Stable organic radical stacked by in situ coordination to rare earth cations in MOF materials†
Felipe
Gándara
a,
Natalia
Snejko
a,
Alicia de
Andrés
a,
Jesús Rodriguez
Fernandez
b,
José C.
Gómez-Sal
b,
Enrique
Gutierrez-Puebla
a and
Angeles
Monge
*a
aInstituto de Ciencia de Materiales de Madrid, Cantoblanco, 20849, Madrid, Spain
bUniversidad de Cantabria, Avda. de los Castros s/n, 39005, Santander, Spain. E-mail: amonge@icmm.csic.es
First published on 24th November 2011
Abstract
With the correct choice of the solvothermal conditions, we have achieved the unprecedented in situ formation of the free radical form of the anthraquinone-1,5-disulfonate molecule, and its favorable organization. The semiquinone radicals are coordinated to rare-earth cations to produce a 2D framework with a very high charge mobility and electric conductivity through the π–π-interactions. The existence of AQDS3−˙ anion radicals is proven on the base of: i) the electrical neutrality: elemental analyses for the different lanthanide RPF-8 bulks, the maximum residual electron densities in the structure, rule out the existence of any other neutralizing ion, ii) the geometrical modifications in the antraquinone molecules, and iii) although less definitive, due to the low magnetic moment μ = 0.39 μB, the exhibited paramagnetism for the La (3+) with no unpaired electrons.
Introduction
Face-to-face π-stacks of extended aromatics with the π-surfaces located within van der Waals distances have emerged as promising nanowires and are among the best performing organic conductors.1,2,3,4 In this context, great efforts to obtain stacked assemblies of extended electroactive aromatic systems are being carried out, in order to provide efficient pathways for charge migration. To achieve this goal different intermolecular interactions involving aromatic rings5,6,7,8,9,10 and other more directional interactions such as hydrogen bonding11,12 have been utilized. For this reason, the possibility of creating systems, where the aromatic arrangements are incorporated and fixed by means of stronger interactions, is highly desirable. In our group we have recently started to explore a strategy that consists in the coordination of the aromatic containing building-blocks to rare earth cations, resulting, thanks to their high and variable coordination number, in the incorporation of the aromatic molecules to polymeric frameworks at distances that allow π–π stacking interactions.13,14 On the other hand, there are some examples of hybrid polymeric frameworks, in which the organic ligand is a radical species, as for example tetracyanoethylene. These materials exhibit interesting magnetic properties.15,16 In them, the radical species exist prior to their use in the formation of the extended network. Quinones are a well known type of molecule, which exhibit a redox chemistry involving the formation of radical species, known as semiquinones.17,18 As a quinone derivative, the anthraquinone disulfonate (AQDS) molecule has two reduced states,19 corresponding to AQDS3− and AQDS4−, and the disposition of the sulfonic functional group in the 1,5-isomer makes this molecule a good candidate for its incorporation into a coordination polymer. The formation of the semiquinone species in the anthraquinone molecule during the synthesis has not been reported up to date. Nevertheless, with the adequate choice of synthesis conditions, we have succeeded in the formation of the radical species of the anthraquinone-1,5-disulfonate anion (AQDS) during a solvothermal reaction, and the incorporation of this radical into a new type of rare-earth polymeric framework, RPF8, by coordination to the lanthanide cations. The new materials presented in this paper have interesting electric transport properties, with high mobility of charge carriers.Experimental section
The compounds are prepared under solvothermal conditions. In a typical experiment, 0.260 g of Na2AQDS (Sigma-Aldrich, used as received) and 0.130 g of La(NO2)3·6H2O (Sigma-Aldrich, used as received) are dissolved in 5.4 ml of water, and then 16.4 ml of n-BuOH are added. The molar composition of this mixture is La3+:2AQDS:1000H2O:600n-BuOH. The mixture is stirred at room temperature for 5 min, and then placed into a Teflon lined stainless steel autoclave. This is sealed and heated in an oven at 180 °C for 18 h. After the heating time, the autoclave is cooled to room temperature and the product is filtered and thoroughly washed with deionized water and acetone, until no color is appreciated in the filtered liquid. The product is dried in air. Similar synthesis procedure has been employed for the products prepared with the other rare earth elements (Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er), employing the corresponding amount of their hexahydrated nitrate salts, keeping the same molar composition of the initial mixture of reactants.Single-crystal structure determination and refinement
Single crystals suitable for X-ray crystallography were obtained for the La-, Pr-, Gd- and Sm-RPF8 compounds. In each case a prismatic dark (almost black) crystal was selected, glued on a glass fiber, and mounted on a SMART-CCD Bruker diffractometer. The data were collected at room temperature, with MoKα λ = 0.71073 Å. Data were collected over a hemisphere of the reciprocal space by a combination of sets of exposures. Each exposure of 20 s covered 0.3° in ω. Unit cell dimensions were determined by a least-squares fit of 60 reflections with I > 20σ (I). The structures were solved by direct methods. The final cycles of refinement were carried out by full-matrix least-squares analyses with anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms of the water molecules were located in difference Fourier maps. Residual electron density indicative of any other neutralizing anion was not found. Calculations were carried out with SMART software for data collection and data reduction and SHELXTL20 (Table 1).| Identification code | La-RPF8 | Pr-RPF8 | Sm-RPF8 | Gd-RPF8 |
|---|---|---|---|---|
| Empirical formula | C14H12S2LaO11 | C14H12S2PrO11 | C14H12S2SmO11 | C14H12S2GdO11 |
| Formula weight | 559.27 | 561.27 | ||
| Temperature | 295(2) K | |||
| Wavelength | 0.71073 Å | |||
| Crystal system, SG | Orthorhombic, P21212 | |||
| Unit cell dimensions | a = 11.5258(7)Å | a = 11.4498(17) Å | a = 11.465(3) Å | a = 11.3486(17)Å |
| b = 21.064(2)Å | b = 20.923(2) Å | b = 20.956(5) Å | b = 20.756(3) Å | |
| c = 7.1175(4) Å | c = 7.1043(9) Å | c = 7.1395(16) Å | c = 7.0785(11) Å | |
| Volume | 1727.97(17) Å3 | 1702.0(4) Å3 | 1715.3(7)Å3 | 1667.4(4) |
| Z, Calc. density | 4, 2.150 Mg/m3 | 4, 2.190 Mg/m3 | 4, 2.210 Mg/m3 | 4, 2.301 |
| Absorption coeff. | 2.776 mm−1 | 3.170 mm−1 | 3.729 mm−1 | 4.292 mm−1 |
| F(000) | 1092 | 1100 | 1112 | 1120 |
| Crystal size mm | 0.10 × 0.05 × 0.05 | 0.30 × 0.06 × 0.02 | 0.30 × 0.02 × 0.02 mm | 0.30 × 0.02 × 0.02 mm |
| Theta range data collection | 1.93° to 29.25° | 2.03° to 26.37° | 2.01° to 25.67° | 2.66° to 26..6° |
| Limiting indices | −8 ≤ h ≤ 15 | −8 ≤ h ≤ 14 | −13 ≤ h ≤ 13 | −14 ≤ h ≤ 13 |
| −27 ≤ k ≤ 27 | −21 ≤ k ≤ 26 | −25 ≤ k ≤ 21 | −25 ≤ k ≤ 25 | |
| −6 ≤ l ≤ 9 | −8 ≤ l ≤ 8 | −8 ≤ l ≤ 8 | −8 ≤ l ≤ 8 | |
| Reflections collected/unique | 9259/4052 | 6516/3213 | 8913/3091 | 12311/3392 |
| Compl. to 25.00° | 98.4% | 97.7% | 97.1% | 99.4% |
| Max. and min. transmission | 0.8737 and 0.7688 | 0.9393 and 0.4497 | 0.9637 and 0.4009 | 0.9583 and 0.3592 |
| Data/restraints/parameters | 4052/0/253 | 3213/132/253 | 3091/84/242 | 3392/0/247 |
| Goodness-of-fit on F2 | 1.030 | 0.977 | 0.975 | 0.976 |
| Final R indices [I > 2σ (I)] | R 1 = 0.0558 | R 1 = 0.0539, | R 1 = 0.0589, | R 1 = 0.0493 |
| wR2 = 0.1270 | wR2 = 0.0795 | wR2 = 0.0977 | wR2 = 0.0787 | |
| R indices (all data) | R 1 = 0.0759, | R 1 = 0.0820 | R 1 = 0.1003 | R 1 = 0.0812 |
| wR2 = 0.1394 | wR2 = 0.0867 | wR2 = 0.1083 | wR2 = 0.0864 | |
| Flack parameter | −0.02(3) | 0.01 (3) | 0.42 (3) | 0.02 (2) |
| Largest diff. peak and hole e·Å−3 | 1.481 and −3.038 | 0.940 and −0.945 | 0.878 and −1.153 | 0.854 and −1.270 |
PXRD for the RPF8 compounds, including rietveld refinements
X-Ray powder patterns were collected on Bruker D8 diffractometer equipped with a PSD detector. Patterns were collected in the 2θ° range 5–60, with a step size of 0.02°, and an exposure time of 2 s per step. Rietveld refinements were performed using the single crystal data obtained as starting point. The Fullprof programs suite21 was employed for the refinement.Magnetic measurements
Magnetic measurements were carried out using a Quantum Design superconducting quantum interference device SQUID magnetometer in the range 2 to 300 K and fields up to 50 kOe. The samples in the form of powder (typically 20 mg) were closed in diamagnetic polycarbonate capsules. The background signal from the straw and capsule were measured separately and subtracted from the data to give the pure magnetic signal of the samples. For each sample, the susceptibility has been measured in a field of 10 kOe, first decreasing the temperature down to 2 K and then increasing it up to 300 K. The temperature dependence of the molar magnetic susceptibility (χ) and its inverse (1/χ) is depicted in Fig. S4–1 for all the studied compounds.Additionally, the field dependence of the magnetization has been measured at T = 10 K and magnetic fields up to 50 kOe. As an example, the curves of some members of the family are compared in Fig. S4–2. Finally, in order to discard a possible long-range order below 2 K in the Ho and Dy-based compounds, which have the largest magnetic moments of the whole family, we performed M(H) measurements at different temperatures down to 2.5 K and fields up to 85 kOe at a Physical Properties Measurement System (PPMS).
IR spectra were recorded on a FTIR Nicolet Magna-550 spectrophotometer with samples as KBr pellets in the 4000–400 cm−1 region.
Results and discussions
The presence of both water and butanol solvents is necessary for the formation of the products. Experiments carried out in water in absence of butanol resulted in the formation of entirely amorphous solids. A series of experiments were carried out with initial mixtures of molar composition of La : AQDS : 500·H2O : x n-BuOH (x = 0, 10, 20, 100, 200, 300). As the amount of butanol in the initial mixture increases, more crystals of the new material appear in the product. With x = 300, the target compound appears as a major phase, but with low yield, and mixed with an amorphous unidentified red solid in the product. The only way to obtain the pure desired compound as the lone phase and in good yield is to adjust the initial mixture composition to a ratio of AQDS/Ln = 2. With this initial mixture, the RPF8 product is purely obtained with good yield (60% on La basis). The results of the elemental analyses for the RPF8 compounds compared with their calculated values are given in Table S1-1† (for Lanthanum compound, % C 30.04 cal. 30.28 exp;% H 2.17 cal., 2.22 exp).It is known that the AQDS molecules suffer self-exchange electron processes when the radical species are formed, and it seems plausible that during the synthesis procedure an excess of AQDS molecules is needed to somehow stabilize the presence of the radical species before they coordinate to the Ln ions, forming the new material.
To obtain the radical species, several factors are decisive: as already mentioned the presence of n-BuOH as a co-solvent is necessary. In addition, the presence of the rare-earth element in the system also has influence not only in the structural type obtained, but also in the formation of the radical species during the synthesis procedure. To prove this fact, several synthesis experiments were performed: first, a synthesis procedure in absence of rare earth salt was carried out. A mixture with the same amounts of Na2AQDS, water and butanol was heated for the same time at the same temperature, in absence of any rare-earth salt. Large, orange crystals were obtained. The structure of one of them was solved, revealed to be a sodium salt of AQDS.22
On the other hand, synthesis experiments employing transition metals were also carried out. Mixtures of molar composition equivalent to the optimized for the rare-earth elements were prepared, now with transition metal salts instead of the rare-earth salts (CoCl2 and FeCl2 were used). The same structural type was obtained in both cases, with the M2+ (M = Co, Fe) cations hexacoordinated to two water molecules and to four O atoms from four sulfonate groups. In this case, the anthraquinone molecules appear as AQDS2− dianion, with no presence of the radical species. The C–O bonds are those expected for the C–O double bond typical in the non reduced form of the AQDS anion. These experiments demonstrated that the presence of the rare-earth elements is necessary to obtain the semiquinone species in the crystalline solid.
In the IR spectra of the La-RPF8 compound two new intense bands, that are absent in the IR spectra of the ligand and can be assigned to a coordinated CO stretch, appear at ∼1430 and 1520 cm−1, which confirms the findings made by structural studies (see Fig. S1.1†).
Crystal structure description
The obtained products are all microcrystalline dark solids formed by small crystals. Single crystals suitable for X-ray crystallography were obtained for La, Pr, Sm and Gd while all other compounds demonstrated to be isostructural by means of powder X-ray diffraction (see ESI, CCDC deposition numbers 728638–72641†).The products crystallize in the orthorhombic system, space groupP21212, with a = 11.526 Å, b = 21.064 Å, c = 7.117 Å and V = 1727.65 Å3 in the case of La compound. The formula of the compounds is C14H12O11S2Ln, which is equivalent to LnL(H2O)3, where L = 1,5-AQDS3−, and (Ln = La, Pr, Nd, Sm, Gd, Eu, Dy, Tb, Ho, Er). Ln atoms are octa-coordinated to three water molecules, and to five oxygen atoms from 1,5-AQDS3−, three of the SO3−groups and two of quinonic groups (Fig. 1).
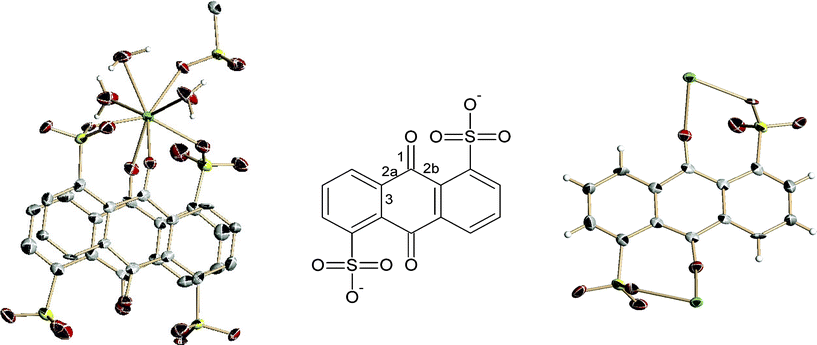 | ||
| Fig. 1 RPF8, ORTEP representation of: left) La coordination environment right) representation of a semiquinone molecule coordinated to the La atoms through the sulfonate groups and the quinonic oxygen atom, and center) numeration scheme for the bonds suffering length changes in the formation of the semiquinone species: green: Ln, grey: carbon, red: oxygen, yellow: sulphur, white: hydrogen. | ||
There are two different SO3−groups regarding their coordination modes. One of them is bonded to two Ln cations in a η2μ2 mode, through two oxygen atoms, while the other is monocoordinated to an Ln cation in a η1 mode. The Ln atoms form zig-zag chains along the a direction, via the bridge η2-μ2 sulfonate group. Two AQDS molecules join these chains along the b direction, giving rise to double layers, which stack in the c axis in an AA sequence. Hydrogen bonds are present between adjacent layers, formed by the coordinated water molecules and the uncoordinated oxygen atoms of the sulfonate groups. The disposition of the Ln atoms in the layers, and their connection through the ligands give rise to a 63 honeycomb topology of the double layers, with the Ln atoms as the three connected nodes, joined by ligand molecules (Fig. 2).
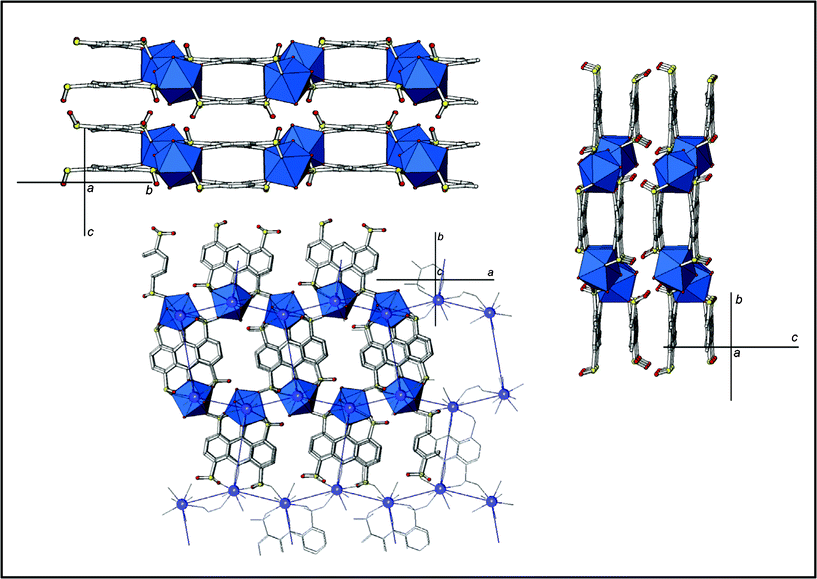 | ||
| Fig. 2 Polyhedral view the RPF8 double layer structure. | ||
The anthraquinone ions in the RPF8 exist as AQDS3−· anion radicals. Among numerous examples of semiquinone-contained structures reported23 , in very few of them the radical is part of the MOF scaffold, and RPF8 is to the best of our knowledge the first case of an anthraquinone radical species formed in situ by coordination to a lanthanide atom. In the semiquinone species, the additional charge is delocalized over the anthraquinone central ring and the quinonic oxygen atoms. The bond distances of these O atoms with their corresponding C atoms suffer an elongation as a consequence of the single bond contribution in the radical species. The C–C bonds in the central ring are also affected by the presence of an additional electron. Thus, the bond 2, as numerated in Fig. 1, decreases in length, since the electronic delocalization implies a partial aromatic component in this central ring, while the bond 3 is elongated. In the four crystal structures here reported, the lengths of the C–O bonds lay in the range 1.260(15)–1.302(13) Å, clearly demonstrating the partial double-bond nature of this bond, due to the reduction of the molecule (Table 2). For each crystal, there are two C–O bonds, corresponding to the two different AQDS ions in each case (the asymmetric units comprise two halves of AQDS ions).
| Compound | Na2AQDS (ref. [23]) | Na2AQDS (ref. [24]) | Co-AQDSc | Fe-AQDSc | LaRPF8 | PrRPF8 | SmRPF8 | GdRPF8 |
|---|---|---|---|---|---|---|---|---|
| Bonda | Quinones | Semiquinones b | ||||||
| a Bonds are numerated according to Fig. 1. b In the RPF8 compounds, two values are given for each bond, since there are two independent molecules in the asymmetric unit. c See ESI.† | ||||||||
| 1 | 1.217 | 1.216 | 1.211 | 1.213 | 1.302 | 1.282 | 1.260 | 1.277 |
| 1.301 | 1.270 | 1.302 | 1.271 | |||||
| 2a | 1.487 | 1.482 | 1.505 | 1.500 | 1.441 | 1.450 | 1.458 | 1.464 |
| 1.426 | 1.454 | 1.426 | 1.464 | |||||
| 2b | 1.498 | 1.496 | 1.498 | 1.492 | 1.451 | 1.455 | 1.508 | 1.438 |
| 1.447 | 1.461 | 1.426 | 1.435 | |||||
| 3 | 1.410 | 1.404 | 1.396 | 1.399 | 1.402 | 1.417 | 1.453 | 1.430 |
| 1.418 | 1.453 | 1.426 | 1.459 | |||||
Magnetic properties
Although other alternatives can be thought to maintain the formula electrical neutrality, for example the presence of some neutralizing anion in the structure, or even the very improvable reduction of the lanthanide (impossible for some of them) by butanol, the formation of the radical semiquinone species is clearly the best, on the basis of : i) the elemental analysis for the different lanthanide RPF-8 bulks, which show no other atoms in the samples, ii) the X-ray structure determinations, that clearly show the anthraquinone deformation with distances and angles present in Table 2, and iii) most definitive, in the structure solutions, the hydrogen atoms were found in the final difference Fourier syntheses, in which no electron density corresponding to any other neutralizing atom appeared.In order to check if the existence of the radical could be detected in some physical property, magnetic susceptibility measurements for the La-RPF8 compound were performed. La (3+) has no unpaired spins, therefore the origin of a paramagnetic signal in the La compound must be unpaired spins associated with the reduced organic ligands AQDS (3−). The dependence of the magnetization with the temperature and with magnetic field at 10 K shows a paramagnetic behavior with an effective magnetic moment of 0.39 μB, obtained from the field dependence of the magnetization at 10 K. To rule out the possibility of some impurity coming from the lanthanide source, measurements on another polymeric framework, La-RPF724, synthesized with the same La(NO3)3 reagent and 2,6-AQDS, in which no radical species is formed, were also performed. As expected for a La3+ compound, a μ value of 0 was obtained (Fig. 3).
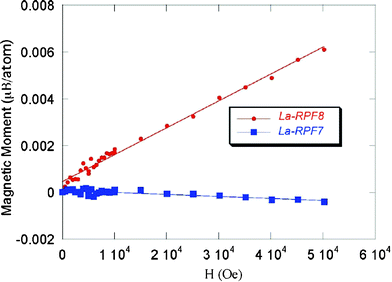 | ||
| Fig. 3 Comparison of the atomic magnetic moment of La-RPF8 with La-RPF-7 (compound without semiquinone radical). | ||
Most Rare-earth compounds are paramagnetic and reach a magnetic order, if any, at very low temperatures. We performed magnetic measurements on the whole series of RPF8 compounds to obtain the magnetic properties, to detect a possible ferromagnetic or antiferromagnetic ordering as well as to identify the possible influence of the radical on the magnetism. The magnetization measurements were performed as a function of temperature and field (see Fig. 4 and ESI for details†). The compounds are paramagnetic and, in general, the inverse of the susceptibility follows a Curie–Weiss law. The obtained Weiss temperature is negative indicating antiferromagnetic interactions between the magnetic moments, except for the Gd-RPF8 compound, where Θ = 0.1
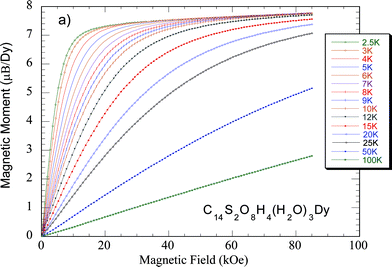 | ||
| Fig. 4 Field dependence of the magnetization for Dy-RPF8 at several temperatures. | ||
Only the Sm and Eu-based compounds deviate from the Curie–Weiss law below 200 K. For compounds with Sm3+ and Eu3+ ions the Van Vleck susceptibility is usual. In the case of the Sm3+ ion, this is due to the fact that the first excited multiplet, with a total angular momentum J = 7/2, is not far in energy from the ground-state (J = 5/2) and, in addition, there is a relatively large matrix element coupling the J = 7/2 and J = 5/2 multiplets. As for Eu compounds, according to the Hund’s rules, the free Eu3+ ion has a nonmagnetic ground state (J = 0); however, under an external magnetic field, the ground state is mixed with the first excited magnetic state (J = 1), thereby leading also to a Van Vleck term. The obtained magnetic moment values were higher than those corresponding to the free Ln3+ cations in all cases. In particular, in Fig. 4, it is depicted that the magnetization curves of the Dy-RPF8 at several temperatures, which show that below 10 K, the magnetization tents to saturate, with a value of 7.3 μB/Dy ions. In addition, the curves follow Brillouin functions for all the temperatures, confirming the paramagnetic behavior.
In the Ln-RPF8 framework the AQDS ions are disposed in a parallel way, giving rise to π–π stacking interactions. These take place along the c axis, with the organic molecules arranged in a perfect face-to-face display. There are two different distances between the AQDS planes; one is for ions in the same layer and the other for ions in adjacent layers. Their respective values are 3.43 Å and 3.69 Å for the La-RPF8 compound, with similar values for the other lanthanide crystals. Besides, the extra electron of the radical semiquinone increases the electronic interactions within the framework. All this favors the possibility of high electric conductivity in RPF8 compounds.
The electric conductivity measurements were carried out in a single crystal of the La-RPF8 compound with dimensions 0.420 × 0.055 × 0.055 mm (Fig. 5). Silver paste at both ends of the crystal was used to contact the sample to gold electrodes. The current was measured versus the applied voltage, in the range 0–100 V along the stacking direction (001). The measured I–V is linear indicating ohmic contacts. The obtained Room Temperature (RT) conductivity for La-RPF8 crystal is 3 × 10−3 S cm−1 (Fig. 5). This value is within the range of other compounds with π–π stacking interaction with shorter distances between aromatic rings,25 or even of that caused by interactions between transition metal ions in 1D MOF nets.26 This seems to indicate that the presence of a delocalized electron in the anthraquinone radical improves the conductivity properties of the materials. Additional measurements were carried out up to 400 V to check the high voltage regime.
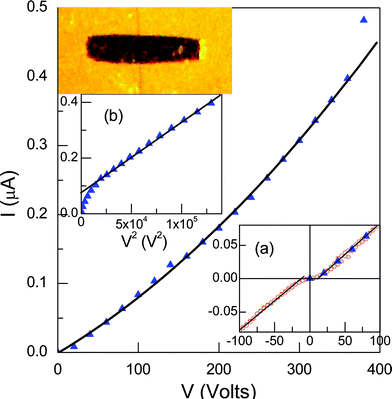 | ||
| Fig. 5 Current versus voltage measured for a La-RPF8 crystal (420 × 55 × 55 μm3) along its longer dimension. The black line is the fit to the SCLC regime and an ohmic component. Inset (a) shows the symmetric and linear low voltage regime, from −100V to 100V, and (b) shows the cuadratic behavior of the current (always in μA) at high voltages (note the V2 scale). | ||
As in the present case, an ideal array of π-stacked radicals with odd number of electrons may give rise to a half filled conduction band and therefore to metallic ground state and conductivity. In fact, the crystals are almost black to the eye indicating small band gap energy. Intermolecular coupling is necessary to produce a large enough bandwidth to overcome onsite Coulomb repulsion and delocalized electrons. However, Peierls distortions may reduce the total energy producing dimers so that the coupling between pairs of adjacent radicals quenches the spin and opens a gap at the Fermi level.27 This occurs in most of the cases so that, in general, low conductivities and activated mechanisms are obtained. In the present compounds the dimerization is proven by the two intermolecular aromatic ring centroid distances (3.43 and 3.69 Å) and the low value of the magnetic moment (0.39 μB) compared to that corresponding to one unpaired electron (1.73 μB). Nevertheless, the conductivity obtained from the low voltage regime (Fig. 5a) for La single crystals (σ = 3 10−3 Ω−1cm−1) is quite high but four orders of magnitude smaller than the corresponding to organic conductors (60 Ω−1cm−1).28
From the solid state voltammetry we obtained an estimation of the LUMO energy of −4.35 eV. Absorption measurements of the La-RPF8 and Na2 ADQS compounds in powder are presented in Fig. 6 in the range from 200 nm to 900 nm. The absorbance evidences the UV bands corresponding to the Na2 AQDS, in which the antraquinone is not in its radical form, and presents a high energy gap. A very efficient transition in the NIR region around 800 nm is detected in the whole series of the RPF8 compounds independently of the rare earth (the figure presents the spectrum corresponding to the La compound), which is most probably related to the radical form of the antraquinone, and indicates that the gap energy is lower than 1.38 eV (900 nm). No emission band, neither from the organic part nor from the rare earth 4f transitions, in photoluminescence experiments was observed. The black colour of the sample is also an indication of a small gap, which is consistent with the picture of the opening of a gap in a half filled band and with the obtained RT conductivity value. The sample is therefore a small gap semiconductor with a considerable density of free charge carriers at RT and its HOMO level is expected to lie lower but close to the LUMO.
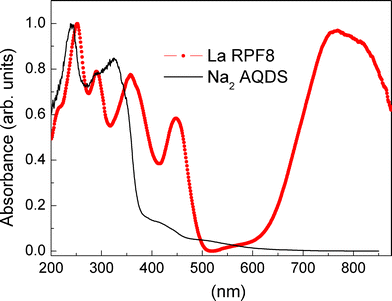 | ||
| Fig. 6 Absorbance spectra of the La-RPF8 and Na2 ADQS compounds at room temperature. | ||
The work function of Ag (−4.2 eV) almost matches that of the LUMO (−4.35 eV) of the compound, therefore no barrier for injection is expected. The geometry of the I–V measurements (high ratio between the contact distance, d = 420 μm, and area of the sample, S = 55 × 55 μm2) and the presence of free carriers require high voltages (above 100 V) to reach high enough electric fields (in the range of 105 V m−1) to enter the space charge limited current (SCLC) regime. The conductivity and electric field for the SCLC regime are comparable to those reported for high electron mobility compounds.29 Above 100 V and up to 360 V a characteristic V2 behaviour of the electric current is observed (Fig. 3). From the Mott Gurney equation for the SCLC observed regime, we obtained an estimation of the mobility of the carriers of μ = 9 cm2 V−1 s−1. The mobility obtained by this method is a lower limit since barriers for injection and traps are not taken into account. From the previously indicated energy levels and Ag work function it seems reasonable that the carriers may be mainly electrons but the presence of holes cannot be discarded. The obtained mobility is found to be among the highest reported values for this type of materials: up to our knowledge, the highest hole mobility obtained with the SCLC technique is 35 cm2 V−1 s−1 and corresponds to ultra-purified pentacene single crystals,30 and the best electron mobility is reported for a diimide derivative to be 6.7 cm−2 V−1 s−1.31,32
In conclusion, with the correct choice of solvothermal conditions, we have achieved the unprecedented in situ formation of the free radical form of the anthraquinone-1,5-disulfonate molecule, and its favorable organization. The existence of anthraquinone ions in RPF8 as AQDS3−˙ anion radicals is proven on the base of: i) the electrical neutrality: the elemental analyses for the different lanthanide RPF-8 bulks, the maximum residual electron densities (from 0.8 to 1.4 e·Å−3), and the R values (R∼0.05), in a structure, in which even the hydrogen atoms were found in final difference Fourier syntheses, demonstrate that no other neutralizing ions exist, ii) the geometrical modifications suffered by the AQDS molecule in the crystal structures, and iii) although less definitive, due to the low magnetic moment, the exhibited paramagnetism in La (3+) with no unpaired spins.
The semiquinone radicals are coordinated to Rare-earth cations to produce a 2D framework with a very high charge mobility and electric conductivity through the π–π-interactions.
Acknowledgements
Authors thank Drs Gomez-Lor, and Iglesias for their helpful discussions. This work has been supported by Spanish MCYT Projects Mat 2007-60822, 2010-17571, and 2008-06542-C04, the Project Consolider-Ingenio CSD2006-2001, and the Project of Comunidad Autónoma de Madrid (S2009/MAT-1756).References
- J. M. Warman, M. P. de Haas, G. Dicker, F. C. Grozema, J. Piris and M. G. Debije, Chem. Mater., 2004, 16, 4600 CrossRef CAS.
- E. C. P. Smits, S. G. J. Mathijssen, P. A. van Hal, S. Setayesh, T. C. T. Geuns, K. A. H. A. Mutsaers, E. Cantatore, H. J. Wondergem, O. Werzer, R. Resel, M. Kemerink, S. Kirchmeyer, A. M. Muzafarov, S. A. Ponomarenko, B. de Boer, P. W. M. Blom and D. M. de Leeuw, Nature, 2008, 455, 956 CrossRef CAS.
- S. Xiao, J. Tang, T. Beetz, X. Guo, N. Tremblay, T. Siegrist, Y. Zhu, M. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2006, 128, 10700 CrossRef CAS.
- S. Xiao, M. Myers, Q. Miao, S. Sanaur, K. Pang, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2005, 44, 7390 CrossRef CAS.
- E. A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1210 CrossRef CAS.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS.
- E. M. Garcia-Frutos, G. Hennrich, E. Gutiérrez-Puebla, A. Monge and B. Gomez-Lor, J. Org. Chem., 2010, 75, 1070 CrossRef CAS.
- S. Klyatskaya, N. Dingenouts, C. Rosenauer, B. Müller and S. Höger, J. Am. Chem. Soc., 2006, 128, 3150 CrossRef CAS.
- Y. Tobe, N. Utsumi, K. Kawabata, A. Nagano, K. Adachi, S. Araki, M. Sonoda, K. Hirose and K. Naemura, J. Am. Chem. Soc., 2002, 124, 5350 CrossRef CAS.
- S. K. Pal, M. E. Itkis, F. S. Tham, R. W. Reed, R. T. Oakley and R. C. Haddon, Science, 2005, 309, 281 CrossRef CAS.
- A. N. Sokolov, T. Frišić and L. R. MacGillivray, J. Am. Chem. Soc., 2006, 128, 2806 CrossRef CAS.
- M. L. Bushey, T.-Q. Nguyen, W. Zhang, D. Horoszewski and C. Nuckolls, Angew. Chem., Int. Ed., 2004, 43, 5446 CrossRef CAS.
- F. Gándara, C. Fortes-Revilla, N. Snejko, E. Gutiérrez-Puebla, M. Iglesias and M. A. Monge, Inorg. Chem., 2006, 45, 9680 CrossRef.
- F. Gándara, M. E. Medina, N. Snejko, E. Gutiérrez-Puebla, D. M. Proserpio and M. A. Monge, CrystEngComm, 2010, 12, 711 RSC.
- H. Zhao, M. J. Bazile-Jr, J. R. Galán-Mascarós and K. R. Dunbar, Angew. Chem., Int. Ed., 2003, 42, 1015 CrossRef CAS.
- N. Roques, D. Maspoch, F. Luis, A. Camon, K. Wurst, A. Datcu, C. Rovira, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2008, 18, 98 RSC.
- M. Kaupp, C. Remenyi, J. Vaara, O. L. Malkina and V. G. Malkin, J. Am. Chem. Soc., 2002, 124, 2709 CrossRef CAS.
- D. Meisel and R. W. Fessenden, J. Am. Chem. Soc., 1976, 98, 7505 CrossRef CAS.
- K. Akiyama and S. Tero-Kubota, J. Phys. Chem. B, 2002, 106, 2398 CrossRef CAS.
- Software for the SMART System V5.04 and SHELXTL V 5.1, Bruker-Siemens Analytical X-ray Instrument Inc., Madison, WI, 1998 Search PubMed.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- F. Gándara, New Rare-earth Coordination Polymers as Multifunctional Materials, PhD Memory, cap., V. 1, 193–195 Search PubMed.
- (a) C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99 CrossRef CAS; (b) S. Roy, B. Sarkar, D. Bubrin, M. Niemeyer, S. Zális, G.K. Lahiri and W. Kaim, J. Am. Chem. Soc., 2008, 130, 15230 CrossRef CAS; (c) D. Herebian, E. Bothe, F. Neese, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 2003, 125, 9116 CrossRef CAS.
- A. Monge, F. Gandara, E. Gutierrez-Puebla and N. Snejko, CrystEngComm, 2011, 13, 5031 RSC.
- S. Q. Liu, T. Kuroda-Sowa, H. Konaka, Y. Suenaga, M. Maekawa, T. Mizutani, G. L. Ning and M. Munakata, Inorg. Chem., 2005, 44, 1031 CrossRef CAS.
- S. Delgado, P. J. Sanz Miguel, J. L. Priego, R. Jiménez-Aparicio, C. J. Gómez-García and F. Zamora, Inorg. Chem., 2008, 47, 9128 CrossRef CAS.
- J. L. Brusso, S. Derakhshan, M. E. Itkis, H. Kleinke, R. C. Haddon, R. T. Oakley, R. W. Reed, J. F. Richardson, C. M. Robertson and L. K. Thompson, Inorg. Chem., 2006, 45, 10958 CrossRef CAS.
- J. Karolak-Wojciechowska, W. Kwiatkowski and A. Socha, Polish J. Chem., 1993, 67, 1159 CAS.
- E. Gamag, B. M. Peake and J. Simpson, Aust. J. Chem., 1993, 46, 1595 CrossRef.
- M. Clemente-León, E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, C. Rovira and V. N. Lauhkin, Synth. Met., 1999, 103, 2339 CrossRef.
- Z. An, J. Yu, B. Domercq, S. C. Jones, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem., 2009, 19, 6688 RSC.
- O. D. Jurchescu, J. Baas and T. T. M. Palstra, Appl. Phys. Lett., 2004, 16, 3063 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00447f |
| This journal is © The Royal Society of Chemistry 2012 |