Physical properties of a molecular conductor (BEDT-TTF)2I3 nanohybridized with silica nanoparticles by dry grinding
Akira
Funabiki
a,
Hiroki
Sugiyama
a,
Tomoyuki
Mochida
*a,
Kunihiro
Ichimura
b,
Takashi
Okubo
cd,
Ko
Furukawa
e and
Toshikazu
Nakamura
e
aDepartment of Chemistry, Graduate School of Science, Kobe University, Kobe, Hyogo 657-8501, Japan. E-mail: tmochida@platinum.kobe-u.ac.jp; Fax: +81-78-803-5679
bFaculty of Science, Toho University, Funabashi, Chiba 274-8510, Japan
cSchool of Science and Engineering, Kinki University, Higashi-Osaka, Osaka 577-8502, Japan
dPRESTO, Japan Science and Technology Agency, Kawaguchi, Saitama 332-0012, Japan
eInstitute for Molecular Science, Okazaki, Aichi 444-8787, Japan
First published on 7th December 2011
Abstract
The dry grinding of a mixture of bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) and silica nanoparticles has produced powdery (BEDT-TTF)–silica nanocomposites. The (BEDT-TTF)–silica nanocomposites are readily doped with iodine in hexane dispersion to give powdery nanocomposites of (BEDT-TTF)2I3–silica. XRD and TEM measurements suggest that (BEDT-TTF)2I3 in the nanocomposite exists as shell layers of core-shell-type nanoparticles and as nanometre-sized crystals incorporated into hollow sites of aggregated silica nanoparticles. Magnetic susceptibility measurements reveal that the nanocomposites accompanied a large number of Curie spins attributable to surface molecules of the core-shell-type nanoparticles. The nanocomposites show a magnetic susceptibility change corresponding to the metal-insulator transition of α-(BEDT-TTF)2I3 in a broad temperature range of 110–140 K, which is attributed to the properties of the nanocrystalline components. Doping in diethyl ether dispersion leads to higher amounts of the nanocrystalline component being obtained. The doping of (BEDT-TTF)–silica nanocomposites by dry grinding produces a paramagnetic powder containing amorphous (BEDT-TTF)2I3, which possesses a Curie spin concentration of 50%. The effects of annealing on these nanocomposites are investigated. The electrical conductivity of the compaction pellets of (BEDT-TTF)–silica nanocomposites is enhanced by iodine doping to reach approximately 10−6 S cm−1, but the value is much lower than that of the bulk crystals (101 S cm−1).
Introduction
Charge-transfer (CT) complexes, including those derived from bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF), exhibit a variety of peculiar electronic properties in the solid state, such as electrical conductivity, magnetism, and phase transitions.1 Nanoscale downsizing of CT complexes is of interest because of its effect on the physical and chemical properties of CT complexes, as a result of the large increases in the fractions of surface atoms or molecules.2 In this context, micrometre- to nanometre-sized crystals of CT complexes have been prepared by self-assembly in solutions,3 electrolysis,4 vapor deposition,5 and reprecipitation.6 Recently, fine nanoparticles of CT complexes with sizes of the order of 10 nm have been fabricated by solution reactions in the presence of ionic liquids.7 The downsizing effect has been observed in the phase transition behavior4 and the optical and electrical properties of CT complexes.6 Extreme downsizing of an organic superconductor has also been investigated.8Ichimura et al. have recently reported that dry grinding of molecular crystals in the presence of silica nanoparticles affords core-shell-type hybrid nanoparticles.9 Unlike conventional grinding techniques, this method enables convenient nanodownsizing of molecular solids such as organic pigments,9a, b molecular crystals,9c–f and polymers.9g In the resultant nanocomposites, surfaces of aggregated primary nanoparticles of silica are covered with ultrathin shell layers of organic solids, whereas a nanometre-sized crystalline fraction fills hollows at the joint sites of aggregated primary silica particles.9b,e We are interested in the application of the nanocomposites to solid-state reactions, as organic solid-state reactions have received much attention because of their simplicity and specific reactivity.10 Based on this idea, we previously prepared nanosized CT complexes of MPc (metal phthalocyanine) with iodine by the reaction of MPc-silica nanocomposites with iodine.11 The enlarged surface areas of the nanocomposites lead to extreme reactivity of the nanocomposite with iodine, whereas bulk MPc is much less reactive.
In this context, we plan to prepare nanosized CT complexes based on these methods and analyze their physical properties. In this study, we focus on (BEDT-TTF)2I3, which is a triiodide salt of BEDT-TTF and is one of the most well-known molecular conductors.1 Bulk crystals of (BEDT-TTF)2I3 exhibit characteristic electronic properties depending on their crystal forms. α-(BEDT-TTF)2I3 exhibits a sharp metal-insulator (M–I) transition at 135 K,12 and the material has drawn special attention as a zero-gap semiconductor.13 The α-form of (BEDT-TTF)2I3 is transformed into the β-form when it is annealed at 376 K;14 the β-form exhibits a superconducting transition at low temperatures.15 A few methods of downsizing (BEDT-TTF)2I3 have been reported; these include vapor deposition to prepare thin films16 and doping of BEDT-TTF dispersed in polymer films with iodine.17
Here, we report the fabrication and characterization of (BEDT-TTF)2I3–silica nanocomposites by iodine doping into (BEDT-TTF)–silica nanocomposites obtained by dry grinding. The physical properties of the nanocomposites, including phase transition, magnetic susceptibility, and electrical conductivity, are presented. The ratios of (BEDT-TTF)2I3 as shell layers and as nanocrystals were estimated on the basis of the magnetic properties.
Experimental
General methods
XRD data were recorded on a Rigaku SmartLab diffractometer using Cu-Kα radiation. Electron micrographs were obtained using a JEOL JEM-2010 transmission electron microscope (TEM) and a JEOL JSM-5610 LVS scanning electron microscope (SEM). Specimens for TEM observation were prepared by dropping an aqueous dispersion of a powdery sample on a copper grid supported by a carbon membrane, followed by drying in vacuo at room temperature. SEM observation was carried out by putting powdery samples on double-sided conductive carbon tape. Magnetic measurements were carried out using a Quantum Design MPMS-XL7 SQUID susceptometer in the applied field 10 kOe. The magnetic susceptibility was estimated by subtracting the Pascal diamagnetic part of the (BEDT-TTF)2I3 molecule (−4.90 × 10−4 emu mol−1) and diamagnetic contribution of silica (−2.96 × 10−6 emu mol−1) from the total susceptibility. FT-IR spectra were recorded on a Perkin Elmer Spectrum 1000 spectrometer using KBr plates. For electrical conductivity measurements, powdery samples were pressed into pellets under 500–600 kg cm−2 using a hydraulic molding press. Au paste or brass plates were attached on both sides of the pellets (thickness: 0.3–0.5 mm) as electrodes. Electrical conductivity was measured using the AC impedance technique at room temperature with an applied voltage of 100 mV using a Solartron 1260 impedance analyzer (four-probe method, frequency range: 10−1–106 Hz). The temperature dependence of the conductivity was measured using a Wayne Kerr 6440B precision component analyzer in a temperature range of 300–100 K.Preparation of iodine-doped (BEDT-TTF)–silica nanocomposites
Stöber silica nanoparticles (Toso Silica; NIPSIL VN3, primary particle diameter of 14.1 nm) were modified with methylhydrogenpolysiloxane to reduce the surface polarity of the powder so as to improve the affinity of the silica surfaces with organic compounds.9a Such surface-modified silica nanoparticles were donated by Toda Kogyo Co., Ltd. BEDT-TTF was purchased from Tokyo Chemical Industry Co., Ltd. and recrystallized from chlorobenzene prior to use. Bulk crystals of α-(BEDT-TTF)2I3 were prepared according to the methods described in the literature.14(BEDT-TTF)–silica nanocomposites were fabricated using the following procedure. Surface-modified silica nanoparticles (100 mg) and a desired amount of BEDT-TTF were placed in a zirconia vessel (12 mL) and milled using a Fritsch P-7 planetary mill with the aid of nylon beads (400 rpm, 4 h). To dope with iodine under wet conditions, the obtained nanocomposites were dispersed in hexane or diethyl ether containing a stoichiometric amount of iodine, and the dispersion was stirred for 1 h. The resulting black powders were collected by filtration, washed with hexane or diethyl ether, and dried under vacuum. Alternatively, iodine doping was carried out under dry conditions. The (BEDT-TTF)–silica nanocomposites and a stoichiometric amount of iodine were milled using nylon beads, and the resulting powders were washed with diethyl ether and dried under vacuum.
Results and discussion
Preparation of (BEDT-TTF)–silica nanocomposites
Dry grinding of the mixtures of BEDT-TTF and surface-modified silica nanoparticles (primary particle diameter of 14.1 nm) produced (BEDT-TTF)–silica nanocomposites. Nanoscale hybridization of the powders was confirmed by TEM observations (Fig. 1a). Averaged diameters of the primary particles of the nanocomposites of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 (w/w) BEDT-TTF/silica ratios are 19.3 nm and 17.2 nm, respectively. These values agree reasonably well with the diameters of 18.2 nm and 16.2 nm, respectively, calculated from the mixing ratios,9a and the enlarged diameters supports the core-shell structures. In contrast, nanoscale hybridization did not proceed when silica nanoparticles without surface modification were used, as indicated by the presence of large particles of irregular shape (Fig. 1b). These results reveal the essential role of surface modification of the silica nanoparticles in hybridization, as has been observed in other cases.9
:1 and 1:2 (w/w) BEDT-TTF/silica ratios are 19.3 nm and 17.2 nm, respectively. These values agree reasonably well with the diameters of 18.2 nm and 16.2 nm, respectively, calculated from the mixing ratios,9a and the enlarged diameters supports the core-shell structures. In contrast, nanoscale hybridization did not proceed when silica nanoparticles without surface modification were used, as indicated by the presence of large particles of irregular shape (Fig. 1b). These results reveal the essential role of surface modification of the silica nanoparticles in hybridization, as has been observed in other cases.9
 | ||
| Fig. 1
TEM images of powders obtained by grinding the 1:2 (w/w) mixtures of (a) BEDT-TTF and surface-modified silica and (b) BEDT-TTF and unmodified silica. | ||
Nanocomposite powders of BEDT-TTF and silica nanoparticles in a 1:2 (w/w) ratio were employed in the following experiments, unless otherwise noted. As shown in Fig. 2A, the (BEDT-TTF)–silica nanocomposite exhibited XRD peaks corresponding to those of bulk BEDT-TTF, showing a decrease in peak intensity and an increase in the peak widths relative to its bulk counterpart. The crystallite size as estimated by Scherrer's equation is 40.7 nm, showing a remarkable reduction relative to that of bulk crystals of approximately 500 nm. As discussed in our previous work,9 the thickness of shell layers consisting of organic crystallites is estimated to be a few nanometres for ideal core-shell nanostructures; therefore, peak widths in the XRD patterns should be extremely broad. Accordingly, the components with a crystal size of a few tens of nanometres can be ascribed to nanocrystalline fractions that filled in hollow sites formed as a result of the dense aggregation of primary silica nanoparticles, as observed in other silica-nanocomposites.9b
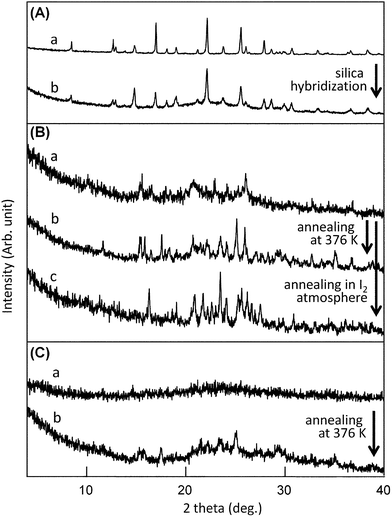 | ||
| Fig. 2 (A) Powder XRD patterns of (a) BEDT-TTF (bulk powders) and (b) (BEDT-TTF)–silica nanocomposites (1:2 w/w). (B) Powder XRD patterns of (a) (BEDT-TTF)2I3–silica nanocomposites prepared by doping in hexane, (b) after annealing the sample at 376 K, and (c) nanocomposites after annealing in iodine atmosphere. (C) Powder XRD patterns of (a) (BEDT-TTF)2I3–silica nanocomposites prepared under dry conditions, and (b) after annealing the nanocomposites at 376 K. | ||
Preparation of (BEDT-TTF)2I3–silica nanocomposites
The iodine doping of (BEDT-TTF)–silica nanocomposites was achieved efficiently under wet conditions by stirring a dispersion of nanocomposite powders for 1 h in hexane containing a stoichiometric amount of iodine. A quantitative reaction occurred readily to produce dark brown powders of (BEDT-TTF)2I3–silica nanocomposite. Since BEDT-TTF is completely insoluble in hexane, the doping reaction likely occurred in the solid state. The TEM image of the 1:2 (w/w) iodine-doped nanocomposite is shown in Fig. 3. The average diameter of primary particles in the nanocomposite is approximately 20 nm, which is slightly greater than that before doping. Fig. 2B (line a) shows the XRD pattern of the 1:2 (w/w) hybrid powders, in which weak diffraction peaks corresponding to α-(BEDT-TTF)2I3 were observed. The crystallite size is estimated to be 30.4 nm according to Scherrer's equation, which is slightly smaller than the size of 40.7 nm before doping. Although a few weak unidentifiable diffraction peaks are observed in the XRD pattern, there are no peaks of β-(BEDT-TTF)2I3 or unreacted BEDT-TTF.
 | ||
| Fig. 3
TEM image of (BEDT-TTF)2I3–silica hybrid (BEDT-TTF:silica = 1:2 w/w) prepared by doping in hexane. | ||
The iodine doping was also carried out in diethyl ether. In this case, the product exhibited stronger XRD peaks corresponding to α-(BEDT-TTF)2I3 than those for the nanocomposites doped in hexane. It seems probable that part of the molecules in the nanocomposites dissolved into diethylether because of their enlarged surface area, leading to the formation of the larger amount of nanocrystalline salts from solution.
Next, the effect of annealing was investigated. The annealing of the (BEDT-TTF)2I3 nanocomposites prepared in hexane at 376 K for 2 h leads to an increase in intensity of XRD diffraction peaks, indicating the development of crystallinity while the crystal form was maintained. Annealing under iodine atmosphere lead to further doping with iodine to yield a component assignable to ξ-(BEDT-TTF)2(I2I3)2,18 as judged from the XRD patterns shown in Fig. 2B (line c).
Iodine doping under dry conditions gave different results. A mixture of (BEDT-TTF)–silica nanocomposite and a stoichiometric amount of iodine was subjected to bead milling to produce a paramagnetic powder that contained no crystalline (BEDT-TTF)2I3. The XRD pattern is shown in Fig. 2C (line a). TEM observation of the powder confirmed that the particulate nanostructures are maintained. When the amorphous powder was annealed at 376 K for 3 h, crystallinity was slightly enhanced to produce an XRD pattern corresponding to β-(BEDT-TTF)2I3, as shown in Fig. 2C (line b). It should be mentioned that all of the (BEDT-TTF)2I3–silica nanocomposites obtained here exhibited high dispersibility in water without any surfactants, similar to other silica-nanocomposites reported previously.9e,g
For comparison, powders of bulk BEDT-TTF, which was prepared by grinding with a mortar in advance, were dispersed and stirred for 1 h in an ether solution of iodine to attempt doping. XRD patterns were essentially unchanged before and after the treatment, and only the surfaces of the powders had apparently reacted with the iodine. The formation of (BEDT-TTF)2I3 by the mechanical grinding of bulk BEDT-TTF with iodine is reported,19 although we found that extreme dry grinding conditions were needed (400 rpm, 8 h) to complete the reaction in the absence of silica nanoparticles. These results confirm the significant role of silica nanoparticles in the nanodownsizing of BEDT-TTF as a consequence of hybridization, which resulted in a large increase in its reactivity. This situation suggests that nanocomposites of organic solids and silica nanoparticles are efficiently applicable to solid-state reactions, even though they are much less reactive in the bulk solid state.
As shown by the TEM observations, (BEDT-TTF)2I3-silica nanocomposites prepared under both wet and dry conditions retained morphological nanostructures. On the other hand, as we have reported recently, the iodine doping of MPc-silica nanocomposite prepared by the same procedure led to drastic morphological changes to yield silica-free nanorods made of the iodine complex of MPc ([MPc]I),11 owing to the self-aggregation of the planar molecules. In contrast, BEDT-TTF may maintain its affinity with silica surfaces, even after complexation, which is likely because of the flexibility of the molecule and its assembled structure. Such contrasting results imply that the morphology of molecular complexes fabricated by the grinding method depends on molecular shapes and structures. Further exploration of materials may lead to interesting nanostructures.
Magnetic susceptibility
The magnetic susceptibility of (BEDT-TTF)2I3-silica nanocomposites was measured to investigate the phase transition characteristic of the α-form of (BEDT-TTF)2I3. Fig. 4 shows the temperature dependence of magnetic susceptibility of the nanocomposite prepared in hexane. The susceptibility slightly decreased at around 110–140 K, corresponding to the M–I transition of α-(BEDT-TTF)2I3. The susceptibility change at the phase transition is about 9.6% that of the bulk α-(BEDT-TTF)2I3.12 This result implies that approximately 10% of the total amount of (BEDT-TTF)2I3 exists as the α-form. Additionally, the observed transition is broad, ranging over approximately 30 K, whereas bulk crystals of α-(BEDT-TTF)2I3 exhibit an abrupt decrease in susceptibility at 135 K.12 This broadening probably stems from the effect of nanodownsizing. The high defect concentration of the nanocomposites may also contribute to the broadening. The magnetic susceptibility accompanied a large amount of Curie fraction, which was estimated at 43% and is primarily attributed to surface molecules. The remarkable increase in the Curie fraction as compared to that of the bulk crystals (approximately 0.3%12) evidently results from highly enlarged surface areas. The remaining fraction of 47% is probably assigned to the molecules inside the shell layer (excluding the surface molecules), which no longer exhibit bulk properties owing to reduced crystal sizes. Annealing at 376 K resulted in an increase in the α-form fraction to 14.2% and the reduction the Curie fraction to 31%. This result is consistent with the growth of the XRD peaks of the α-form by annealing, as described above.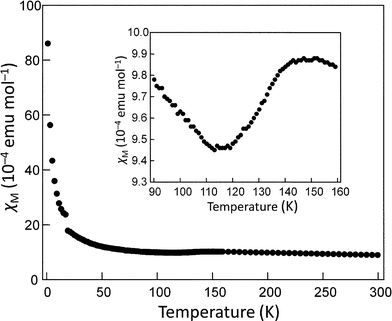 | ||
| Fig. 4 Temperature dependence of the magnetic susceptibility of (BEDT-TTF)2I3–silica nanocomposites (BEDT-TTF:silica = 1:2 w/w) prepared by doping in hexane. Inset shows the temperature dependence around the metal-insulator transition temperature of the α-form. | ||
On the other hand, in the 1:2 (w/w) nanocomposite doped in diethyl ether, the α-form fraction and the Curie fraction are estimated to be 47% and 12%, respectively, from the magnetic susceptibility data. This result is consistent with the larger amount of nanocrystalline salts and concomitant decrease in surface molecules. By annealing the sample, the α-form fraction was partly converted into the β-form, as confirmed by XRD and magnetic susceptibility measurements.
The (BEDT-TTF)2I3–silica nanocomposite prepared under dry conditions showed simple paramagnetic behavior and no signs of phase transition, which is consistent with the absence of crystalline components. The number of Curie spins was approximately 50% of the total, and this fraction was reduced to 31% by annealing.
Electrical conductivities
The electrical conductivities of the nanocomposites were measured at room temperature using their compaction pellets. As expected, the (BEDT-TTF)–silica nanocomposite (1:2 w/w) was as an insulator before iodine doping, showing a conductivity of 2.9 × 10−12 S cm−1. After doping in hexane, the conductivity remarkably increased to 1.7 × 10−6 S cm−1. However, the value is 10−6 times smaller than the pellet conductivity of bulk (BEDT-TTF)2I3 (9 S cm−1, literature value 15 S cm−120). As the (BEDT-TTF)2I3–silica nanocomposite prepared under dry conditions exhibited a very low conductivity of 1.7 × 10−8 S cm−1, it is likely that the nanocrystalline components substantially contribute to the conducting properties of the nanocomposite. The electrical conductivity of the 1:4 (w/w) nanocomposite (9.2 × 10−7 S cm−1) was lower than that of the 1:2 (w/w) nanocomposite. The remarkable reduction in conductivity in these nanocomposites compared to the bulk is attributed to the presence of grain boundaries, defects, and the core-shell nanoparticles. The conductivity of the 1:2 (w/w) nanocomposite doped in diethyl ether was 4.0 × 10−5 S cm−1, one order of magnitude larger than that of the nanocomposite doped in hexane, which is consistent with the increase in crystalline components. The electrical conductivity of a compaction pellet of a mixture of bulk (BEDT-TTF)2I3 and silica powder (1:2 w/w) was 8.5 × 10−6 S cm−1, which indicates that silica greatly reduces conductivity.
The temperature dependence of the electrical conductivity of the 1:2 (w/w) nanocomposite doped in diethyl ether was measured between 300 K and 100 K. Semiconducting behavior with an activation energy of 0.077 eV was observed over the entire temperature region. The apparent semiconducting behavior is probably attributed to the use of compaction pellets. No change of conductivity in the M–I transition temperature was observed.
Conclusion
A solid mixture of BEDT-TTF and silica nanoparticles has been subjected to dry bead milling to yield a powdery nanocomposite. In the nanocomposite, hybridized BEDT-TTF exists in two states, namely, ultrathin shell layers of core-shell-type nanoparticles and nanometre-sized crystals incorporated into hollow sites of aggregated silica nanoparticles. Iodine doping in a suspension of (BEDT-TTF)–silica nanocomposite in hexane or diethyl ether occurs readily to yield a (BEDT-TTF)2I3–silica nanocomposite. The crystalline component of the α-form, detected for the iodine complex doped in the wet condition, is assigned to the nanocrystalline component. The amount of each component depends on the dispersion media (hexane or diethylether). On the other hand, the dry grinding of the (BEDT-TTF)–silica nanocomposite with iodine results in the formation of a powdery nanocomposite of the iodine complex without nanocrystalline components.Magnetic susceptibility measurements reveal that the nanocomposite doped under wet conditions consists of three types of (BEDT-TTF)2I3, including the α-form component, the Curie component, and another silent fraction. The first two fractions are assigned to the nanocrystalline component and surface molecules of shell layers, respectively, while the third fraction is assumed to be the component inside the shell layers. Therefore, the ratio of the core-shell component and nanocrystalline component could be estimated based on the magnetic properties of (BEDT-TTF)2I3. The electrical conductivity of the nanocomposite is much lower than that of bulk (BEDT-TTF)2I3.
This study has shown that nanocomposites of organic solids and silica nanoparticles are efficiently applicable to solid-state reactions, even though the organic solids are much less reactive in the bulk solid state. Furthermore, even compounds that are insoluble in solvents may be applied to chemical reactions in this manner. In this study, the hybridization method allowed for the simple synthesis of electrically functional fine powders. While conventional investigation of CT salts has focused predominately on single crystals or films, access to nanosized composite materials expands the scope and applications of the research on CT salts, because these nanosized composite materials have high dispersibility in liquids21 and high processability.
Acknowledgements
We thank Prof. T. Uchino (Kobe University) for useful discussions and his help with the grinding experiments, Dr T. Sakurai and Prof. H. Ohta (Kobe University) for SQUID measurements, Prof. D. Kuwahara (The University of Electro-communications) for his help with instrumental analyses, and Dr H. Hasegawa (Kobe Advanced ICT Research Center, National Institute of Information and Communications Technology) for his help with the SEM measurements. We also thank M. Nakama (WarpStream Tokyo Co., Ltd.) for providing Web-based database systems. This work was supported by KAKENHI (No. 23110719) and the Joint Studies Program (2009–2010) of the Institute for Molecular Science.References
- (a) T. Mori, Chem. Rev., 2004, 104, 4947–4969 CrossRef CAS; (b) R. P. Shibaeva and E. B. Yagubskii, Chem. Rev., 2004, 104, 5347–5378 CrossRef CAS; (c) Special issue C. R. Chimie, Chem. Rev., 2004, 104 Search PubMed; (d) Special issue Y. H. Dai and D. H. Li, J. Phys. Soc, Jpn., 2006, 75 Search PubMed; (e) T. Ishiguro, K. Yamaji, G. Saito, Organic superconductors, 2nd Ed.; Springer-Verlag: Berlin, 1998 Search PubMed.
- (a) G. Schmid, ed. Nanoparticles: From Theory to Application; Wiley-VCH, 2004 Search PubMed; (b) G. Cao, ed. Nanostructures & Nanomaterials: synthesis, properties & applications; Imperial College Press: London, 2004 Search PubMed; (c) H. S. Nalwa, Nanostructured Materials and Nanotechnology; Academic Press: New York, 2002 Search PubMed; (d) A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- (a) K. Naka, D. Ando, X. Wang and Y. Chujo, Langmuir, 2007, 23, 3450–3454 CrossRef CAS; (b) K. Tanaka, T. Kunita, F. Ishiguro, K. Naka and Y. Chujo, Langmuir, 2009, 25, 6929–6933 CrossRef CAS.
- (a) H. Hasegawa, T. Kubota and S. Mashiko, Thin Solid Films, 2003, 438–439, 352–355 CrossRef CAS; (b) H. Hasegawa, Y. Noguchi, R. Ueda, T. Kubota and S. Mashiko, Thin Solid Films, 2008, 516, 2491–2494 CrossRef CAS; (c) H. M. Yamamoto, H. Ito, K. Shigeto, K. Tsukagoshi and R. Kato, J. Am. Chem. Soc., 2006, 128, 700–701 CrossRef CAS.
- (a) K. Xiao, I. N. Ivanov, A. A. Puretzky, Z. Liu and D. B. Geohegan, Adv. Mater., 2006, 18, 2184–2188 CrossRef CAS; (b) K. Xiao, J. Tao, Z. Pan, A. A. Puretzky, I. N. Ivanov, S. J. Pennycook and D. B. Geohegan, Angew. Chem., Int. Ed., 2007, 46, 2650–2654 CrossRef CAS.
- (a) K. Hiraishi, A. Masuhara, T. Yokoyama, H. Kasai, H. Nakanishi and H. Oikawa, J. Cryst. Growth, 2009, 311, 948–952 CrossRef CAS; (b) K. Hiraishi, A. Masuhara, H. Kasai, H. Nakanishi and H. Oikawa, Jpn. J. Appl. Phys., 2010, 49, 01AE08 CrossRef.
- D. de Caro, K. Jacob, C. Faulmann, J.-P. Legros, F. Senocq, J. Fraxedas and L. Valade, Synth. Met., 2010, 160, 1223–1227 CrossRef CAS.
- K. Clark, A. Hassanien, S. Khan, K.-F. Braun, H. Tanaka and S.-W. Hla, Nat. Nanotechnol., 2010, 5, 261–265 CrossRef CAS.
- (a) K. Hayashi, H. Morii, K. Iwasaki, S. Horie, N. Horiishi and K. Ichimura, J. Mater. Chem., 2007, 17, 527–530 RSC; (b) S. Horiuchi, S. Horie and K. Ichimura, ACS Appl. Mater. Interfaces, 2009, 1, 977–981 CrossRef CAS; (c) K. Ichimura, Chem. Commun., 2010, 46, 3295–3297 RSC; (d) K. Ichimura, A. Funabiki and K. Aoki, Chem. Lett., 2010, 39, 586–587 CrossRef CAS; (e) K. Ichimura, K. Aoki, H. Akiyama, S. Horiuchi, S. Nagano and S. Horie, J. Mater. Chem., 2010, 20, 4312–4320 RSC; (f) K. Ichimura, Chem. Lett., 2010, 39, 614–615 CrossRef CAS; (g) K. Ichimura, K. Aoki, H. Akiyama, S. Horiuchi and S. Horie, J. Mater. Chem., 2010, 20, 4784–4791 RSC.
- (a) G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC; (b) D. Braga and F. Grepioni, Angew. Chem., Int. Ed., 2004, 43, 4002–4011 CrossRef CAS; (c) A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846–855 RSC; (d) T. Friščić, E. Meštrović, D. Š. Šamec, B. Kaitner and L. Fábián, Chem.–Eur. J., 2009, 15, 12644–12652 CrossRef; (e) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025–1074 CrossRef CAS; (f) D. R. Weyna, T. Shattock, P. Vishweshwar and M. Zaworotko, Cryst. Growth Des., 2009, 9, 1106–1123 CrossRef CAS.
- A. Funabiki, T. Mochida, H. Hasegawa, K. Ichimura and S. Kimura, New J. Chem., 2011, 35, 483–488 RSC.
- B. Rothaemel, L. Forro, J. R. Cooper and J. S. Schilling, Phys. Rev. B, 1986, 34, 704–712 CrossRef CAS.
- (a) N. Tajima, S. Sugawara, R. Kato, Y. Nishio and K. Kajita, Phys. Rev. Lett., 2009, 102, 176403 CrossRef; (b) A. Kobayashi, S. Katayama and Y. Suzumura, Sci. Technol. Adv. Mater., 2009, 10, 024309 CrossRef; (c) T. Takahashi, Y. Nogami and K. Yakushi, J. Phys. Soc. Jpn., 2006, 75, 051008 CrossRef.
- H. Müller, A. N. Fitch, M. Lorenzen, S. O. Svensson, S. Wanka and J. Wosnitza, Adv. Mater., 1999, 11, 541–546 CrossRef.
- (a) E. B. Yagubskii, I. F. Shchegolev, V. N. Laukin, P. A. Kononovich, M. V. Kartsovnik, A. V. Zvarykina and L. I. Buravov, JETP Lett., 1984, 39, 12–15 CAS; (b) V. F. Kaminskii, T. G. Prokhorova, R. P. Shibaeva and E. B. Yagubskii, JBTP Lett., 1984, 39, 15–18 CAS; (c) J. M. Williams, T. J. Emge, H. H. Wang, M. A. Beno, P. T. Copps, L. N. Hall, K. D. Carlson and G. W. Crabtree, Inorg. Chem., 1984, 23, 2558–2560 CrossRef CAS; (d) G. W. Crabtree, K. D. Carlson, L. N. Hall, P. T. Copps, H. Ii. Wang, T. J. Emge, M. A. Beno and J. M. Williams, Phys. Rev. B, 1984, 30, 2958–2960 CrossRef CAS.
- U. Niebling, J. Steinl, D. Schweitzer and W. Strunzb, Solid State Commun., 1998, 106, 505–507 CrossRef CAS.
- E. E. Laukhina, V. A. Merzhanov, S. I. Pesotskii, A. G. Khomenko, E. B. Yagubskii, J. Ulanski, M. Kryszewski and J. K. Jeszka, Synth. Met., 1995, 70, 797–800 CrossRef CAS.
- M. A. Beno, U. Geiser, K. L. Kostka, H. H. Wang, K. S. Webb, M. A. Firestone, K. D. Cadson, L. Nuñez, M.-H. Whangbo and J. M. Williams, Inorg. Chem., 1987, 26, 1912–1920 CrossRef CAS.
- (a) I. Smirani, A. Brau and J. P. Farges, Synth. Met., 1998, 93, 203–206 CrossRef CAS; (b) I. Smirani, P. Auban-Senzier, D. J. Crome, A. Brau and J. P. Farges, Synth. Met., 1999, 102, 1259–1260 CrossRef CAS; (c) I. Smirani, S. Flandrois, N. B. Chanh, A. Brau and J.-P. Farges, Synth. Met., 1999, 102, 1261–1262 CrossRef CAS; (d) I. Smirani, V. N. Semkin, A. Graja, A. Brau and J.-P. Farges, Synth. Met., 1999, 102, 1263 CrossRef CAS.
- D. Schweitzer, S. Gärtner, H. Grimm, E. Gogu and H. J. Keller, Solid State Commun., 1989, 69, 843–845 CrossRef CAS.
- (a) S. Hara, H. Tanaka, T. Kawamoto, M. Tokumoto, M. Yamada, A. Gotoh, H. Uchida, M. Kurihara and M. Sakamoto, Jpn. J. Appl. Phys., 2007, 46, L945–L947 CrossRef CAS; (b) H. Shiozaki, T. Kawamoto, H. Tanaka, S. Hara, M. Tokumoto, A. Gotoh, T. Satoh, M. Ishizaki, M. Kurihara and M. Sakamoto, Jpn. J. Appl. Phys., 2008, 47, 1242–1244 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |