BF2-bound chromophore-containing N⁁NPt(II) bisacetylide complex and its application as sensitizer for triplet–triplet annihilation based upconversion†
Yifan
Liu
,
Qiuting
Li
,
Jianzhang
Zhao
* and
Huimin
Guo
*
State Key Laboratory of Fine Chemicals, School of Chemical Engineering, Dalian University of Technology, E-208 West Campus, 2 Ling-Gong Raod, Dalian, 116024, P. R. China. E-mail: zhaojzh@dlut.edu.cn; guohm@dlut.edu.cn; Web: http://finechem.dlut.edu.cn/photochem
First published on 7th December 2011
Abstract
Difluoroboron (BF2) bound phenylacetylide was attached to the Pt(II) center of N⁁NPt(II) bisacetylide complex (Pt-1). Enhanced absorption in the visible region (ε = 1.37 × 104 M−1 cm−1 at 434 nm) and emissive 3IL excited state (τ = 0.92 μs, Φ = 3.3%) were observed for Pt-1, compared to the model complex dbbpy Pt(II) bisphenylacetylide (Pt-2, τ = 0.90 μs, Φ = 40.0%. dbbpy = 4,4′-di-tert-butyl-2,2′-bipyridine). Pt-1 was used as the triplet sensitizer for triplet–triplet annihilation (TTA) based upconversion and an upconversion quantum yield (ΦUC) of 8.9% was observed with 9,10-diphenylanthracence (DPA) as the triplet acceptor. In comparison, no upconversion was observed for the model complex Pt-2.
Introduction
N⁁NPt(II) bisacetylide complexes have attracted much attention, due to their phosphorescence upon photoexcitation (τ is up to μs).1–11Pt(II) bisacetylide complexes have been used for photovoltaics or photoinduced charge separation,12,13 photocatalysis,14 luminescent molecular probes,15–17 optical limitations,2,4,18 luminescent oxygen sensing,19,20 and recently for triplet–triplet annihilation (TTA) based upconversion.20–22The photophysical properties of Pt(II) bisacetylide complexes, such as the UV-vis absorption and the emission wavelength, as well as the lifetime of the triplet excited state, can be tuned by variation of the arylacetylide ligands,1 which is feasible from synthesis perspective. The Pt(II) bisacetylide complexes are usually prepared with arylacetylides, such as phenyl acetylides, or other polycyclic aromatic hydrogen moieties.6,7,23–26 Previously we attached naphthalimide,19,27coumarin,20 and naphthalenediimide moieties to the Pt(II) center.22 Greatly improved photophysical properties were observed for these new complexes, such as red-shifted absorption,22 room temperature (RT) near-IR emission and long-lived triplet excited states (lifetime up to 124 μs).22 However, we noticed that the structural diversity of the arylacetylides attached to the Pt(II) center in these complexes are still limited and much room is left to investigate the effect of the organic chromophores on photophysical properties of the complexes.
On the other hand, boron-containing fluorophores, such as the boron-dipyrromethene (BODIPY) and the BF2-bound fluorophores, have attracted much attention.28–30 These dyes usually show strong absorption in the visible region, high fluorescence quantum yield and high photostability.31 Thus BF2 or boron-containing fluorophores have been extensively used for molecular probes and light harvesting molecular arrays.32–38 To the best of our knowledge, however, no BF2 bound chromophores have been attached to the Pt(II) center of the Pt(II) bisacetylide complexes to investigate their effect on photophysical properties.
Herein for the first time a BF2 chromophore was attached to the Pt(II) center of Pt(II) bisacetylide complex (Pt-1, Scheme 1). We observed significant influence on the photophysical properties of the complex compared to the parent complex dbbpy Pt(II) bisphenylacetylide (Pt-2, Scheme 1. Where dbbpy = 4, 4′-di-tert-butyl-2,2′-bipyridine). For example, the UV-vis absorption of Pt-1 in the visible region was intensified. Furthermore, different from the 3MLCT (metal-to-ligand-charge-transfer) excited state of Pt-2,13IL (intraligand excited state) was assigned as the major component of the emissive triplet excited state of Pt-1, supported by emission at 77 K (small thermally induced Stokes shift, ΔES) and DFT calculations (spin density analysis). Pt-1 was used as the triplet sensitizer for TTA upconversion, where much higher upconversion quantum yield was observed than that with Pt-2. We propose that weakly phosphorescent complexes can be used as triplet sensitizers for TTA upconversion, and currently most of the triplet sensitizers are phosphorescent. Our findings may greatly increase the availability of the triplet sensitizers.
![Synthesis of Pt-1. The model complex Pt-2 and the triplet acceptor 9,10-diphenylanthracene (DPA) used in the TTA upconversion, as well as the model triplet sensitizer Ru(dmb)3[PF6]2, are presented. (i) Pd(PPh3)4, CuI, NEt3, ethynyltrimethylsilane, argon atmosphere, 80 °C, 3 h, then THF, TBAF, room temperature, 30 min, 36.8%; (ii) aniline, ethanol, 50 °C, 4 h, 95% (iii) CH2Cl2, NEt3, BF3–Et2O, room temperature, 20 min, 71%. (iv) CH2Cl2, i-Pr2NH, room temperature, 24 h, 54%.](/image/article/2012/RA/c1ra00434d/c1ra00434d-s1.gif) | ||
| Scheme 1 Synthesis of Pt-1. The model complex Pt-2 and the triplet acceptor 9,10-diphenylanthracene (DPA) used in the TTA upconversion, as well as the model triplet sensitizer Ru(dmb)3[PF6]2, are presented. (i) Pd(PPh3)4, CuI, NEt3, ethynyltrimethylsilane, argon atmosphere, 80 °C, 3 h, then THF, TBAF, room temperature, 30 min, 36.8%; (ii) aniline, ethanol, 50 °C, 4 h, 95% (iii) CH2Cl2, NEt3, BF3–Et2O, room temperature, 20 min, 71%. (iv) CH2Cl2, i-Pr2NH, room temperature, 24 h, 54%. | ||
Experimental
Materials and reagents
Dbbpy is a product of Aldrich. Other chemicals are analytically pure and were used as received without further purification. Solvents were dried or distilled before being used for synthesis or spectroscopic studies.Apparatus
NMR spectra were recorded on a 400 MHz Varian Unity Inova spectrophotometer. Mass spectra were recorded with a Q-TOF Micro spectrometer and MALDI micro MX. UV-Vis absorption spectra were recorded on a HP8453 UV-vis spectrophotometer. Fluorescence spectra were recorded on a Shimadzu RF5301PC or a Sanco 970 CRT spectrofluorometer. Fluorescence or phosphorescence lifetimes were measured on a Horiba Jobin Yvon Fluoro Max-4 (TCSPC). Nanosecond time-resolved transient absorption spectroscopy was obtained with LP-920 laser flash photolysis spectrometer (Edinburgh Instruments, U.K.). The emission at 77 K was measured with an Oxford Optistat DN™ cryostat (with liquid nitrogen filling) and FS920 fluorospectrometer (Edinburgh Instruments Ltd., U.K.).4-Ethynyl-2-((E)-(phenylimino)methyl)phenol (3)
The mixture of compound 2 (140.0 mg, 0.96 mmol), aniline (112.0 mg, 1.2 mmol), and ethanol (10 mL) was stirred at 50 °C for 4 h. After removal of solvent under reduced pressure, the residue was purified by column chromatography (silica gel; dichloromethane/n-hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) to give 201 mg of yellow crystal. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.57 (d, 1H, J = 2 Hz), 7.51 (d, 1H, J = 8.8 Hz), 7.44 (t, 2H, J = 8.0 Hz), 7.28−7.33 (m, 3H), 7.00 (d, 1H, J = 8.4 Hz). ESI-HRMS ([C15H10NO]+): calcd m/z = 220.0762, found m/z = 220.0767.
:1, v/v) to give 201 mg of yellow crystal. Yield: 95%. 1H NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 7.57 (d, 1H, J = 2 Hz), 7.51 (d, 1H, J = 8.8 Hz), 7.44 (t, 2H, J = 8.0 Hz), 7.28−7.33 (m, 3H), 7.00 (d, 1H, J = 8.4 Hz). ESI-HRMS ([C15H10NO]+): calcd m/z = 220.0762, found m/z = 220.0767.
Compound 4
The mixture of compound 3 (100.0 mg, 0.48 mmol), triethylamine (0.12 mL), boron fluoride ethyl ether (0.4 mL) and CH2Cl2 (5 mL) was stirred at room temperature for 30 min. After removal of solvent in vacuo, the residue was purified with column chromatography (silica gel; dichloromethane) to give 92 mg of yellow green solid. Yield: 71.0%. 1H NMR (400 MHz, CDCl3) δ 8.42 (s, 1H), 7.77 (d, 1H, J = 8.8 Hz), 7.68 (s, 1H), 7.51−7.57 (m, 5H), 7.15 (d, 1H, J = 8.8 Hz), 3.08 (s, 1H). ESI-HRMS ([C15H10BF2NO]+): calcd m/z = 269.0824, found m/z = 269.0827.Pt-1
Compound 4 (30.0 mg, 0.11 mmol) and Pt(dbbpy)Cl2 (31.0 mg, 0.06 mmol) were dissolved in dichloromethane (5 mL) under a argon atmosphere. Diisopropylamine (1.0 mL) was added to the above solution and CuI (2.0 mg, 0.001 mmol) was added. The mixture was stirred at 25 °C for 24 h. After removal of solvent under reduced pressure, the residue was purified with column chromatography (silica gel; dichloromethane/methanol, 50:1, v/v) to give 30.0 mg orange solid. Yield: 54%. 1H NMR (400 MHz, CDCl3) δ 9.56 (s, 1H), 9.28 (d, 1H, J = 6.0 Hz), 8.12 (s, 1H), 7.79 (s, 1H), 7.60 (d, 1H, J = 6.0 Hz),7.48 (d, 2H, J = 8.4 Hz), 7.24 (s, 1H), 6.84 (d, 1H, J = 8.8 Hz), 6.62 (s, 3H), 1.56 (s, 9H). 13C NMR (100 MHz, CDCl3/CD3OD) δ 165.2, 163.6, 157.9, 156.0, 150.8, 142.1, 137.3, 128.0, 127.4, 124.6, 123.6, 120.4, 118.7, 116.1, 91.2, 86.8, 37.2, 36.1, 34.0, 30.6. ESI-HRMS ([C48H42B2F4N4O2Pt+H]+): calcd m/z = 1000.3156, found m/z = 1000.3168.
Upconversion
Diode pumped solid state laser (DPSSL) with 445 nm output was used as the excitation source for the upconversion experiments. The output power of the DPSS laser can be adjusted continuously. The laser power was measured with a phototube. For the upconversion experiments, the mixed solution of the complex (triplet sensitizer) and DPA (triplet acceptor) was degassed for at least 15 min with N2 or Ar. Then the solution was excited with a laser. The upconverted fluorescence of DPA was recorded with a fluorospectrometer. The upconversion quantum yields were calculated with eqn 1, where ΦUC, Aunk, Iunk and ηunk represent the quantum yield, absorbance, integrated photoluminescence intensity and the refractive index of the samples and the solvents, respectively (eqn 1). The equation is multiplied by factor of 2 in order to make the maximum quantum yield to be unity (100%).39a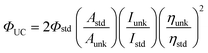 | (1) |
Computational details
The ground state structures of the complexes were optimized using density functional theory (DFT) with B3LYP functional and 6-31G(d) basis set (considered as the gas phase). The spin density was calculated based on the optimized triplet state geometry, the calculations were carried out on the 6-31G(d)/LanL2DZ level. All these calculations were performed with Gaussian 09W.40Results and discussions
Synthesis of Pt-1
Our design rationale is to directly attach the π-conjugated core of the BF2 chromophore to the Pt(II) center. Thus the heavy atom effect of Pt(II) on the BF2 chromophore can be maximized. At the same time, the emissive triplet excited state of the Pt(II) bisacetylide complex can be perturbed significantly with this approach. 4-Bromosalicylaldehyde was used as the starting material for Pt-1. Acetylide (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) was introduced by a Sonogashira coupling reaction. Condensation with aniline leads to Schiff base 3. The complexation with BF3 was carried out in the presence of triethylamine. Pt-1 was obtained in a satisfying yield.
C) was introduced by a Sonogashira coupling reaction. Condensation with aniline leads to Schiff base 3. The complexation with BF3 was carried out in the presence of triethylamine. Pt-1 was obtained in a satisfying yield.
UV-vis absorption and emission at room temperature
The UV-vis absorption of Pt-1 was compared with the parent complex Pt-2 (Fig. 1). We found the absorption of Pt-1 was enhanced compared to Pt-2. For example, the molar extinction coefficient (ε) of Pt-1 is 1.37 × 104 M−1 cm−1 at 434 nm, vs. 5.80 × 103 M−1 cm−1 at 424 nm for Pt-2. Intense absorption of visible light is beneficial for the photochemical related applications of the Pt(II) complexes, such as for photovoltaics or TTA upconversions.20–22,41–44 Intense absorption at 300 nm was also observed for Pt-1, which is attributed to the dbbpy related absorption, supported by the similar absorption band of Pt-2.7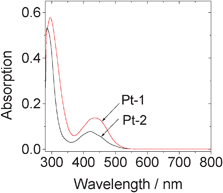 | ||
| Fig. 1 Absorption spectra of Pt-1 and Pt-2. In toluene, 1.0×10−5 M. 20 °C. | ||
The emission of Pt-1 was studied (Fig. 2). We found similar emission wavelength for Pt-1 and Pt-2. However, the phosphorescence quantum yield of Pt-1 (3.3%) is much smaller than Pt-2 (40.0%). The emission of Pt-1 can be significantly quenched by O2, thus supporting the assignment of the emission as phosphorescence. The phosphorescence lifetime of Pt-1 (τ) is 0.92 μs. For Pt-2, τ is 0.90 μs (Table 1).
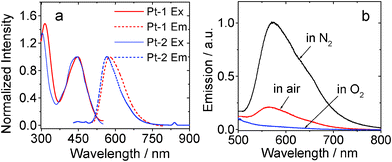 | ||
| Fig. 2 (a) Excitation and normalized emission spectra of Pt-1 and Pt-2. (b) Emission (λex = 434 nm) of Pt-1 under different atmosphere. In toluene, 1.0 × 10−5 M. 20 °C. | ||
| Compounds | λ abs/nma | ε b | λ em/nma | Φ (%)d | τ (μs)e | K sv a [103 M-1] | Φucf |
|---|---|---|---|---|---|---|---|
| a Result of compounds in toluene solution (1.0 × 10−5 M). b Molar extinction coefficient at the absorption maxima. ε: 104/cm−1 mol−1 dm3. c Result in MeCN. d Result in toluene, with [Ru(dmb)3][PF6]2 as the standard (ΦP = 0.073 in CH3CN). e Luminescence lifetime. 5.0 × 10−5 mol dm−3 in toluene. f Upconversion quantum yields. g Not applicable. | |||||||
| Pt-1 | 434 | 1.37 | 575 | 3.3 | 0.92 μs | 524.0 | 0.089 |
| Ligand 4 | 390 | 0.78 | 392 | 19.0 | 2.0 ns | − g | − g |
| Pt-2 | 424 | 0.58 | 570 | 40.0 | 0.90 μs | 4.5 | 0.000 |
| Ru-dmb c | 465 | 2.35 | 611 | 1.7 | 0.84 μs | 3.6 | 0.034 |
77 K emission spectra
Since Pt-1 shows slightly longer phosphorescence lifetime than the model complex Pt-2 and the phosphorescence quantum yield of Pt-1 is much smaller than that of Pt-2, we propose a different emissive excited state for Pt-1 compared to Pt-2. In order to study the features of the emissive excited state of Pt-1, the emission at 77 K was studied (Fig. 3). The emission band become narrower at 77 K. FWHM (full width at half maximum) for the emission at RT is 4519 cm−1. At 77 K, the FWHM is 3082 cm−1.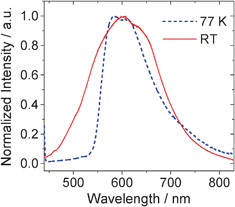 | ||
| Fig. 3
Phosphorescence spectra of Pt-1 at RT and 77 K (in EtOH:MeOH, 4:1, v/v). λex = 430 nm. | ||
Furthermore, the emission peak shows a very small blue shift upon cooling from RT to 77 K, the thermally induced Stokes shift (ΔES) is 570 cm−1. By comparison, the model complex Pt-2 was reported to show a significant ΔES value of 2470 cm−1.45 It is known that large thermally induced Stokes shifts indicate a MLCT feature,20,45–47 which is the case for Pt-2. For Pt-1, however, we propose the emissive triplet excited state is mainly in an 3IL feature.
In order to support the assignment of the 3IL feature of the emissive triplet excited state of Pt-1 from a theoretical perspective, the spin density surface of the T1 state of Pt-1 was compared (Fig. 4).48,49 For Pt-2, the spin density is distributed on the phenylacetylide ligand, the Pt(II) center and the dbbpy ligand.7 This is in line with the known fact that the emissive triplet excited state of Pt-2 is 3MLCT/3LLCT (ligand to ligand charge transfer).
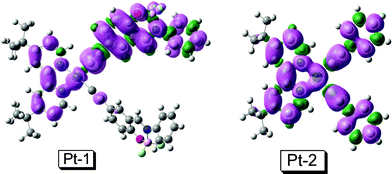 | ||
| Fig. 4 Isosurfaces of spin density of Pt-1 and Pt-2 at the optimized triplet state (isovalue: 0.0004). The calculation was performed at B3LYP/6-31G/LANL2DZ level with Gaussian 09W. | ||
For the new complex Pt-1, however, the spin density is more significantly distributed on the BF2 bound phenylacetylide ligand which takes a coplanar geometry with the Pt(II) coordination center, that is, the 3IL feature of the emissive triplet excited state of Pt-1 is more significant than that of Pt-2. This is in line with its small thermally induced Stokes shift (Fig. 3). The population of the 3IL state of Pt-1 may be due to slightly lower energy level of the 3IL state than the 3MLCT state. We also made an attempt to study the nanosecond time-resolved transient difference absorption spectra to reveal the feature of the triplet excited state,20,22,50 but no satisfying transients can be observed.
Triplet–triplet annihilation based upconversion
As Pt-1 shows enhanced UV-vis absorption and long-lived triplet excited state at the μs scale, thus Pt-1 can be used as a triplet sensitizer for some photophysical processes that require a triplet excited state to initiate. Recently TTA based upconversion has attracted much attention due to its readily tunable excitation/emission wavelength.20–22,39,44,51,52 Furthermore, the excitation power density required for TTA based upconversion is much lower than the other upconversion techniques, such as two-photon-absorption dyes (MW cm−2).53 It has been demonstrated that excitation power density similar to sunlight is sufficient for excitation of TTA upcovnersion.54,55 Triplet sensitizers are required in TTA upconversion to harvest the excitation light and transfer the energy to the triplet acceptor, via the triplet–triplet-energy-transfer process (TTET, see Scheme 2 for the Jablonski diagram of the TTA upconversion).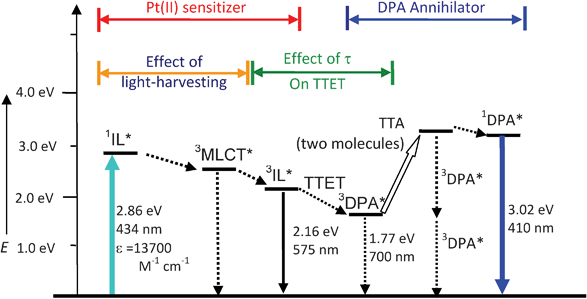 | ||
| Scheme 2 Qualitative Jablonski diagram illustrating the sensitized TTA upconversion process between Pt-1 and DPA. The effect of the light-harvesting ability and the luminescence lifetime of the Pt(II) sensitizer on the efficiency of the TTA upconversion is also shown. E is energy. GS is ground state (S0). 1IL* is intraligand singlet excited state. IC is inner conversion. ISC is intersystem crossing. 3MLCT* is the Pt(II) based metal-to-ligand-charge-transfer triplet excited state. 3IL* is intraligand triplet excited state. TTET is triplet–triplet-energy-transfer. 3DPA* is the triplet excited state of DPA. TTA is triplet–triplet annihilation. 1DPA* is the singlet excited state of DPA. The emission bands observed for the sensitizers alone is the 3IL emissive excited state. The emission bands observed in the TTA experiment is the simultaneous 3IL* emission (phosphorescence) and the 1DPA* emission (fluorescence). | ||
Currently the triplet sensitizers for TTA upconversion are limited to a large extent to the Pt(II) porphyrin complexes.39 However, it is difficult to tune the T1 state energy levels of these sensitizers by chemical modification. Thus it is highly desired to develop new triple sensitizers for which the T1 state energy level can be readily tuned. N⁁NPt(II) bisacetylide complexes can fulfil this purpose because the photophysical properties of these complexes are largely dependent on the structure of the arylacetylide.1,6,7
A N⁁C⁁NPt(II) acetylide complex has been used as the triplet sensitizer for TTA upconversion, but the upconversion quantum yield is not satisfying (<1.5%).21 Recently we used a coumarin acetylide or naphthalenediimide (NDI)-containing Pt(II) bisacetylide complex and coumarin containing cyclometalated Ir(III) complexes as the triplet sensitizer for the TTA upconversion.20,22,44,52 However, to the best of our knowledge, no BF2 bound acetylide-containing Pt(II) complexes have been used as a triplet sensitizer for TTA upconversion. Herein for the first time we studied the application of a Pt(II) complex with BF2 bound chromophore for TTA upconversion, since we have shown that Pt-1 is with enhanced absorption in the visible region and long-lived triplet excited state. These photophysical properties are ideal for TTA upconversion.
First the emission of Pt-1 and Pt-2 upon 445 nm laser excitation were recorded (Fig. 5a). Pt-1 shows much weaker emission than Pt-2, due to its smaller phosphorescence quantum yield than Pt-2 (Table 1). The emission of a benchmark complex Ru(dmb)3[PF6]2 was presented for comparison because this complex shows absorption in the same region with that of Pt-1 and Pt-2.56 Second, the emission of the complexes were studied in the presence of triplet acceptor DPA (Fig. 5b). The characteristic blue emission of DPA was observed in the range 400–500 nm upon selective excitation of triplet sensitizers (Pt-1) in the presence of DPA. Irradiation of DPA alone with a 445 nm laser does not produce the blue emission, thus proving that the blue emission in the range 400 nm–500 nm with sensitizer Pt-1 is due to the upconverted fluorescence of DPA.
![TTA upconversion with the complexes as the sensitizers and DPA as the triplet acceptor. (a) Phosphorescence profile of sensitizers without DPA. (b) Upconverted emission in the presence of DPA. (c) Photography of the upconversion with Pt-1 and (d) with Pt-2. Excited with 445 nm laser. The asterisks in a and b indicated the scattered laser. c [sensitizers] = 1.0 × 10−5 M. c [DPA] = 4.3 × 10−5 M. In deaerated toluene. 20 °C.](/image/article/2012/RA/c1ra00434d/c1ra00434d-f5.gif) | ||
| Fig. 5 TTA upconversion with the complexes as the sensitizers and DPA as the triplet acceptor. (a) Phosphorescence profile of sensitizers without DPA. (b) Upconverted emission in the presence of DPA. (c) Photography of the upconversion with Pt-1 and (d) with Pt-2. Excited with 445 nm laser. The asterisks in a and b indicated the scattered laser. c [sensitizers] = 1.0 × 10−5 M. c [DPA] = 4.3 × 10−5 M. In deaerated toluene. 20 °C. | ||
The triplet sensitizers demonstrated drastically different upconversion efficiency. The most significant upconversion was observed with Pt-1. For Pt-2, however, weak upconversion was observed. The Ru(dmb)3[PF6]2 gives a moderate upconversion.56 We attribute the significant upconversion of Pt-1 to its intense absorption and long-lived T1 excited state, with which the TTET, the key step of TTA upconversion, can be enhanced (Scheme 2).
Interestingly, we found that the quenched phosphorescence peak area is much smaller than the upconverted fluorescence emission peak area. This is abnormal since the currently accepted TTA upconversion mechanism, as well as the experimental results that have been observed, requires that the quenched phosphorescence peak area must be at least two fold of the upconverted fluorescence peak areas.39 In order to rationalize this discrepancy between the experimental observation and the conventional understanding of the TTA upconversion mechanism, we propose that the sensitizer molecules at the triplet excited state that otherwise non-emissive were involved in the TTET process.20,22,44
These results indicate that the triplet excited state of Pt-1 is actually more efficiently populated upon photoexcitation than the phosphorescence quantum yield suggested (Table 1). It should be pointed out that a wide variety of applications of transition metal complexes are actually not luminescence related, such as photovoltaics, photocatalysis and upconversions, etc. Thus, we propose that weakly phosphorescent, or even non-phosphorescent transition metal complexes, can be used as the triplet sensitizers for TTA upconversion, or any other appropriate photophysical processes. This concept will greatly increase the availability of the triplet sensitizers for TTA upconversion.
The upconversion is clearly visible with un-aided eyes (Fig. 5c and 5d). For the sensitizers alone, red (Pt-1) or yellow (Pt-2) emission was observed. In the presence of the triplet acceptor DPA, strong blue emission was observed for Pt-1, but for Pt-2, however, the yellow emission is persistent, due to the lack of upconversion with Pt-2 (Fig. 5b).
The role of the dark excited state of Pt-1 played in the TTA upconversion is clearly demonstrated in Fig. 6. With increasing the DPA concentration, the phosphorescence of Pt-1 shows only minor changes, but the upconverted fluorescence emission was enhanced significantly. The intensified upconverted emission is derived from the dark excited state of the sensitizer.
![Variation of the upconversion intensity with increasing DPA concentration: the role of the dark excited state in upconversion. λex[Pt-1] = 445 nm, phosphorescence was measured as a function of DPA concentration. In toluene. C [Pt-1] = 1.0 × 10−5 M, 20 °C.](/image/article/2012/RA/c1ra00434d/c1ra00434d-f6.gif) | ||
| Fig. 6 Variation of the upconversion intensity with increasing DPA concentration: the role of the dark excited state in upconversion. λex[Pt-1] = 445 nm, phosphorescence was measured as a function of DPA concentration. In toluene. C [Pt-1] = 1.0 × 10−5 M, 20 °C. | ||
The key step of the TTA upconversion, that is, the TTET process, was studied quantitatively by monitoring the quenching of the phosphorescence of the sensitizers while increasing the DPA concentration (Fig. 7). The results show that the phosphorescence of Pt-1 was significantly quenched by DPA, but the quenching effect with Pt-2 and Ru(dmb)3[PF6]2 was much smaller.
![Efficiency of the TTET process: quenching of the phosphorescence of (a) Pt-1 (λex = 450 nm) and (b) Pt-2 (λex = 420 nm) with increasing the DPA concentration in deaerated toluene. c[complex] = 1.0 × 10−5 M, 20 °C.](/image/article/2012/RA/c1ra00434d/c1ra00434d-f7.gif) | ||
| Fig. 7 Efficiency of the TTET process: quenching of the phosphorescence of (a) Pt-1 (λex = 450 nm) and (b) Pt-2 (λex = 420 nm) with increasing the DPA concentration in deaerated toluene. c[complex] = 1.0 × 10−5 M, 20 °C. | ||
The Stern–Volmer quenching curves were constructed (Fig. 8). The Stern–Volmer quenching constant of Pt-1 is ca. 100-fold of that of Pt-2 and Ru(bmd)3[PF6]2 (Table 1), unambiguously demonstrating the efficient TTET process with Pt-1 compared to that with Pt-2 or Ru(bmd)3[PF6]2. We noticed the Pt-1 and Pt-2 show similar phosphorescence lifetimes. We propose that the difference between the 3IL and the 3MLCT excited states may be responsible for the different TTET efficiencies of Pt-1 and Pt-2.
![The Stern–Volmer quenching of the phosphorescence in the presence of DPA. c [sensitizers] = 1.0 × 10−5 M. λex [Pt-1] = 450 nm, λex[Pt-2] = 420 nm, λex[Ru(dmb)3] = 440 nm, phosphorescence was measured as a function of DPA concentration. In toluene. 20 °C.](/image/article/2012/RA/c1ra00434d/c1ra00434d-f8.gif) | ||
| Fig. 8 The Stern–Volmer quenching of the phosphorescence in the presence of DPA. c [sensitizers] = 1.0 × 10−5 M. λex [Pt-1] = 450 nm, λex[Pt-2] = 420 nm, λex[Ru(dmb)3] = 440 nm, phosphorescence was measured as a function of DPA concentration. In toluene. 20 °C. | ||
For example, the 3IL spin density surface is more localized on the ligand. This may make the collision between the sensitizer and the acceptor, i.e. the TTET, more efficient for Pt-1 than that of Pt-2. The spin density surface of Pt-2 is more de-localized (Fig. 4). This de-localized spin density surface of Pt-2 may make the TTET between Pt-2 and the acceptor less efficient. The efficient TTET process is responsible for the significant upconversion with Pt-1.
The photophysics of Pt-1 sensitized TTA upconversion is summarized in Scheme 2. The photoexcitation of the sensitizer will produce the singlet excited state, then the triplet excited state will be populated via the heavy atom effect of the Pt(II) (intersystem crossing, ISC). TTET between the triplet sensitizer and the triplet acceptor will produce the acceptor at the triplet excited state. Annihilation of the acceptor at the triplet excited state produces the acceptor at the singlet excited state, thus the radiative decay of the acceptor as the singlet excited state produces the upconverted fluorescence of DPA. The two key properties of the sensitizers for TTA upconversion are the light harvesting ability of the sensitizer and the lifetime of the T1 excited state of the sensitizer. Intense absorption will potentially produce more excited sensitizer molecules, long-lived T1 excited state of the sensitizer will enhance the TTET process, both effects will improve the upconverted fluorescence emission. Pt-1 shows intensified absorption than the model complex Pt-2, thus Pt-1 is a desired triplet sensitizer for TTA upconversion.
Conclusions
In summary, difluoroboron (BF2) bound phenylacetylide was attached to the Pt(II) center of NˆNPt(II) bisacetylide complex (Pt-1). Enhanced absorption in the visible region (ε = 1.37 × 104 M−1 cm−1) and room temperature phosphorescence (ΦP = 3.3%) was observed. The long-lived emissive triplet excited state of Pt-1 was assigned as the 3IL excited state (τ = 0.92 μs), which was supported by the emission at 77 K (thermally induced Stokes shift is 570 cm−1) and the DFT calculations (spin density surface of the triplet excited state is mainly distributed on the BF2 bound acetylide ligand). Pt-1 was used as a triplet sensitizer for triplet–triplet annihilation (TTA) based upconversion and upconversion quantum yield of was observed as 8.9% with 9,10-diphenylanthracence as the triplet acceptor. Our result will be useful for design of new phosphorescent Pt(II) complexes and for their applications in photovoltaics, upconversions, etc.Acknowledgements
We thank the NSFC (20972024 and 21073028), the Fundamental Research Funds for the Central Universities (DUT10ZD212 and DUT11LK19), Ministry of Education (SRFDP-200801410004 and NCET-08-0077), the Royal Society (UK) and NSFC (China-UK Cost-Share Science Networks, 21011130154), the Education Department of Liaoning Province (2009T015) and Dalian University of Technology for financial support.References
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- G. Zhou, W. Wong, S. Poon, C. Ye and Z. Lin, Adv. Funct. Mater., 2009, 19, 531–544 CrossRef CAS.
- W. Wong, Dalton Trans., 2007, 4495–4510 RSC.
- G. Zhou and W. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- F. N. Castellano, I. E. Pomestchenko, E. Shikhova, F. Hua, M. L. Muro and N. Rajapakse, Coord. Chem. Rev., 2006, 250, 1819–1828 CrossRef CAS.
- C. E. Whittle, J. A. Weinstein, M. W. George and K. S. Schanze, Inorg. Chem., 2001, 40, 4053–4062 CrossRef CAS.
- M. Hissler, W. B. Connick, D. K. Geiger, J. E. McGarrah, D. Lipa, R. J. Lachicotte and R. Eisenberg, Inorg. Chem., 2000, 39, 447–457 CrossRef CAS.
- E. O. Danilov, I. E. Pomestchenko, S. Kinayyigit, P. L. Gentili, M. Hissler, R. Ziessel and F. N. Castellano, J. Phys. Chem. A, 2005, 109, 2465–2471 CrossRef CAS.
- T. Dube, J. W. Faller and R. H. Crabtree, Inorg. Chem., 2002, 41, 5561–5565 CrossRef CAS.
- P. P. Elliott, Annu. Rep. Prog. Chem., Sect. A, 2010, 106, 526–552 RSC.
- (a) S. C. F. Kui, Y. Law, G. Tong, W. Lu, M. Yuen and C. Che, Chem. Sci., 2011, 2, 221–228 RSC; (b) D. Nolan, B. Gil, F. A. Murphy and S. M. Draper, Eur. J. Inorg. Chem., 2011, 3248–3256 CrossRef CAS.
- J. E. McGarrah, Y. Kim, M. Hissler and R. Eisenberg, Inorg. Chem., 2001, 40, 4510–4511 CrossRef CAS.
- Q. Wang and W. Wong, Polym. Chem., 2011, 2, 432–440 RSC.
- X. Wang, S. Goeb, Z. Ji, N. A. Pogulaichenko and N. Castellano Felix, Inorg. Chem., 2011, 50, 705–707 CrossRef CAS.
- Q. Zhao, F. Li and C. Huang, Chem. Soc. Rev., 2010, 39, 3007–3030 RSC.
- J. Ni, L. Zhang, H. Wen and Z. Chen, Chem. Commun., 2009, 3801–3803 RSC.
- P. Lanoë, H. L. Bozec, J. A. Gareth Williams, J. Fillaut and V. Guerchais, Dalton Trans., 2010, 39, 707–710 RSC.
- G. Zhou, W. Wong, Z. Lin and C. Ye, Angew. Chem., Int. Ed., 2006, 45, 6189–6193 CrossRef CAS.
- H. Guo, S. Ji, W. Wu, W. Wu, J. Shao and J. Zhao, Analyst, 2010, 135, 2832–2840 RSC.
- H. Sun, H. Guo, W. Wu, X. Liu and J. Zhao, Dalton Trans., 2011, 40, 7834–7841 RSC.
- P. Du and R. Eisenberg, Chem. Sci., 2010, 1, 502–506 RSC.
- Y. Liu, W. Wu, J. Zhao, X. Zhang and H. Guo, Dalton Trans., 2011, 40, 9085–9089 RSC.
- X. Han, L. Wu, G. Si, J. Pan, Q. Yang, L. Zhang and C. Tung, Chem.–Eur. J., 2007, 13, 1231–1239 CrossRef CAS.
- A. A. Rachford, S. Goeb and F. N. Castellano, J. Am. Chem. Soc., 2008, 130, 2766–2767 CrossRef CAS.
- Y. Sun, Z. M. Hudson, Y. Rao and S. Wang, Inorg. Chem., 2011, 50, 3447–3457 CrossRef.
- F. Nastasi, F. Puntoriero, S. Campagna, J. Olivierb and R. Ziessel, Phys. Chem. Chem. Phys., 2010, 12, 7392–7402 RSC.
- H. Guo, M. L. Muro-Small, S. Ji, J. Zhao and F. N. Castellano, Inorg. Chem., 2010, 49, 6802–6804 CrossRef CAS.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS.
- R. Ziessel and A. Harriman, Chem. Commun., 2011, 47, 611–631 RSC.
- G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS.
- X. Zhang, Y. Xiao and X. Qian, Org. Lett., 2008, 10, 29–32 CrossRef CAS.
- A. Coskun and E. U. Akkaya, J. Am. Chem. Soc., 2006, 128, 14474–14475 CrossRef CAS.
- H. Sunahara, Y. Urano, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 5597–5604 CrossRef CAS.
- Y. Wu, X. Peng, B. Guo, J. Fan, Z. Zhang, J. Wang, A. Cui and Y. Gao, Org. Biomol. Chem., 2005, 3, 1387–1392 CAS.
- T. Cheng, Y. Xu, S. Zhang, W. Zhu, X. Qian and L. Duan, J. Am. Chem. Soc., 2008, 130, 16160–16161 CrossRef CAS.
- H. Guo, Y. Jing, X. Yuan, S. Ji, J. Zhao, X. Li and Y. Kan, Org. Biomol. Chem., 2011, 9, 3844–3853 CAS.
- J. Shao, H. Guoa, S. Ji and J. Zhao, Biosens. Bioelectron., 2011, 26, 3012–3017 CrossRef CAS.
- (a) T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560–2573 CrossRef CAS; (b) J. Zhao, S. Ji and H. Guo, RSC Adv., 2011, 1, 937 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. FoxGaussian 09 (Revision A.1), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- W. Wu, W. Wu, S. Ji, H. Guo, P. Song, K. Han, L. Chi, J. Shao and J. Zhao, J. Mater. Chem., 2010, 20, 9775–9786 RSC.
- W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2010, 4470–4482 CrossRef CAS.
- W. Wu, C. Cheng, W. Wu, H. Guo, S. Ji, P. Song, K. Han, J. Zhao, X. Zhang, Y. Wu and G. Du, Eur. J. Inorg. Chem., 2010, 4683–4696 CrossRef CAS.
- (a) W. Wu, W. Wu, S. Ji, H. Guo and J. Zhao, Dalton Trans., 2011, 40, 5953–5963 RSC; (b) S. Ji, W. Wu, W. Wu, H. Guo and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 1626–1629 CrossRef CAS; (c) S. Ji, H. Guo, W. Wu, W. Wu and J. Zhao, Angew. Chem., Int. Ed., 2011, 50, 8283–8286 CrossRef CAS.
- I. E. Pomestchenko and F. N. Castellano, J. Phys. Chem. A, 2004, 108, 3485–3492 CrossRef CAS.
- S. Develay, O. Blackburn, A. L. Thompson and J. A. Gareth Williams, Inorg. Chem., 2008, 47, 11129–11142 CrossRef CAS.
- C. She, A. A. Rachford, X. Wang, S. Goeb, A. El-Ballouli, F. N. Castellano and J. T. Hupp, Phys. Chem. Chem. Phys., 2009, 11, 8586–8591 RSC.
- E. Baranoff, S. Fantacci, F. De Angelis, X. Zhang, R. Scopelliti, M. Grätzel and M. Nazeeruddin, Inorg. Chem., 2011, 50, 451–462 CrossRef CAS.
- K. Hanson, A. Tamayo, V. V. Diev, M. T. Whited, P. I. Djurovich and M. E. Thompson, Inorg. Chem., 2010, 49, 6077–6084 CrossRef CAS.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963 RSC.
- A. Monguzzi, J. Mezyk, F. Scotognella, R. Tubino and F. Meinardi, Phys. Rev. B, 2008, 78, 195112–1 CrossRef.
- J. Sun, W. Wu, H. Guo and J. Zhao, Eur. J. Inorg. Chem., 2011, 3165–3173 CrossRef CAS.
- J. Lee, C. Lim, Y. Tian, J. Han and B. Cho, J. Am. Chem. Soc., 2010, 132, 1216–1217 CrossRef CAS.
- A. Monguzzi, R. Tubino and F. Meinardi, Phys. Rev. B, 2008, 77, 155122–1 CrossRef.
- S. Baluschev, V. Yakutkin, T. Miteva, Y. Avlasevich, S. Chernov, S. Aleshchenkov, G. Nelles, A. Cheprakov, A. Yasuda, K. Müllen and G. Wegner, Angew. Chem., Int. Ed., 2007, 46, 7693–7696 CrossRef CAS.
- R. R. Islangulov, D. V. Kozlov and F. N. Castellano, Chem. Commun., 2005, 3776–3778 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: more structural characterization data and Z-matrix of the compounds for DFT calculations. See DOI: 10.1039/c1ra00434d/ |
| This journal is © The Royal Society of Chemistry 2012 |