 to the 1H peaks at 6.1 and 4.4 ppm, corresponding to hydrogen bonded silanols and hydrated silanols respectively (Fig. 8, right panel). Upon dehydration, the Q3 sites still show strong correlations with 1H peaks at 5.6 ppm and 4.3 ppm, this latter testifying the presence of residual water (ca. 0.5 wt%, detected by thermogravimetric analysis, carried out on the as-synthesized SBA-16 sample after isothermal treatment at 80 °C for 8 h in inert conditions (Fig. S3†)) in the sample, not detected in the 1H MAS spectrum (Fig. 8, left).
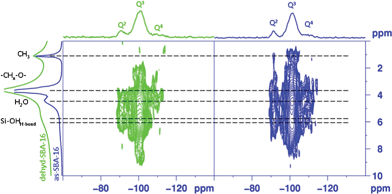 | ||
| Fig. 8 2D 29Si–1H CPLG HETCOR NMR spectra of as-SBA-16 (right) and dehyd-SBA-16 (left). | ||
The presence of strong surface interactions between the copolymer and various silicon sites is hinted by the overlapping cross peak pairs generated between protons of the alkyloxy units and silicon nuclei of the SBA-16. Indeed, the signal at a proton chemical shift of ∼3.7 ppm, associated to CH2O and/or CHO groups in PEO and PPO blocks, is strongly correlated to Q4, Q3 and Q2silicon sites at −111, −101 and −90 ppm, respectively. Interestingly, the cross-peaks that arise from Q4 sites, although smaller in intensity compared to Q3 and Q2 sites, are a strong indication of a high degree of intimacy between the structure directing copolymer chains and the silica surface. In addition, for as-SBA-16 correlation peaks appeared between Q3 and Q2 sites 29Si resonances and methyl protons at ∼1 ppm. The presence of such cross peaks in as-SBA-16 hints at an interpenetration of the F127 polymer blocks into the inner silica walls in this case, as the expected connecting site to the silanol is likely to be the etheric oxygen. Since no such cross peak is observed for the dehyd-SBA-16, in which the reduced mobility should enhance the CP transfer, it may be inferred that a reorganization of the PEO and PPO blocks of the copolymer takes place during the dehydration.
Fig. 9 shows the 13C–1H CP HETCOR experiments recorded at room temperature at a MAS rate of 13840 Hz for both as-SBA-16 (Fig. 9 right) and dehyd-SBA-16 (Fig. 9 left), with a contact time of 2 ms. The corresponding MAS 1H and 13C spectra are reported above the respective axis for clarity of the comparison. Both experiments revealed the presence of cross-peaks corresponding to directly bound C–H pairs (PPO: CH3 0.9–1.1/16.3 ppm, CHO 3.4/76 ppm, CH2O 3.6 ppm/73.5 ppm; PEO: CH2O 3.7 ppm/70.6 ppm).
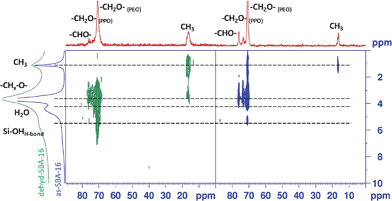 | ||
| Fig. 9 2D 13C–1H CPMAS HETCOR NMR spectra of as-SBA-16 (right) and dehyd-SBA-16 (left). | ||
Consistently with the 13C CPMAS spectrum, as-SBA-16 shows a narrower line width for the correlation peaks than dehyd-SBA-16. The overall intensity of the correlation signal is somewhat higher in the dehyd-SBA-16 sample, despite the broader lines, probably due to a larger value of the dipolar coupling, which is partially averaged by motion in the as-SBA-16. An interesting feature of the as-synthesized material is the correlation between the water proton signal and the PPO CHO carbon at 76.1 ppm, of an intensity comparable to the one stemming from the direct C–H coupling, suggesting a location of water close to this section of the polymer. It should be noted in passing that a correlation of this kind for the PEO section, could be masked by its broader cross-peak. Conversely, the only additional feature (other than the directly bonded pair correlations) in the dehyd-SBA-16 is a weak but clear cross-peak between the PEO CH2O (70.7 ppm) and the 1H signal at 5.7 ppm, confirming an interaction with this part of F127 and the silica framework. Another striking feature of this experiment is the presence of a unique cross peak between the methyl protons (∼1 ppm) of the PPO block and the methylenoxy carbons (∼70.6 ppm) of the PEO block in as-SBA-16. Interestingly, no such correlations are observed in dehyd-SBA-16 even though the same experimental conditions are used to record the data. The absence of such dipolar couplings between different blocks of the copolymers reinforces the hypothesis of a reorganization of the guest species during dehydration. A dehydration–rehydration experiment revealed that the water-dependent reorganization of the guest molecules is a reversible process (Fig. S4†).
These results are consistent with a cooperative assembly of the structure directing block copolymer aggregates and silica species at their interface, mediated by water. On one hand the presence of water induces mobility in the copolymer, probably by competing towards the surface silanols, while on the other it serves as an interface fabricating platform between block copolymers and inorganic silica. Moreover, the separation of a hydrophobic phase appears to be induced by hydration, as suggested by the folding of the PPO block into the PEO one for as-SBA-16.
Summarizing, thermogravimetric analysis along with spectroscopic studies allowed to estimate the presence of water in the as-SBA-16 and its influence on the interactions originated at the interface between polymer and silica surface.
SS-NMR studies allowed to derive that in hydrated conditions F127 co-polymer interacts with SiOH groups through CH2O, CH–O as found by the presence of cross-peaks in 2D 29Si–1H HETCOR NMR spectra in which the proton chemical shift (associated to CH2O and/or CHO groups in PEO and PPO blocks, at ∼3.7 ppm) is strongly correlated to Q4, Q3 and Q2silicon sites at −111, −101 and −90 ppm, respectively.
The dehydration process, carried out in vacuum at 80 °C, results in a reorganization of the PEO and PPO blocks of the co-polymer as suggested by the absence of the 2D 13C–1H NMR cross peak between the methyl protons (∼1 ppm) of the PPO block and the methylenoxy carbons (∼70.6 ppm) of the PEO block observed in as-SBA-16. In addition, SS-NMR indicates that PEO groups of F127 are interacting with silanols of SBA-16, as suggested by the presence of a cross-peak between the PEO CH2O (70.7 ppm) and the 1H signal at 5.7 ppm.
These findings confirm the IR variable temperature ones, where the presence of two broad bands with maxima at 3450 and 3550 cm−1 are an indication of the H-bonding interactions mainly occurring between SiOH and etheric oxygen atoms of F127 co-polymer with different local accessibility.
4. Conclusions
In the present work, the physico-chemical properties of SBA-16/Pluronic F127 composite material have been derived by correlating the information obtained through different experimental techniques. The complex interactions present at the interface between SBA-16 and SDA have been studied by IR and NMR spectroscopy, and all analyses indicated that in the solid state a significant interaction between the siliceous framework and the copolymer occurred from the silanol species and the hydrophilic moieties of Pluronic F127 when sample is dehydrated. In detail, both 13C–1H and 29Si–1H SS-NMR experiments showed cross-correlations between the CH2O groups of PEO blocks and SiOH surface species. IR spectroscopy allowed to derive that this interaction mainly occurred through the formation of H-bonding between SiOH and the etheric oxygen atoms of PEO.Nevertheless, in the presence of water, the F127 organization is different and also the hydrophobic domains (PPO blocks) of the co-polymer become close with the surface thanks to water mediation, as observed from the presence of a cross-peak between methyl group of PPO and Q2, Q3silanols in 29Si–1H HETCOR NMR spectra. These results suggested that both the hydrophobic and hydrophilic parts of the co-polymer may interact in some extent with the silica walls of SBA-16 material.
NMR experiments allowed to determine that the co-polymer has a different structural organization depending on the hydration degree of the SBA-16 sample. The comprehension of these structural differences and of the host–guest interactions present at the silica surface of hydrated sample will be helpful in clarifying the formation mechanisms of SBA-16 material.
References
- F. Hoffmann, M. Cornelius, J. Morell and M. Froba, Angew. Chem. Int. Ed., 2006, 45, 3216 CrossRef.
- Q. Yang, J. Liu, L. Zhang and C. Li, J. Mater. Chem., 2009, 19, 1945 RSC.
- H. Xiong, Y. Zhang, S. Wang, K. Liew and J. Li, J. Phys. Chem. C, 2008, 112, 9706 Search PubMed.
- W. Zhou, J. M. Thomas, D. S. Shephard, B. F. G. Johnson, D. Ozkaya, T. Maschmeyer, R. G. Bell and Q. Ge, Science, 1998, 280, 705 CrossRef CAS.
- B. J. Scott, G. Wirnsberger and G. D. Stucky, Chem. Mater., 2001, 13, 3140 CrossRef CAS.
- X. Gao and S. Nie, J. Phys. Chem. B, 2003, 107, 11575 CrossRef CAS.
- B. Lei, B. Li, H. Zhang, L. Zhang and W. Li, J. Phys. Chem. C, 2007, 111, 11291 CrossRef CAS.
- J. Feng, H. J. Zhang, C. Y. Peng, J. B. Yu, R. P. Deng, L. N. Sun and X. M. Guo, Microporous Mesoporous Mater., 2008, 113, 402 CrossRef CAS.
- S. Wang, Microporous Mesoporous Mater., 2009, 117, 1 CrossRef CAS.
- M. Vallet-Regi, F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548 CrossRef CAS.
- J. Kim, Y. Piao and T. Hyeon, Chem. Soc. Rev., 2009, 38, 372 RSC.
- F. Carniato, L. Tei, W. Dastru`, L. Marchese and M. Botta, Chem. Commun., 2009, 1246 RSC.
- F. Carniato, C. Bisio, G. Paul, G. Gatti, L. Bertinetti, S. Coluccia and L. Marchese, J. Mater. Chem., 2010, 20(26), 5504 RSC.
- J. Y. Ying, C. P. Mehnert and M. S. Wong, Angew. Chem., Int. Ed., 1999, 38, 56 CrossRef CAS.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024 CrossRef CAS.
- Y. Wan, Y. Shia and D. Zhao, Chem. Commun., 2007, 897 RSC.
- F. Kleitz, L. A. Solovyov, G. M. Anilkumar, S. H. Choi and R. Ryoo, Chem. Commun., 2004, 1536 RSC.
- Y. K. Hwang, J.-S. Chang, Y.-U. Kwon and S.-E. Park, Microporous Mesoporous Mater., 2004, 68, 21 CrossRef CAS.
- M. Mesa, L. Sierra, J. Patarin and J.-L. Guth, Solid State Sci., 2005, 7, 990 CrossRef CAS.
- D. Li, X. Guan, J. Song, Y. Di, D. Zhang, X. Ge, L. Zhao and F.-S. Xiao, Colloids Surf., A, 2006, 272, 194 Search PubMed.
- K. Flodstrom, H. Wennerstrom, C. V. Teixeira, H. Amenitsch, M. Linden and V. Alfredsson, Langmuir, 2004, 20, 10311 CrossRef.
- T.-W. Kim, R. Ryoo, M. Kruk, K. P. Gierszal, M. Jaroniec, S. Kamiya and O. Terasaki, J. Phys. Chem. B, 2004, 108, 11480 CrossRef CAS.
- M. Mesa L. Sierra and J.-L. Guth, Microporous Mesoporous Mater., 2008, 112, 338 CrossRef CAS.
- (a) E. J. van Rossum, H. Forster and H. J. M. de Groot, J. Magn. Reson., 1997, 124, 516 CrossRef CAS; (b) G. Paul, S. Steuernagel and H. Koller, Chem. Commun., 2007, 5194 RSC.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies. Chichester: Wiley, 2001 Search PubMed.
- A. Zecchina, S. Bordiga, G. Spoto, L. Marchese, G. Petrini, G. Leofanti and M. Padovan, J. Phys. Chem., 1992, 96, 4991 CrossRef CAS.
- A. Novak, Hydrogen Bonding in Solids. Correlation of Spectroscopic and Crystallographic data, from Structure and Bonding, Vol 18, Springer-Verlag, Berlin-Heidelberg-New York, 1974 Search PubMed.
- N. A. Melosh, P. Lipic, F. S. Bates, F. Wudl, G. D. Stucky, G. H. Fredrickson and B. F. Chmelka, Macromolecules, 1999, 32, 4332 CrossRef CAS.
- S. Kirmayer, E. Dovgolevsky, M. Kalina, E. Lakin, S. Cadars, J. D. Epping, A. Fernandez-Arteaga, C. Rodriguez-Abreu, B. F. Chmelka and G. L. Fray, Chem. Mater., 2008, 20, 3745 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00460c |
| This journal is © The Royal Society of Chemistry 2012 |