Titanium and zirconium amido complexes supported by imidazole-containing ligands: syntheses, characterization and catalytic activities†
Yang
Zhao
a,
Miaoshui
Lin
a,
Zhou
Chen
b,
Hao
Pei
a,
Yahong
Li
*ac,
Yanmei
Chen
b,
Xiufang
Wang
b,
Lei
Li
a,
Yanyuan
Cao
a,
Yong
Zhang
a and
Wu
Li
b
aKey Laboratory of Organic Synthesis of Jiangsu Province, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215006, China. E-mail: liyahong@suda.edu.cn
bCAS Key Laboratory of Salt Lake Resources and Chemistry, Qinghai Institute of Salt Lakes, Chinese Academy of Sciences, Xining, 810008, China
cState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, 730000, China
First published on 26th October 2011
Abstract
A series of new titanium and zirconium amido complexes, incorporating 4,5-diphenyl-2-(2-pyridinyl)-1H-imidazole (HL1), 4,5-bis(4-chlorophenyl)-2-(2-pyridinyl)-1H-imidazole (HL2), 4,5-bis(4-methoxyphenyl)-2-(2-pyridinyl)-1H-imidazole (HL3) and 4,5-bis(2-methoxyphenyl)-2-(2-pyridinyl)-1H-imidazole) (HL4) as the ancillary ligands, have been prepared and are shown to be pre-catalysts for the hydroamination of alkynes. Treatment of Ti(NMe2)4 with 1 equivalent of HL1, HL2, HL3 and HL4, respectively, results in transamination with one dimethylamide group providing Ti(L1)(NMe2)3 (1), Ti(L2)(NMe2)3 (2), Ti(L3)(NMe2)3 (3), and Ti(L4)(NMe2)3·THF (4·THF). Reaction of Ti(NMe2)4 with 2 equivalents of HL1, HL2, HL3 and HL4, respectively, leads to production of titanium bisamido complexes Ti(L1)2(NMe2)2·THF (5·THF), Ti(L2)2(NMe2)2·THF (6·THF), Ti(L3)2(NMe2)2·THF (7·THF), and Ti(L4)2(NMe2)2 (8). Similarly, complexes Zr(L1)(NMe2)3 (9), Zr(L1)2(NMe2)2·1.5THF (10·1.5THF), Zr(L2)2(NMe2)2·0.5THF (11·0.5THF), and Zr(L3)2(NMe2)2·0.5THF (12·0.5THF) are generated by addition of 1 or 2 equivalents of the corresponding ligands to Zr(NMe2)4. All compounds have been characterized by elemental and spectroscopic analyses. The solid-state structures of compounds 2 and 6·THF have been further established by single X-ray diffraction analyses. The catalytic activities of 2 and 6·THF towards the hydroamination of alkynes were explored. Complexes 2 and 6·THF were found to be active pre-catalysts for the hydroamination reactions. Complex 2 showed higher catalytic activity and gave highly Markovnikov selective hydroamination of terminal alkynes.
Introduction
To a large extent, investigation of titanium complexes has been driven by studies of the hydroamination reaction, in which an N–H bond is added across an unsaturated C–C bond to form imines, enamines and N-containing heterocycles in a single step.1 Since this highly atom economic process allows direct access to industrially and biologically relevant classes of compounds from cheap and readily available starting materials, both inter- and intramolecular hydroamination have continuously been the subject of intensive research and attracted ever-increasing attention during the past 20 years.1A plethora of complexes from across the periodic table, including alkali and alkaline earth metal complexes,2 early3 and late transition metal complexes,4 and lanthanide complexes,5 have been developed for hydroamination transformation. Among which titanium complexes have been proved most useful thus far for the hydroamination of alkynes due to their low cost, low toxicity, enhanced stability and improved functional group tolerance. To date, a variety of catalytic systems have been developed, initially focusing on Cp-based ligand systems and then moving into non-Cp-based catalysts by the groups of Odom,6 Bergman,7Zi,8 Doye,9 Beller,10 Schafer11 and others.12 The research of these groups has indicated that the ligand chelating to the metal center plays a key role in controlling the regioselectivity of the hydroamination products. In general, anti-Markovnikov products are preferentially obtained with titanocene catalysts; in contrast, Markovnikov isomers are found with pyrrolyl ligands. As a continuation of our ongoing efforts in studying the hydroamination of alkynes catalyzed by pyrrolyl ligand-chelated titanium compounds,13 we expand our efforts to synthesizing titanium pre-catalysts coordinated by imidazole-containing ligands, which contain an imidazole heterocycle analogous to a pyrrolyl ring, and studying their catalytic activities. Although a handful of titanium complexes supported by various imidazole ligands have been reported,14 the catalytic properties of these complexes towards hydroamination reactions have never been explored. Driven by the impetus of exploring the catalytic properties of the titanium complexes chelated by imidazole-containing ligands, and comparing the catalytic behaviours of the pyrrolyl ligand-coordinated titanium complexes with those chelated by imidazole-containing ligands, we carried out the reactions of imidazole-containing ligands (Scheme 1) with Ti(NMe2)4 and Zr(NMe2)4, and twelve complexes were prepared. Herein we report the syntheses and characterizations of these complexes. The catalytic activities of two of the complexes towards intermolecular hydroamination of alkynes are also presented.
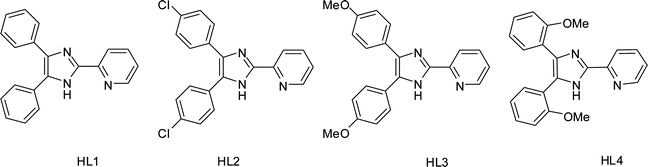 | ||
| Scheme 1 Structures of the ligands. | ||
Results and discussion
Syntheses of the ligands
The ligands HL1, HL2 and HL3 were prepared by heating the 2.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5 mixtures of 2-cyanopyridine, substituted benzaldehyde, and NH4OAc in HOAc solvent at 170 ˚C.15HL4 is a new compound and was synthesized by using 2-methoxybenzaldehyde as the aldehyde source (Scheme 1). The structure of HL4 was confirmed from 1H and 13C NMR spectra.
:1:5 mixtures of 2-cyanopyridine, substituted benzaldehyde, and NH4OAc in HOAc solvent at 170 ˚C.15HL4 is a new compound and was synthesized by using 2-methoxybenzaldehyde as the aldehyde source (Scheme 1). The structure of HL4 was confirmed from 1H and 13C NMR spectra.
Syntheses of the titanium and zirconium amido complexes
Treatment of a Ti(NMe2)4 solution with 1 equiv. of HL1, HL2, HL3 and HL4, respectively (Scheme 2), leads to near quantitative transamination generating Ti(L1)(NMe2)3 (1), Ti(L2)(NMe2)3 (2), Ti(L3)(NMe2)3 (3), and Ti(L4)(NMe2)3·THF (4·THF). Trace amounts of the second products which were suspected to be the bisligand-chelated bisamido complexes were found in the samples of 1, 2, 3 and 4·THF, respectively. Sufficiently pure compounds 1, 2, 3 and 4·THF could be obtained by recrystallization from THF/hexane.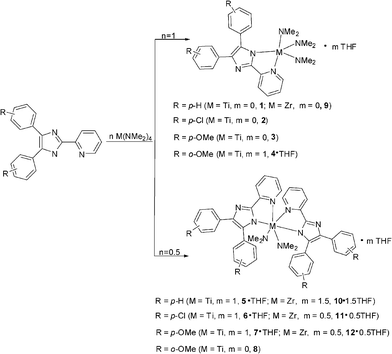 | ||
| Scheme 2 Syntheses of the complexes. | ||
Because trace amounts of impurities were observed in the products of 1, 2, 3 and 4·THF, syntheses of the bisligand-chelated bisamido titanium complexes were conducted. Reaction of Ti(NMe2)4 with 2 equiv. of HL1, HL2, HL3 and HL4, respectively, in THF, followed by recrystallization from THF/hexane, gives the bisligand-chelated titanium bisamido complexes Ti(L1)2(NMe2)2·THF (5·THF), Ti(L2)2(NMe2)2·THF (6·THF), Ti(L3)2(NMe2)2·THF (7·THF) and Ti(L4)2(NMe2)2 (8) in good yields. They have been characterized by 1H and 13C NMR spectroscopy and elemental analyses. The 1H NMR spectra of 5·THF, 6·THF, 7·THF and 8 support a ratio of amido group NMe2 and ligand of 2:2.
The reactions of Zr(NMe2)4 with 1 equiv. of HL1 and 2 equiv. of HL1–HL3 were also explored, and Zr(L1)(NMe2)3 (9), Zr(L1)2(NMe2)2·1.5THF (10·1.5THF), Zr(L2)2(NMe2)2·0.5THF (11·0.5THF) and Zr(L3)2(NMe2)2·0.5THF (12·0.5THF) were generated in good yields. They were also characterized by 1H and 13C NMR spectroscopy and elemental analyses.
Crystals suitable for X-ray diffraction of compounds 2 and 6·THF were grown from a toluene/THF solution left standing at room temperature in a vibration-free environment for a week.
Structure descriptions of complexes 2 and 6·THF
The molecular structures of 2 and 6·THF in the solid state have been confirmed by X-ray analysis and are shown in Fig. 1 and 2, respectively. The crystallographic data and experimental details for structural analyses are summarized in Table 1. Selected bond distances and angles are listed in Table 2.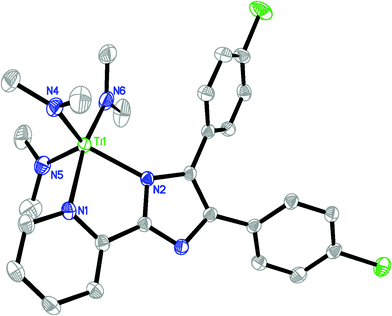 | ||
| Fig. 1 ORTEP structural drawing of 2. Ellipsoids are drawn at the 30% probability level, and hydrogen atoms are omitted for clarity. | ||
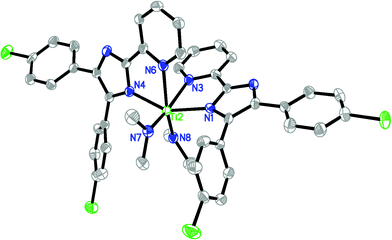 | ||
| Fig. 2 ORTEP structural drawing of 6·THF. Ellipsoids are drawn at the 30% probability level. Hydrogen atoms and the solvated molecule are omitted for clarity. | ||
| 2 | 6·THF | |
|---|---|---|
| a Including solvate molecules. b Mo-Kα radiation. c R 1 = Σ(|Fo| − |Fc|)/Σ(|Fo|) for observed reflections. d w = 1/[σ2(Fo2) + (αP)2 + bP] and P = [max(Fo2,0)+2Fc2]/3. e wR 2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2 for all data. | ||
| Formulaa | C26H30Cl2N6Ti | C44H36Cl4N8Ti |
| M/g mol−1a | 545.36 | 866.51 |
| Temperature/K | 223(2) | 223(2) |
| Wavelengthb/Å | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | P212121 |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
| a/Å | 11.829(2) | 8.9628(18) |
| b/Å | 12.517(3) | 13.517(3) |
| c/Å | 18.391(4) | 19.262(4) |
| α/° | 90.00 | 73.96(3) |
| β/° | 90.00 | 87.86(3) |
| γ/° | 90.00 | 74.76(3) |
| V/Å3 | 2723.2(9) | 2162.3(8) |
| ρ/g cm−3 | 1.330 | 1.331 |
| Z | 4 | 2 |
| F(000) | 1136 | 892 |
| Crystal size/mm | 0.20 × 0.15 × 0.15 | 0.40 × 0.20 × 0.20 |
| θ range/° | 3.24 to 25.30 | 3.08 to 25.00 |
| Limiting indices | −12 < h < 13 | −8 < h < 10 |
| −10 < k < 15 | −16 < k < 15 | |
| −20 < l < 22 | −22 < l < 22 | |
| Reflections collected/unique | 8641/4561 | 17888/7536 |
| Data/restraints/parameters | 4561/0/323 | 7536/7/518 |
| GOF | 0.999 | 1.099 |
| R 1, wR2 [I > 2σ(I)] | R 1 = 0.0406 | R 1 = 0.0879 |
| wR2 = 0.0871 | wR 2 = 0.1709 | |
| R 1 c, wR2d,e (all data) | R 1 = 0.0452 | R 1 = 0.1400 |
| wR 2 = 0.0906 | wR 2 = 0.1971 | |
| Largest diff. peak and hole/e Å3 | 0.222 and −0.231 | 0.269 and −0.390 |
| 2 | |||
| Ti1-N4 | 1.880(3) | Ti1-N5 | 1.914(3) |
| Ti1-N6 | 1.902(3) | Ti1-N2 | 2.142(2) |
| Ti1-N1 | 2.292(2) | ||
| N4-Ti1-N5 | 110.46(12) | N6-Ti1-N5 | 95.24(13) |
| N4-Ti1-N2 | 124.57(11) | N6-Ti1-N2 | 96.20(10) |
| N5-Ti1-N2 | 121.06(11) | N4-Ti1-N1 | 91.29(11) |
| N6-Ti1-N1 | 168.59(10) | N5-Ti1-N1 | 87.28(11) |
| N2-Ti1-N1 | 73.08(8) | N4-Ti1-N6 | 98.21(13) |
| 6·THF | |||
| Ti2-N1 | 2.124(4) | Ti2-N8 | 1.880(5) |
| Ti2-N7 | 1.899(6) | Ti2-N4 | 2.152(4) |
| Ti2-N3 | 2.308(4) | Ti2-N6 | 2.314(5) |
| N8-Ti2-N7 | 104.9(2) | N8-Ti2-N1 | 97.18(18) |
| N7-Ti2-N1 | 100.82(18) | N8-Ti2-N4 | 100.11(18) |
| N7-Ti2-N4 | 96.49(17) | N1-Ti2-N4 | 151.44(16) |
| N8-Ti2-N3 | 87.00(17) | N7-Ti2-N3 | 167.93(18) |
| N1-Ti2-N3 | 75.20(15) | N4-Ti2-N3 | 83.16(15) |
| N8-Ti2-N6 | 160.54(17) | N7-Ti2-N6 | 94.23(18) |
| N1-Ti2-N6 | 82.41(16) | N4-Ti2-N6 | 73.77(16) |
| N3-Ti2-N6 | 74.04(15) | ||
Single-crystal X-ray diffraction studies reveal that complex 2 crystallizes in the orthorhombic crystal system of the P212121 space group. The overall structure of 2 is remarkably close to a distorted trigonal bipyramid, with one pyridine nitrogen atom and one amide nitrogen atom axial and the other equatorial. Angles between equatorial nitrogens add to 356.09°. The axial position occupied by dimethylamide is nearer to perpendicular with respect to the equatorial plane, having angles of 98.21(13)°, 95.24(13)°, and 96.20(10)° relative to those equatorial nitrogens. The Ti-N distances of complex 2 display interesting features. As expected, the Ti-N(pyridine) bond length of 2.292(2) Å is the longest and in the range which has been previously reported for Ti-N(pyridine) bond containing compounds (min: 2.126 Å, max: 2.450 Å for 317 examples in the CSD). A larger difference between Ti-N(imidazole) and Ti-N(dimethylamide) bond lengths is observed. The Ti-N(imidazole) bond distance is found to be 0.243 Å longer than the average distance of the Ti-N(dimethylamide) bonds. An analysis of known Ti-N(NMe2) crystallographically determined bond lengths reveals that the Ti-N(NMe2) distances in 2 are relatively short, averaging 1.900(3) Å.
Single crystal X-ray analysis reveals 6·THF crystallizes in the triclinic crystal system of the P space group. Complex 6·THF is a bisligand-chelated titanium bisamido compound. The titanium atom is pseudo-octahedral, with two dimethylamides being in a cis-orientation. The plane constituted by two nitrogen atoms of one L1− ligand is almost perpendicular to the plane formed by the coordination site of the another L1− ligand, with nitrogen atoms of imidazole being trans to each other, and the two pyridine nitrogens being in a cis-orientation. Due to this pseudo-octahedral geometry, the two Ti-N(NMe2) bond lengths are nearly identical (1.880(5) and 1.899(6) Å), and remarkably shorter than the average Ti-N(imidazole) (2.139 Å) and Ti-N(pyridine) (2.314 Å) bond distances.
Complexes 2 and 6·THF are members of a family of transition metal complexes with a 2-(imidazol-2-yl) pyridine ligand.16 Compounds 2 and 6·THF are among the rare examples of titanium complexes incorporating the 2-(imidazol-2-yl) pyridine ligand.14a,b
Hydroamination of alkynes catalyzed by 2 and 6·THF
The successful determination of the crystal structures of the titanium trisamido complex 2 and bisamido complex 6·THF prompted us to explore the catalytic activities of 2 and 6·THF in the hydroamination of alkynes. Initially, we investigated the reaction of aniline with a selection of alkynes (phenylacetylene, 1-octyne, 3-hexyne, diphenylacetylene and trimethylsilylacetylene) catalyzed by 10 mol% of 2 or 6·THF. For comparison purposes, the catalytic reactions were carried out at 100 °C in toluene for 24 h with a 2:3 molar ratio of alkyne and aniline. Because the resulting imines were not stable to column chromatography, the hydroamination products were directly reduced to amines by LiAlH4. The results are shown in Table 3. As we can see in Table 3, both 2 and 6·THF could promote the hydroamination of most of the alkynes and afforded relatively low (Table 3, entry 1, entries 3–4 and 6–9) to moderate (Table 3, entry 2) yields. No hydroamination products were determined for the trimethylsilylacetylene (Table 3, entries 5 and 10) when 2 or 6·THF was employed as the pre-catalyst. The yield for the reaction of 1-octyne with aniline is the highest for 2 catalyzed reactions (58%). The Markovnikov product is the favored or exclusive product of the hydroamination of nearly all of the tested alkynes. This is in contrast to utilizing Me2TiCp2 as the pre-catalyst,17 and is consistent with employing the pyrrolyl ligand-chelated titanium complexes as the pre-catalyst.6
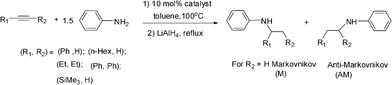









The yields of 2 catalyzed hydroamination reactions are much higher than those of 6·THF. The lower activity shown by complex 6·THF might be attributed to the two rigid and bulky ligands around the titanium atom making the coordination sphere of the central metal much more crowded. The six-coordinated titanium atom is relatively stable, and hard to be attacked by amine substrates.18
We further probed the utility and regioselectivity of the catalyst 2 through the hydroamination of 1-octyne by a number of different amines (Table 4). In all cases, the Markovnikov product was favored, often in excess of 91:9, over the anti-Markovnikov product. 4-Nitroaniline led to no hydroamination product (Table 4, entry 5); 4-methoxyaniline (Table 4, entry 4), 4-bromoaniline (Table 4, entry 1) and 2,4-dichloroaniline (Table 4, entry 2) hydroaminated 1-octyne with high regioselectivities and yields; when 2-fluoroaniline and naphthalene-1-amine were employed as the amine source, low yields with high regioselectivities were observed (Table 4, entries 3 and 6).
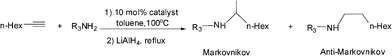



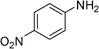

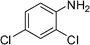

Complex 2 catalyzed hydroaminations of phenylacetylene with alkyl amines and secondary amines, such as tertiary amine, N-methyl aniline and diphenyl amine, were also examined and no hydroamination products were detected.
For comparison purposes, the hydroamination reactions between phenylacetylene and 2,4-dichloroaniline and 4-methoxyaniline catalyzed by Ti(NMe2)4 were also conducted and the results are listed in Table 4 too. The results of the Ti(NMe2)4 catalyzed reactions were different from those utilizing 2 as pre-catalyst. Hydroamination of phenylacetylene by 4-methoxyaniline gave nearly 1:1 Markovnikov/anti-Markovnikov product (M:anti-M = 54:46) with lower yield. Exclusive Markovnikov product with higher yield was observed for the hydroamination of phenylacetylene by 2,4-dichloroaniline under similar reaction conditions. Comparison of 2 and Ti(NMe2)4 as hydroamination pre-catalysts indicates that the active species in the Ti(NMe2)4 catalyzed reactions is not the same as that for 2, for the regioselectivities and activities of these two complexes are different.
Conclusions
In summary, twelve new compounds were synthesized and characterized. The catalytic behaviour of complexes 2 and 6·THF towards the hydroamination of alkynes was investigated. 2 and 6·THF were able to catalyze the hydroamination of alkynes. Complex 2 showed higher catalytic activity and gave highly Markovnikov selective hydroamination of terminal alkynes.Experimental section
General considerations
All manipulations of air-sensitive compounds were carried out in a Mikrouna glovebox under a purified nitrogen atmosphere. Ti(NMe2)4 and Zr(NMe2)4 were purchased from Acros and used without further purification. Amines were distilled from CaH2. Alkynes were degassed, flushed with argon and stored over molecular sieves (4 Å). Tetrahydrofuran, toluene, and hexane were refluxed over sodium benzophenone ketyl for at least 4 days. CDCl3 was distilled from P2O5 under a nitrogen atmosphere. 1H and 13C spectra were recorded on Innova-400 spectrometers at ambient temperature using TMS as an internal standard, and chemical shifts were reported in ppm. Elemental analyses were determined using a Carlo-Erba EA1110 CHNOS microanalyzer. The ligands HL1, HL2 and HL3 were prepared according to literature procedures.15X-Ray crystallography
Crystals grown from concentrated solutions at room temperature were quickly selected and mounted on a glass fiber in wax. The data collections were carried out on a Rigaku Mercury CCD detector equipped with graphite-monochromated Mo-Kα radiation by using the φ/ω scan technique at room temperature. The structures were solved by direct methods with SHELXS-97.19,20 The hydrogen atoms were assigned with common isotropic displacement factors and included in the final refinement by use of geometrical restraints. A full-matrix least-squares refinement on F2 was carried out using SHELXL-97.General procedure for hydroamination reactions
To a 50 mL pressure tube was added the pre-catalyst (0.3 mmol), amine (4.5 mmol), alkyne (3 mmol) and toluene (5 mL) in a dry box. The pressure tube was sealed with a Teflon screw cap, taken out of the dry box and heated at 100 °C for 24 h. Then at 0 °C the reaction solution was carefully added to a suspension of LiAlH4 in toluene and the mixture was refluxed for 3 h. After cooling the solution to 0 °C, the excess LiAlH4 was hydrolyzed with aqueous NaOH (3 mol L−1). The mixture was then extracted with CH2Cl2 (3 × 30 mL), and the combined organic layers were dried with MgSO4 and concentrated under vacuum. Column chromatography of the residue on silica gel afforded the pure amine derivatives.Syntheses of the ligand HL4 and complexes
Acknowledgements
The authors appreciate the financial support of the Hundreds of Talents Program (2005012) of CAS, the Natural Science Foundation of China (20872105), the “Qinglan Project” of Jiangsu Province (Bu109805) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- For selected reviews on hydroamination, see: (a) S. Doye, in Science of Synthesis, ed. D. Enders, Thieme, Stuttgart, 2009, vol. 40a, p. 241 Search PubMed; (b) J. F. Hartwig, Nature, 2008, 455, 314 CrossRef CAS; (c) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef; (d) A. L. Odom, Dalton Trans., 2005, 225 RSC; (e) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (f) J. L. Klinkenberg and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 86 CrossRef CAS.
- For selected examples, see: (a) A. G. M. Barrett, C. Brinkmann, M. R. Crimmin, M. S. Hill, P. Hunt and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 12906 CrossRef CAS; (b) J. S. Wixey and B. D. Ward, Chem. Commun., 2011, 47, 5449 RSC; (c) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS; (d) A. Mukherjee, S. Nembenna, T. K. Sen, S. P. Sarish, P. K. Ghorai, H. Ott, D. Stalke and H. W. Roesky, Angew. Chem., Int. Ed., 2011, 50, 3968 CrossRef CAS; (e) J. Jenter, R. Köppe and P. W. Roesky, Organometallics, 2011, 30, 1404 CrossRef CAS; (f) M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS; (g) M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Organometallics, 2011, 30, 1291 CrossRef CAS; (h) J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680 CrossRef CAS.
- For selected examples, see: (a) M. R. Maurya, A. Arya, U. Kumar, A. Kumar, F. Avecilla and J. C. Pessoa, Dalton Trans., 2009, 9555 RSC; (b) L. L. Anderson, J. A. R. Schmidt, J. Arnold and R. G. Bergman, Organometallics, 2006, 25, 3394 CrossRef CAS; (c) C. Lorber, R. Choukroun and L. Vendier, Organometallics, 2004, 23, 1845 CrossRef CAS.
- For selected examples, see: (a) B. M. Cochran and F. E. Michael, J. Am. Chem. Soc., 2008, 130, 2786 CrossRef CAS; (b) Z. Liu and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 1570 CrossRef CAS; (c) H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100 CrossRef CAS; (d) K. Aikawa, M. Kojima and K. Mikami, Angew. Chem., Int. Ed., 2009, 48, 6073 CrossRef CAS; (e) K. D. Hesp, S. Tobisch and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 413 CrossRef CAS; (f) K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026 CrossRef CAS; (g) Z. Liu, H. Yamamichi, S. T. Madrahimov and J. F. Hartwig, J. Am. Chem. Soc., 2011, 133, 2772 CrossRef CAS; (h) M. J. Gainer, N. R. Bennett, Y. Takahashi and R. E. Looper, Angew. Chem., Int. Ed., 2011, 50, 684 CrossRef CAS.
- For selected examples, see: (a) G. Zi, Dalton Trans., 2009, 9101 RSC; (b) A. L. Reznichenko, H. N. Nguyen and K. C. Hultzsch, Angew. Chem., Int. Ed., 2010, 49, 8984 CrossRef CAS; (c) L. J. E. Stanlake and L. L. Schafer, Organometallics, 2009, 28, 3990 CrossRef CAS; (d) P. Benndorf, J. Jenter, L. Zielke and P. W. Roesky, Chem. Commun., 2011, 47, 2574 RSC; (e) S. B. Amin and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 10102 CrossRef CAS; (f) S. Seo and T. J. Marks, Chem.–Eur. J., 2010, 16, 5148 CrossRef CAS; (g) T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550 RSC; (h) F. Lauterwasser, P. G. Hayes, W. E. Piers, L. L. Schafer and S. Bräse, Adv. Synth. Catal., 2011, 353, 1384 CrossRef CAS; (i) H. Kim, T. Livinghouse, J. H. Shim, S. G. Lee and P. H. Lee, Adv. Synth. Catal., 2006, 348, 701 CrossRef CAS; (j) B. D. Stubbert and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 4253 CrossRef CAS.
- (a) D. L. Swartz II and A. L. Odom, Organometallics, 2006, 25, 6125 CrossRef; (b) Y. Shi, C. Hall, J. T. Ciszewski, C. Cao and A. L. Odom, Chem. Commun., 2003, 586 RSC; (c) Y. Shi, J. T. Ciszewski and A. L. Odom, Organometallics, 2001, 20, 3967 CrossRef CAS; (d) C. Cao, J. T. Ciszewski and A. L. Odom, Organometallics, 2001, 20, 5011 CrossRef CAS; (e) S. Majumder and A. L. Odom, Organometallics, 2008, 27, 1174 CrossRef CAS; (f) D. L. Swartz II and A. L. Odom, Organometallics, 2007, 26, 6684 CrossRef; (g) C. Cao, Y. Li, Y. Shi and A. L. Odom, Chem. Commun., 2004, 2002 RSC.
- (a) J. S. Johnson and R. G. Bergman, J. Am. Chem. Soc., 2001, 123, 2923 CrossRef CAS; (b) B. F. Straub and R. G. Bergman, Angew. Chem., Int. Ed., 2001, 40, 4632 CrossRef CAS; (c) L. Ackermann, R. G. Bergman and R. N. Loy, J. Am. Chem. Soc., 2003, 125, 11956 CrossRef CAS; (d) L. Ackermann and R. G. Bergman, Org. Lett., 2002, 4, 1475 CrossRef CAS.
- (a) G. Zi, J. Organomet. Chem., 2011, 696, 68 CrossRef CAS; (b) Q. Wang, H. Song and G. Zi, J. Organomet. Chem., 2010, 695, 1583 CrossRef CAS; (c) G. Zi, F. Zhang, L. Xiang, Y. Chen, W. Fang and H. Song, Dalton Trans., 2010, 39, 4048 RSC; (d) L. Xiang, H. Song and G. Zi, Eur. J. Inorg. Chem., 2008, 1135 CrossRef CAS.
- (a) T. Janssen, R. Severin, M. Diekmann, M. Friedemann, D. Haase, W. Saak, S. Doye and R. Beckhaus, Organometallics, 2010, 29, 1806 CrossRef CAS; (b) C. Müller, R. Koch and S. Doye, Chem.–Eur. J., 2008, 14, 10430 CrossRef; (c) K. Gräbe, B. Zwafelink and S. Doye, Eur. J. Org. Chem., 2009, 5565 CrossRef; (d) K. Gräbe, F. Pohlki and S. Doye, Eur. J. Org. Chem., 2008, 4815 CrossRef; (e) K. Marcseková, B. Wegener and S. Doye, Eur. J. Org. Chem., 2005, 4843 CrossRef; (f) D. Mujahidin and S. Doye, Eur. J. Org. Chem., 2005, 2689 CrossRef CAS.
- (a) A. Tillack, V. Khedkar, H. Jiao and M. Beller, Eur. J. Org. Chem., 2005, 5001 CrossRef CAS; (b) A. Tillack, H. Jiao, I. G. Castro, C. G. Hartung and M. Beller, Chem.–Eur. J., 2004, 10, 2409 CrossRef CAS; (c) V. Khedkar, A. Tillack and M. Beller, Org. Lett., 2003, 5, 4767 CrossRef CAS; (d) A. Tillack, I. G. Castro, C. G. Hartung and M. Beller, Angew. Chem., Int. Ed., 2002, 41, 2541 CrossRef CAS; (e) N. Schwarz, K. Alex, I. A. Sayyed, V. Khedkar, A. Tillack and M. Beller, Synlett, 2007, 1091 CAS.
- (a) J. A. Bexrud, C. Li and L. L. Schafer, Organometallics, 2007, 26, 6366 CrossRef CAS; (b) Z. Zhang, D. C. Leitch, M. Lu, B. O. Patrick and L. L. Schafer, Chem.–Eur. J., 2007, 13, 2012 CrossRef CAS; (c) A. V. Lee, M. Sajitz and L. L. Schafer, Synthesis, 2009, 97 CAS; (d) Z. Zhang and L. L. Schafer, Org. Lett., 2003, 5, 4733 CrossRef CAS.
- (a) M. L. Buil, M. A. Esteruelas, A. M. López, A. C. Mateo and E. Oñate, Organometallics, 2007, 26, 554 CrossRef CAS; (b) M. L. Buil, M. A. Esteruelas, A. M. López and A. C. Mateo, Organometallics, 2006, 25, 4079 CrossRef CAS; (c) M. A. Esteruelas, A. M. López, A. C. Mateo and E. Oñate, Organometallics, 2006, 25, 1448 CrossRef CAS; (d) H. Shen and Z. Xie, J. Am. Chem. Soc., 2010, 132, 11473 CrossRef CAS.
- (a) F. Zhou, M. Lin, L. Li, X. Zhang, Z. Chen, Y. Li, Y. Zhao and W. Li, Organometallics, 2011, 30, 1283 CrossRef CAS; (b) X. Han, Y. Li, H. Wei and Y. Zhang, Chin. J. Chem., 2007, 25, 1334 CrossRef CAS.
- (a) S. Liu, W. Zuo, S. Zhang, P. Hao, D. Wang and W. Sun, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3411 CrossRef CAS; (b) W. Zuo, S. Zhang, S. Liu, X. Liu and W. Sun, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3396 CrossRef CAS; (c) A. L. Beauchaup, D. Cozak and A. Mardhy, Inorg. Chim. Acta, 1984, 92, 191 CrossRef; (d) A. K. Verma, J. H. Chou and T. B. Rauchfuss, Inorg. Chem., 1998, 37, 5960 CrossRef CAS; (e) K. Hensen, A. Lemke and C. Nather, Z. Anorg. Allg. Chem., 1997, 623, 1973 CrossRef CAS; (f) T. Irrgang and R. Kemle, Eur. J. Inorg. Chem., 2005, 4382 CrossRef CAS.
- X. Wu, R. Jiang, X. Xu, X. Su, W. Lu and S. Ji, J. Comb. Chem., 2010, 12, 829 CrossRef CAS.
- (a) A. O. Eseola, M. Zhang, J. Xiang, W. Zuo, Y. Li, J. A. O. Woods and W. Sun, Inorg. Chim. Acta, 2010, 363, 1970 CrossRef CAS; (b) A. O. Eseola, W. Li, O. G. Adeyemi, N. O. O. Egbedi and J. A. O. Woods, Polyhedron, 2010, 29, 1891 CrossRef CAS.
- E. Haak, I. Bytschkov and S. Doye, Angew. Chem., Int. Ed., 1999, 38, 3389 CrossRef CAS.
- Two mechanisms for the titanium- and zirconium-catalyzed hydroamination of alkynes and alkenes have been proposed: (1) 2+2 cycloaddition of the unsaturated bond of alkynes or alkenes to the M
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, and (2) insertion of the unsaturated bond of alkynes or alkenes into the MN bond. According to these two mechanisms, the hydroamination reaction was initiated via attack of the metal center by the amine. For the detailed mechanisms, see:
(a) D. C. Leitch, C. S. Turner and L. L. Schafer, Angew. Chem., Int. Ed., 2010, 49, 6382 CrossRef CAS;
(b) P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708 CrossRef CAS;
(c) A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753 CrossRef CAS.
N bond, and (2) insertion of the unsaturated bond of alkynes or alkenes into the MN bond. According to these two mechanisms, the hydroamination reaction was initiated via attack of the metal center by the amine. For the detailed mechanisms, see:
(a) D. C. Leitch, C. S. Turner and L. L. Schafer, Angew. Chem., Int. Ed., 2010, 49, 6382 CrossRef CAS;
(b) P. J. Walsh, A. M. Baranger and R. G. Bergman, J. Am. Chem. Soc., 1992, 114, 1708 CrossRef CAS;
(c) A. M. Baranger, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1993, 115, 2753 CrossRef CAS. - G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallography, 1H and 13C NMR and IR and HRMS data. CCDC reference numbers 783591 and 804537 for complexes 2 and 6·THF, respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00408e |
| This journal is © The Royal Society of Chemistry 2012 |