A new approach for the preparation of variable valence rare earth alloys from nano rare earth oxides at a low temperature in molten salt†
Milin
Zhang
*a,
Yongde
Yan
*ab,
Wei
Han
a,
Xing
Li
a,
Zhiyao
Hou
a,
Yang
Tian
a,
Ke
Ye
a,
Lihong
Bao
c,
Xiaodong
Li
c and
Zhijian
Zhang
b
aKey Laboratory of Superlight Materials and Surface Technology, Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, China. E-mail: zhangmilin@hrbeu.edu.cn; y5d2006@hrbeu.edu.cn; Fax: +86 (0)451 82533026; Tel: +86 (0)451 82533026
bCollege of Nuclear Science and Technology, Harbin Engineering University, Harbin 150001, China
cDepartment of Mechanical Engineering, University of South Carolina, 300 Main Street, Columbia, South Carolina 29208, USA
First published on 22nd December 2011
Abstract
The solubility of RE2O3 (RE = Eu, Sm, and Yb) with variable valence in molten salts is extremely low. It is impossible to directly obtain variable valence metals or alloys from RE2O3 using electrolysis in molten salts. We describe a new approach for the preparation of variable valence rare earth alloys from nano rare earth oxide. The excellent dispersion of nano–Eu2O3 in LiCl–KCl melts was clearly observed using a luminescent feature of Eu3+ as a probe. The ratio of solubility of nano-Sm2O3/common Sm2O3 is 16.98. Electrochemical behavior of RE2O3 on a molybdenum and Al electrode in LiCl–KCl melts containing AlCl3 at 480 °C was investigated by different electrochemical techniques, such as cyclic voltammetry (CV), square wave voltammetry, and chronopotentiometry. Prior to the reduction peak of Al, the reduction peaks of Sm(III)/Sm(II), Yb(III)/Yb(II), and Eu(III)/Eu(II) were observed at about −0.85, −0.45, and 0.39 V in square wave voltammetry, respectively. The underpotential deposition of RE on pre-deposited aluminum leads to the formation of Al–RE alloy. The structure, morphology, and energy dispersion analysis of the deposit obtained by potentiostatic electrolysis are analyzed. Al2Sm and Al3Sm alloys were successfully obtained from nano–Sm2O3.
1. Introduction
It has been long thought that rare earth (RE = Eu, Sm, and Yb) metals with variable valencies can not be obtained viareduction of their halides using Ca or Li as a reducer, because these RE metals can form stable bivalent halides. In principle, since the reduction potential of RE(II) to RE(0) is even more negative than the solvent in the chloride system, it is impossible to prepare variable valence RE metals directly by electrolysis from molten salt chlorides. The only technique for making variable valence RE metals is through reduction by lanthanum, cerium or their mixture with low vapor tension, and distillation at high temperature in a vacuum. However, this methodology has many drawbacks, such as low throughput, a complex and discontinuous production processes, and high energy consumption. In such a process, lanthanum and cerium prepared by electrolysis are essential reduction materials, but incur a high cost. A better approach to produce these variable valence RE metals is alloying directly with other metals.1–7In the past decades, the raw materials for the preparation of variable valence RE alloys are RE chlorides and fluorides, obtained via the chlorination and fluorination of RE oxides. However, toxic and corrosive HCl and HF have to be bubbled into the melts to avoid the formation of RE2O3 precipitates during the electrolysis process, because RE(III) is very sensitive to O2− ions. In order to overcome the drawbacks of the route using chlorides and fluorides as precursors, some researchers have achieved metals, nonmetals, or alloys from their respective oxidesvia electrochemical reduction in molten salts. Chen and Fray8 reported the direct electro-deoxidation of titanium dioxide to titanium in calcium chloride. Chen9 further investigated the electrochemical metallization of solid terbium oxide to terbium metal under Ca deposition in molten CaCl2. The electroreduction of solid titanium and terbium oxide to their respective metals can only proceed under the conditions for Ca deposition and predominantly electro-deoxidize with little contribution of Ca reduction. Nohira et al.10 succeeded in removing oxygen from the surfaces of solid SiO2 plates in a molten CaCl2 electrolyte at 850 °C. Chen et al.11 developed an electrochemical preparation of nonmetallic silicon and its alloys from solid oxides in the same melt. However, the electrochemical preparation of variable valence RE metals or alloys from solid oxides in molten salts has hitherto not been reported. Here we present a new approach for making RE alloys from nanosized RE2O3 by taking advantage of nano effects for the first time. This new technique overcomes the limitation of low RE2O3 solubility in molten salts, and opens up unprecedented opportunities for making RE and other alloys which have been thought impossible before. Compared with direct electro-deoxidation (FFC Cambridge process), this technique offers an alternative route to fabricate alloys from oxide using the reduction of metallic ions dissolved in molten salts.
2. Experimental
2.1 Nanoparticle synthesis
Nano particles of Eu2O3, Sm2O3, Yb2O3 and Y2O3:Eu3+ were all prepared by sol–gel process. The doping concentration of Eu3+ is 5 mol% that of Y3+ in Y2O3 host, which has been optimized previously.12Eu2O3, Sm2O3, Yb2O3, and stoichiometric amounts of Y2O3 and Eu2O3 (all with purity of 99.999%, Science and Technology Parent Company of Changchun Institute of Applied Chemistry, China) were dissolved in HNO3 (A. R.) and then mixed with a water-ethanol (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7) solution containing citric acid (A. R.) as a chelating agent for the metal ions. The molar ratio of metal ions to citric acid was 1:2. Polyethylene glycol (PEG, molecular weight = 10000, A. R.) was added to adjust the viscoelasticity of the solution (a final concentration of PEG is 0.08 g mL−1 in the water–ethanol solution). The resultant mixtures were stirred for 1 h to obtain a homogeneous hybrid sol and heated at 75 °C in a water bath until homogeneous gels formed. After being dried in an oven at 110 °C for 10 h, the gels were ground and prefired at 500 °C for 3 h in air. Finally the samples were fully ground and fired to 700 °C for 3 h.
:7) solution containing citric acid (A. R.) as a chelating agent for the metal ions. The molar ratio of metal ions to citric acid was 1:2. Polyethylene glycol (PEG, molecular weight = 10000, A. R.) was added to adjust the viscoelasticity of the solution (a final concentration of PEG is 0.08 g mL−1 in the water–ethanol solution). The resultant mixtures were stirred for 1 h to obtain a homogeneous hybrid sol and heated at 75 °C in a water bath until homogeneous gels formed. After being dried in an oven at 110 °C for 10 h, the gels were ground and prefired at 500 °C for 3 h in air. Finally the samples were fully ground and fired to 700 °C for 3 h.
2.2 Purification of the melts and solubility measurement of RE2O3
The mixture of LiCl–KCl (50:50 wt%, analytical grade) was first dried under vacuum for more than 72 h at 473 K to remove excess water. Metal ion impurities in the melts were removed by pre-electrolysis at −2.0 V (vs.Ag/AgCl) for 4 h.13,14Aluminum and samarium, ytterbium and europium ions were introduced into the bath in the form of AlCl3, Sm2O3, Yb2O3, and Eu2O3 powders. To determine the solubility of RE2O3 (wt%) in the LiCl–KCl melts at 480 °C, the nanosized and common particles were added into the LiCl–KCl melts and held for 3 h at 480 °C, and then the samples were taken from the clear supernatant fluid in the molten salt mixtures. After the clear supernatant fluid samples were solidified, each sample was dissolved in distilled water for analysis. The solution was diluted and analyzed using an inductively coupled plasma atomic emission spectrometer (ICP-AES, IRIS Intrepid II XSP, Thermo Elemental). The contents of K+, Li+, and RE3+ measured by ICP were converted to the masses of KCl, LiCl, and RE2O3, and then the solubility of RE2O3 in LiCl–KCl melts was calculated.
2.3 Electrochemical apparatus and electrodes
All electrochemical measurements were carried out using an electrochemical workstation (Im6eX, Zahner Co., Ltd.) with THALES 3.08 software package. A silver wire with a diameter of 1 mm, which was dipped into a solution of AgCl (1 wt%) in LiCl–KCl melts contained in a Pyrex tube was used as a reference electrode. A spectrally pure graphite rod with a diameter of 6 mm served as the counter electrode. The working electrodes were molybdenum wires (diameter 1 mm, 99.99% purity), which were polished using SiC paper to remove the surface oxides, then cleaned ultrasonically with ethanol prior to use. The active electrode surface area was determined after each experiment by measuring the immersion depth of the electrode in the molten salts.2.4 Preparation and characterization of Al–RE alloys
The Al–RE alloys were prepared by potentiostatic electrolysis under different conditions. After electrolysis, all samples were washed with hexane (99.8% purity) in an ultrasonic bath to remove salts and then stored in a glove box for analysis. These deposits were analyzed by X-ray diffraction (XRD, X' Pert Pro; Philips Co., Ltd.) using Cu-Kα radiation at 40 kV and 40 mA. The microstructure of the Al–RE alloys was measured by scanning electron microscopy (SEM) and energy dispersive spectrometry (EDS) mapping analysis (JSM-6480A; JEOL Co., Ltd.).3. Results and discussion
3.1 Characterization of nanosized RE2O3
In the first step, we prepared nanosized Eu2O3, Sm2O3 and Yb2O3 particles by a sol–gel process. Fig. 1 shows the XRD spectra of the as-prepared Eu2O3, Sm2O3 and Yb2O3 samples together with the JCPDS cards for Eu2O3 (No. 34–0392), Sm2O3 (No. 15–0813) and Yb2O3 (No. 65–3173), respectively. All the diffraction peaks of these samples can be readily indexed to their corresponding standard patterns. No diffraction peaks of any impurity were observed, indicating high purity of these final products. These results suggest that the precursor samples were crystallized into Eu2O3, Sm2O3 and Yb2O3 at the heating temperature of 700 °C. | ||
| Fig. 1 X-ray diffraction spectra for Eu2O3 (a), Sm2O3 (b) and Yb2O3 (c) samples after annealing at 700 °C for 3 h in air, the JCPDS card 34–0392 for Eu2O3, 15–0813 for Sm2O3 and 65–3173 for Yb2O3. | ||
Fig. 2a, d and g illustrate a representative overview of the morphologies of the Eu2O3, Sm2O3 and Yb2O3 samples, respectively. The obtained samples are composed of aggregated spherical nanoparticles with a size range from 10 nm to 50 nm. The HRTEM images and corresponding FFT diffraction patterns (Fig. 2b and c, 2e and f, 2h and i) reveal that individual nanoparticles of Eu2O3, Sm2O3, and Yb2O3 are single crystals. No defects were observed in the nanoparticles.
 | ||
| Fig. 2 TEM images of Eu2O3 (a) with its HRTEM (b, c), Sm2O3 (d) with its HRTEM (e, f) and Yb2O3 (g) with its HRTEM (h, i). | ||
The bulk density of the nano-particles (RE2O3) is readily compared with normal particles, as shown in Fig. 3a–c. The bulk density ratios of nano to normal for Eu2O3, Sm2O3, Yb2O3 are 0.75, 0.77 and 0.85, respectively. Compared with common materials, nano-particles in nano-materials interleave each other and form a porous network structure, resulting in greater porosity. Therefore, the bulk density of the aggregated nanosized Sm2O3 is lower compared to common materials. The solubility values of normal and nano Sm2O3, measured in the LiCl–KCl melts at 480 °C, were 0.007981 and 0.1355 wt%, respectively, as shown in Fig. 3d–f. The ratios of solubility of nano RE2O3 (RE = Eu, Sm, Yb)/common RE2O3 (wt%) are 13.81, 16.98, and 17.29, respectively. The large increase of solubility for nano-materials can be ascribed to the following reasons. According to the view presented by Ma,15 the decrease in particle size from normal RE2O3 to the nano size leads to a larger dangling-bond density. The existence of a mass of dangling bonds will principally generate two results: On one hand, the dangling bonds increase the surface energy, which consequently decreases the critical temperatures of thermodynamic processes like the melting point,16crystallization temperature,17sintering temperature.18 On the other hand, dangling bonds have a tendency of binding to other atoms. Accordingly, they become the active centers for adatom adsorption and interaction, making some thermodynamic processes and chemical reactions easier.15 The nanoparticles have smaller dimensions and larger specific surface areas, which facilitate greater contact of RE2O3 with Cl−. The nanoparticles, therefore, possess more active atoms than normal ones, which are highly active to absorb large numbers of Cl−. These factors facilitate the diffusion of RE2O3 nanoparticles into the melts.
 | ||
| Fig. 3 bulk density (a–c); solubility (d–f); dispersion of Eu2O3, Sm2O3, Yb2O3 (left: nano, right: common RE2O3) (g–i); and luminescent photograph of nano (j) and normal (k) Y2O3:Eu3+ in the LiCl–KCl melts. | ||
The practical states of dispersion of common and nanosized RE2O3 are plotted in Fig. 3g–i. To further illuminate the dispersion of the Eu2O3 sample, we also added nanosized Y2O3:Eu3+ phosphors (prepared by sol–gel process) to the LiCl–KCl melt system due to their luminescent feature. As the main and unsurpassed red emitting materials in fluorescent lamps and flat panel devices, Y2O3:Eu3+ phosphors inevitably gather more attention because of their good luminescent characteristics, acceptable atmospheric stability, reduced degradation under applied voltages, and the lack of hazardous constituents as opposed to sulfide phosphors.19,20 The Eu3+ ion is an important RE ion that acts as an activator, which mainly shows emission due to transitions of 5D0–7FJ (J = 4, 3, 2, 1, 0) in the red regions.21 The LiCl–KCl–Y2O3:Eu3+ system clearly exhibits a red emission upon 254 nm excitation under a UV lamp (see Fig. 3j). Under short wavelength UV irradiation, Y2O3:Eu3+ phosphors exhibit the characteristic red emission of Eu3+, dominated by the hypersensitive 5D0–7F2 transition of Eu3+ at about 612 nm.20 The red emission of the melts system in the quartz tube, with the addition of Y2O3:Eu3+, indicates that Y2O3:Eu3+ phosphors disperse in the system homogeneously. Different from nano–RE2O3 uniformly dispersing in the melts, common RE2O3 sinks to the bottom of the quartz tube (Fig. 3k).
In the vicinity of RE2O3 atoms, RE3+ ions are in higher concentration than in the bulk solution. RE2O3 atoms and RE3+ ions form a positively charged complex, of which there is abundance in the melts. Electrostatic repulsion between these complexes promotes suspension, which could explain the even dispersion of particles in melts. This could explain why nano–RE2O3 particles can disperse well in melts. Small size, high solubility and even dispersion of nano-particles are prerequisites for electrolyzing RE alloys from RE2O3 in melts.
3.2 Electrochemical behavior of nano–RE2O3 on Mo and Al electrodes
The element Al was selected as an alloying element in the variable valence RE alloys due to its excellent mechanical properties. The electrochemical behavior of LiCl–KCl containing AlCl3 and RE2O3 (Sm, Yb, Eu) on a molybdenum electrode was studied. Fig. 4 shows the representative CVs obtained on molybdenum electrodes (S = 0.322cm2) before and after the addition of 2 wt% AlCl3 and 1wt% nano-Sm2O3, 1wt% nano-Yb2O3, and 1wt.% nano-Eu2O3 in LiCl–KCl melts at 480 °C. The dotted curve represents the voltammogram before the addition of AlCl3 and RE2O3. Only one couple of the cathodic/anodic signals D/D′ is observed, corresponding to the deposition and dissolution of liquid Li, since no alloys or intermetallic compounds exist in the Mo–Li binary system at 480 °C. The solid curves show the voltammograms measured after the addition of AlCl3 and RE2O3. Since all three kinds of melts contain AlCl3 and their deposition potentials are almost the same, cathodic peak A is observed from approximate −1.00 V (vs Ag/AgCl) which corresponds to the reduction of Al(III) in LiCl–KCl–AlCl3–RE2O3 melts. An anodic current background or wave on the neck of anodic peak A′ (the oxidation of deposited metal Al) is likely to be associated with the formation of Al–Mo alloy in the positive-going scan (Fig. S1, ESI†). It is not feasible to study the redox RE(II)/RE(0) system because RE(II) is reduced to RE(0) at a high negative value close to that of lithium reduction, which agrees with data in the literature.22–24 However, because nano RE2O3 can be well dispersed in molten LiCl–KCl salt at low temperature, the underpotential deposition of RE on pre-deposited aluminum leads to the formation of Al–RE (RE = Sm, Yb, and Eu) alloys, marked by cathodic current peaks B1, B2, and B3, respectively. The peak of alloy formation was observed in melts containing nano RE2O3. Nano effects, responsible for this behavior, give rise to high solubility and even dispersion in melts, and subsequently result in the deposition of RE on Al and formation of Al–RE alloys. After peaks B2 and B3, a current wave C is observed, which is associated with the underpotential deposition of Li(I) on pre-deposited Al forming Al–Li alloys. On the anodic sweep, A′, B′, and C′ are related to the oxidation of metallic Al, Al–RE and Al–Li alloys. From the inset graph in Fig. 4(a) and (b), B1′ and B2′ are split into two oxidation peaks B1′′ and B1′′′, and B2′′ and B2′′′, respectively. According to the phase diagram of Al–Sm and Al–Yb system,25 the oxidation peaks Bn′′ and Bn′′′ (n = 1 and 2) correspond to two intermetallic compounds of Al–Sm and Al–Yb alloys, respectively. The redox peaks of REs(III) to REs(II), which should have occurred prior to Al signals, are not clearly observed. This is probably because the concentration of RE2O3 is relatively low and the redox peaks are not easily found in comparison with the larger-scaled peak A. Another possible explanation is that the CV experiments did not cover a wider potential range. These points were proved in square wave voltammetry and Fig. S1, ESI.† The reduction signals of REs(III) to REs(II) with the same concentration are detected in square wave voltammetry (see Fig. 5), which is a more sensitive method than cyclic voltammetry. Prior to the reduction peak of Al, the reduction peaks of Sm(III)/Sm(II), Yb(III)/Yb(II), and Eu(III)/Eu(II) were observed at about −0.85, −0.45, and 0.39 V, respectively. These values are in agreement with the data reported in literature,4,7,22 when temperature and melt system differences are taken into account. The formation signal of Al–Mo is also observed between the reduction peaks of Al and RE(III) to RE(II) (RE = Eu and Yb). We do not observe the peak (or shoulder) corresponding to Al–Mo formation in LiCl–KCl–AlCl3–Sm2O3 melts. The signal is probably hidden by a larger-scale reduction current of Sm(III) to Sm(II) due to the slight difference of their formation potential. Two additional cathodic peaks after the reduction peak of Al correspond to the underpotential deposition of RE (Sm, Yb, and Eu) and Li on pre-deposited Al forming Al–RE and Al–Li alloys, respectively.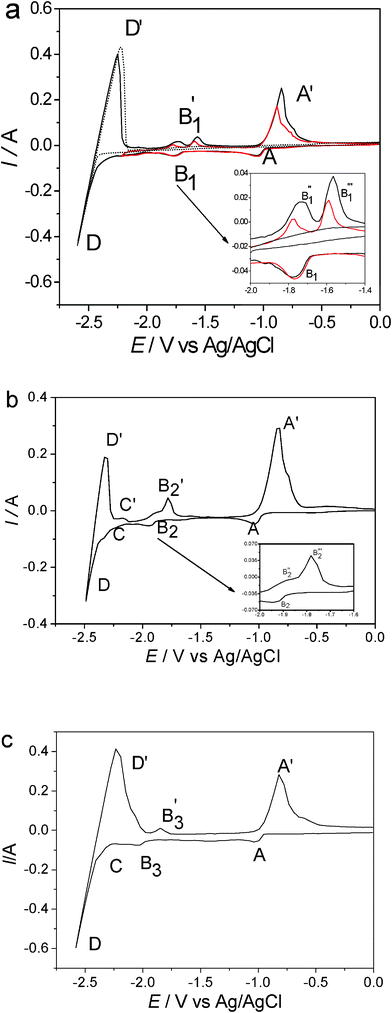 | ||
| Fig. 4 CVs of the LiCl–KCl melts before (dotted line) and after (solid line) the addition of 2 wt% AlCl3 and 1wt% (a) nano-Sm2O3 (b) nano-Yb2O3 (c) nano-Eu2O3 on molybdenum electrodes (S = 0.322 cm2) at different cathodic limits at 480 °C. Scan rate: 0.1 V s−1. | ||
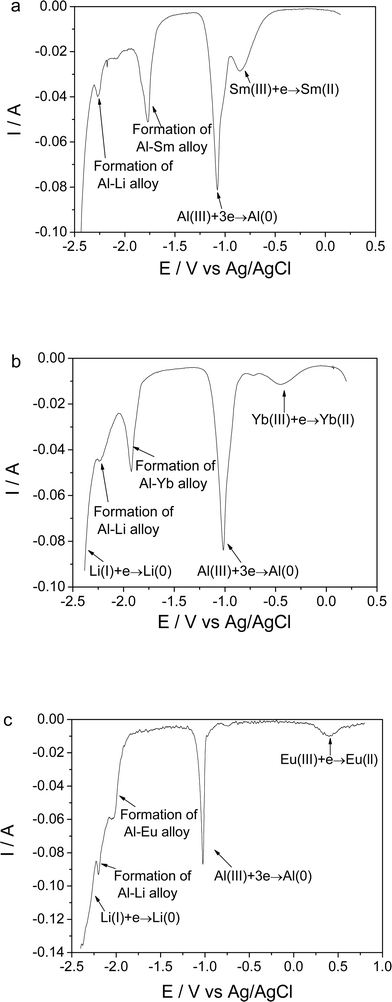 | ||
| Fig. 5 Square wave voltammograms of the LiCl–KCl–AlCl3 (2 wt%) melts after the addition of 1 wt% nano-materials (a) Sm2O3 (b) Yb2O3 (c) Eu2O3 on molybdenum electrodes (S = 0.322 cm2) at 480 °C. F = 20 Hz. | ||
In the previous section, the electro-deposition of Al–RE alloys from nano-RE2O3 was studied on an inert electrode. The electrochemical behavior of nano-RE2O3 was also investigated on an active Al electrode using CVs and open-circuit chronopotentiometry (Fig. 6). The element Sm was selected to represent an RE element with variable valency. The shape of the curve obtained at the Mo electrode is remarkably different from the curves obtained at the Al electrode. The electrochemical window of the Al electrode is limited cathodically by Li(I) reduction forming a Li–Al alloy, and anodically by the oxidation of the Al electrode, marked as C and A′, respectively. In this electroactive range, a group of signals B/B′, were observed at a more positive potential than that at the Mo electrode. Since this potential value is more positive than the potential of Sm metal deposition, the cathodic peak is thought to be caused by the formation of an Al–Sm alloy. We can also see that the reduction potential value of Li(I) is more positive at the Al electrode than that at the Mo electrode.
 | ||
| Fig. 6 (a) Cyclic voltammograms of the LiCl–KCl–AlCl3 (2 wt%) melts containing 1wt% nano-Sm2O3 at Mo (dotted line) (S = 0.322 cm2) and Al (solid line) (S = 0.598 cm2) electrodes. Scan rate:0.1 V s−1.(b) Open-circuit chronopotentiogram for Al electrode (S = 0.598 cm2) after electrodepositing Li metal by galvanostatic electrolysis at −200 mA for 5 min in the LiCl–KCl melts containing 1wt% nano–Sm2O3 at 480 °C. | ||
Cyclic voltammetry cannot be used to clearly identify the intermetallic compounds. So, open-circuit potentiometry was carried out to investigate the formation potential of Al–Sm alloys (Fig. 6b). The measurements were conducted as follows: after a short cathodic polarisation at a high cathodic over potential, the open-circuit potential of the electrode was measured versus time. Since the deposited Sm metal reacts with Al and diffuses into the bulk of the electrode, the electrode potential gradually shifts to more positive values. During this process, the evolution of the potential consists of successive plateaus, of increasing potential, each plateau typical of equilibrium in the solid state between two intermetallic compounds. The diffusion of Sm within the substrate explains the increase at potential of the cathode.
For 100 s in the beginning, the potential was kept at −2.15 V, assessed as the formation of Al–Li alloy. Potential plateaus, observed at (2) −1.80 V; (3) −1.60 V, are considered to correspond to the coexisting phase states of Al–Sm alloys. Finally, a potential plateau observed at −1.00 V is the potential abandon of the Al electrode.
3.3 Potentiostatic electrolysis and characterization of the deposits
Based on the results obtained by CVs and chronopotentiogram, potentiostatic electrolysis was carried out in LiCl–KCl melts containing 1 wt% nano-Sm2O3 and 2 wt% AlCl3 with different concentrations on aluminum electrodes at 480 °C. The structure, morphology, and energy dispersion analysis of deposit obtained by electrolysis are shown in Fig. 7. Fig. 7a presents the XRD patterns of Al–Sm alloys obtained by potentiostatic electrolysis from LiCl–KCl–AlCl3 (2 wt%)–Sm2O3(1 wt%) at different potentials for 2 h. The sample (i) obtained at −1.7 V was identified as an Al3Sm and Al substrate. Thus, the alloy formed on the Al electrode is identified as the Al3Sm alloy. The sample (ii) was identified as a mixture of Al3Sm + Al2Sm + KCl at a more negative potential of −2.0 V. The morphology of the deposit obtained at a more negative potential −2.2 V is shown in Fig. 7b. Some white particles form on the Al substrate. To figure out the composition of the particles, EDS analysis was carried out. EDS of points A (Fig. 7c) and B (Fig. 7d) indicate that the Sm content in the alloys is relatively high. These Al–Sm-rich intermetallic compounds are mainly composed of Al and Sm elements with Al:Sm atomic ratios of 1.80 and 2.51, respectively, which are in good agreement with the stoichiometric ratio (2:1 or 3:1). This data suggests that Al2Sm and Al3Sm are formed during electrolysis.
 | ||
| Fig. 7 (a) XRD patterns of Al–Sm alloys obtained at different potentials (i) −1.7 (ii) −2.0 V. (b) SEM and (c, d) EDS quantitative analysis of the deposit by potentiostatic electrolysis at −2.2 V from the LiCl–KCl–AlCl3(2 wt%)−Sm2O3(1 wt%) melts for 2 h at 480 °C. | ||
4. Conclusions
The RE (RE = Sm, Eu, and Yb) alloys with variable valent were prepared for the first time by electrolysis from the corresponding nano-rare earth oxides at 480 °C in LiCl–KCl melts. Size, solubility and dispersion of the nano-particles in the LiCl–KCl melts were examined. The bulk density ratios and solubility of nano–Sm2O3/normal Sm2O3 are 0.77 and 16.98, respectively. Electrochemical behavior of RE2O3 on a molybdenum and Al electrode in LiCl–KCl melts containing AlCl3 at 480 °C was investigated by cyclic voltammetry, square wave voltammetry, and chronopotentiometry. The results of cyclic voltammetry indicates that the underpotential deposition of RE on pre-deposited aluminum leads to the formation of Al–RE (RE = Sm, Yb, and Eu) alloy. The formation signals of Al–RE alloys also are detected in square wave voltammetry and open-circuit chronopotentiometry. The structure, morphology, and energy dispersion analysis of deposit obtained by potentiostatic electrolysis were analyzed, and the formation of Al–Sm alloys was confirmed.Acknowledgements
The work was financially supported by the National 863 Project of the Ministry of Science and Technology of China (2009AA050702 and 2011AA03A409), the National Natural Science Foundation of China (50871033, 21103033, and 21101040), the Heilongjiang Postdoctoral Fund (LBH-Z10196 and LBH-Z10207), China Postdoctoral Science Foundation (20100480974) and Fundamental Research funds for the Central Universities. The authors are particularly grateful to Dr Tom Mann for kindly discussing various parts of the manuscript.References
- T. Iida, T. Nohira and Y. Ito, Electrochim. Acta, 2001, 46, 2537–2544 Search PubMed.
- T. Iida, T. Nohira and Y. Ito, Electrochim. Acta, 2003, 48, 1531–1536 CrossRef CAS.
- T. Iida, T. Nohira and Y. Ito, Electrochim. Acta, 2003, 48, 2517–2521 CrossRef CAS.
- M. R. Bermejo, F. de la Rosa, E. Barrado and Y. Castrillejo, J. Electroanal. Chem., 2007, 603, 81–95 Search PubMed.
- Y. Castrillejo, P. Fernández, M. R. Bermejo, E. Barrado and A. M. Martínez, Electrochim. Acta, 2009, 54, 6212–6222 Search PubMed.
- S. A. Kuznetsov and M. Gaune-Escard, Electrochim. Acta, 2001, 46, 1101–1111 CrossRef CAS.
- V. Smolenski, A. Novoselova, A. Osipenko, C. Caravacac and G. Córdoba, Electrochim. Acta, 2008, 54, 382–387 CrossRef CAS.
- G. Z. Chen, D. J. Fray and T. W. Farthing, Nature, 2000, 407, 361–364 CrossRef CAS.
- D. H. Wang, G. H. Qiu, X. B. Jin, X. H. Hu and G. Z. Chen, Angew. Chem., Int. Ed., 2006, 45, 2384–2388 CrossRef CAS.
- T. Nohira, K. Yasuda and Y. Ito, Nat. Mater., 2003, 2, 397–401 CrossRef CAS.
- X. B. Jin, P. Gao, D. H. Wang, X. H. Hu and G. Z. Chen, Angew. Chem., 2004, 751–754 Search PubMed Angew. Chem. Int. Ed. 2004, 43, 733-736.
- M. Yu, J. Lin, Z. Wang, J. Fu, S. Wang, H. J. Zhang and Y. C. Han, Chem. Mater., 2002, 14, 2224–2231 CrossRef CAS.
- Y. D. Yan, M. L. Zhang, Y. Xue, W. Han, X. Y. Jing, L. Y. He and Y. Yuan, Phys. Chem. Chem. Phys., 2009, 11, 6148–6155 RSC.
- Y. D. Yan, M. L. Zhang, Y. Xue, W. Han, D. X. Cao and S. Q. Wei, Electrochim. Acta, 2009, 54, 3387–3393 Search PubMed.
- F. Ma and K. W. Xu, Surf. Interface Anal., 2007, 39, 611–614 CrossRef CAS.
- S. L. Lai, J. Y. Guo, V. Petrova, G. Ramanath and L. H. Allen, Phys. Rev. Lett., 1996, 77, 99–102 CrossRef CAS.
- K. K. Nanda, F. E. Kruis and H. Fissan, Phys. Rev. Lett., 2002, 89, 256103–1-256103-4 CrossRef CAS.
- M. Hirasawa, T. Orii and T. Seto, Appl. Phys. Lett., 2006, 88, 093119–1-093119-3 CrossRef.
- J. Yang, Z. W. Quan, D. Y. Kong, X. M. Liu and J. Lin, Cryst. Growth Des., 2007, 7, 730–735 CrossRef CAS.
- H. Wang, M. Yu, C. K. Lin, X. M.Liu and J. Lin, J. Phys. Chem. C, 2007, 111, 11223–11230 CrossRef CAS.
- Z. Y. Hou, H. Z. Lian, M. L. Zhang, L. L. Wang, M. F. Lv, C. M. Zhang and J. Lin, J. Electrochem. Soc., 2009, 156, J209–J214 Search PubMed.
- G. Cordoba and C. Caravaca, J. Electroanal. Chem., 2004, 572, 145–151 Search PubMed.
- J. A. Plambeck (ed.:A. J. Bard), Encyclopedia of Chemistry of the Elements, vol. 10, Marcel Dekker, New York, 1976 Search PubMed.
- A. V. Bychkov and O. V. Skiba, Pyrochemical Separations, Workshop Proceeding OECD/NEA, Avignon, France, 2000 Search PubMed.
- T. B. Massalski, J. L. Murray, L. H. Benett and H. Baker, Binary alloy Phase Diagrams, American Society for Metals, 1990 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c1ra00285f/ |
| This journal is © The Royal Society of Chemistry 2012 |