Random copolymer of styrene and diene derivatives via anionic living polymerization followed by intramolecular Friedel–Crafts cyclization for high-performance thermoplastics†
Atsuhiro
Nakahara
ab,
Kotaro
Satoh
a and
Masami
Kamigaito
*a
aDepartment of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: kamigait@apchem.nagoya-u.ac.jp
bKurashiki Research Centre, Kuraray Co. Ltd, 2045-1, Sakazu, Kurashiki, Okayama, 710-0801, Japan
First published on 9th November 2011
Abstract
A series of random copolymers of styrene and diene derivatives prepared by living anionic copolymerization, using tetrahydrofuran (THF) as a randomizer, were treated with various Brönsted and Lewis acids to cationically cyclize the adjacent diene–styrene units into tetrahydronaphthyl bicyclic main-chain structures via intramolecular Friedel–Crafts alkylation. This reaction produced high-performance plastics with a high glass transition temperature and robust mechanical properties. Of the various Friedel–Crafts catalysts, CF3SO3H and BF3·2AcOH/benzyl chloride proved to be the most efficient in terms of their reactivity and product selectivity. All of the random styrene–diene-based copolymers, including styrene–isoprene (r-SIR), styrene–butadiene (r-SBR), p-methylstyrene–isoprene (r-pMSIR), and styrene–isoprene–butadiene (r-SIBR) copolymers, underwent efficient intramolecular cyclization by CF3SO3H despite the different monomer units and microstructures of the diene units, and yielded high-performance plastics (Tg ≥ 130 °C). The Tg value of the cyclized r-SBR was slightly lower than the Tg values observed for r-SIR and r-pMSIR. The cyclized random copolymers exhibited relatively high flexural moduli and more strength than the polystyrenehomopolymer. Thus, the styrene–diene-based random copolymer provides a novel platform for the production of high-performance thermoplastics that can be easily prepared from commercially available styrene and diene derivatives via living anionic polymerization followed by Friedel–Crafts alkylation.
Introduction
Because the properties of polymers significantly depend on their backbone structures, the post-polymerization reactions that change main-chain structures can lead to new applications for existing polymers. Copolymers based on conjugated diene derivatives are generally recognized as elastomers or rubbers, and are employed as soft materials in many fields because of their low glass transition temperatures (Tg's). For example, 1,3-butadiene and isoprene are used to produce various industrially important rubbers, such as styrene–1,3-butadiene (SBR), styrene–isoprene (SIR), and acrylonitrile–1,3-butadiene (NBR) rubbers. Of these, the styrene and dienes can be efficiently copolymerized by either radical polymerization in an emulsion or anionic polymerization in solution. The anionic copolymerization of these non-polar conjugated hydrocarbon monomers is known to proceed in a living fashion when polymerized with an alkyl alkali metalinitiator, such as butyllithium. Living anionic polymerization has been applied to the synthesis of block copolymers, such as the thermoplastic elastomer styrene–1,3-butadiene–styrene (SBS), via sequential monomer additions.In recent years, amorphous cyclic hydrocarbonpolymers possessing rigid cyclic structures in the main chain have been employed as optically transparent materials (especially in optoelectronics) due to their non-hygroscopic nature, good thermal properties, and transparency.1 In addition to the polymerization or copolymerization of cyclic olefin monomers, the intramolecular cyclization between the pendent and/or adjacent functional groups in the main chain, such as the cyclization of unsaturated hydrocarbonpolymers under acidic conditions, is one of the most efficient methods for preparing amorphous hydrocarbonpolymers with cyclic units in the main chain.2–6Cyclization has been extensively studied for soft elastomers based on poly(dienes), such as natural rubber,2 synthetic polyisoprene,3 polybutadiene,4 and their copolymers with styrene, resulting in brittle, amber-colored plastics.5,6
Recently, we found that random copolymers of styrene and isoprene (r-SIR) prepared by living anionic copolymerization can be converted to cycloolefincopolymer analogues with high Tg's via the well-controlled cyclization of random copolymers.7 The sequence composition of the two monomers in the r-SIR was tuned to be nearly equimolar and random, with specific operation-delaying monomer-mixture feeds during living anionic polymerization in the presence of a small amount of THF as the randomizer. The subsequent cationic cyclization of the rubbery r-SIR (Tg = 20 °C) with a strong Brönsted acid (i.e., CF3SO3H) resulted in a polymer with a relatively high Tg (Tg = 130 °C). This high Tg is most likely due to the formation of rigid tetrahydronaphthyl bicyclic structures via intramolecular Friedel–Crafts alkylation, in which the protonated isoprene units predominantly reacted with the ortho position of the benzene ring of the adjacent styrene unit formed viarandom copolymerization.
In this study, we examined the CF3SO3H-induced intramolecular Friedel–Crafts cyclization of a series of styrene–diene-based random copolymers, including styrene–butadiene (r-SBR), p-methylstyrene–isoprene (r-pMSIR), and styrene–isoprene–butadiene (r-SIBR) copolymers and r-SIR with various microstructures prepared by living anionic copolymerizations (Scheme 1). In addition, Brönsted and Lewis acids were also employed as catalysts for the intramolecular Friedel–Crafts reaction with r-SIR.8 The combination of a Lewis acid with a Brönsted acid or an alkyl halide as the cationogen proved to be highly efficient, which is consistent with previous reports using living cationic polymerization.9 The mechanical properties of the obtained cycloolefincopolymer analogues were evaluated as thermoplastics and compared to commercially available polystyrene, which exhibits a lower Tg.
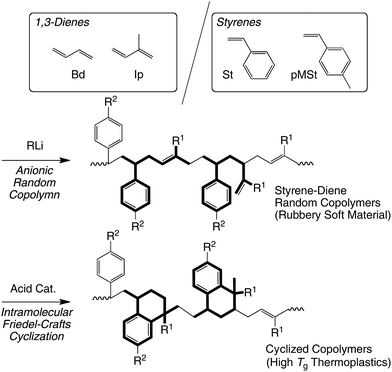 | ||
| Scheme 1 Living anionic copolymerization of 1,3-diene and styrene derivatives, and subsequent intramolecular Friedel–Crafts cationic cyclization of the copolymer. | ||
Experimental
Materials
Styrene (St: Kishida Chemical; purity 99.5%) and 4-methylstyrene (pMeSt: Aldrich; purity >99%) were distilled from calcium hydride and stored at −25 °C. Isoprene (Ip: Tokyo Chemical Industry; purity >99.0%) was dried over calcium chloride before use. 1,3-Butadiene (Bd: Fluka; purity 99.5%) was dried over zeolite 4A before use. Cyclohexane (Wako Pure Chemical Industries; purity >99.5%) and toluene (Wako Pure Chemical Industries; purity >99.5%) were distilled from calcium hydride. THF (tetrahydrofuran; Wako Pure Chemical Industries; purity >99.5%), sec-butyllithium (Kanto Chemical; 1.0 mol L−1 in n-hexane), methanol (Wako Pure Chemical Industries; purity >99.5%), sodium carbonate (Wako Pure Chemical Industries; purity >99.5%), trifluoromethanesulfonic acid (Wako Pure Chemical Industries; purity >98%), chlorosulfonic acid (Wako Pure Chemical Industries; purity >97.0%), sulfuric acid (Wako Pure Chemical Industries; purity >95%), trifluoroacetic acid (Wako Pure Chemical Industries; purity >99.0%), trichloroacetic acid (Wako Pure Chemical Industries; purity >99.0%), and methanesulfonic acid (Wako Pure Chemical Industries; purity >98.0%) were used as received. Aluminium chloride, anhydrous (Aldrich; purity 99.999%), titanium chloride (Kanto Chemical; purity >99.0%), iron(III) chloride, anhydrous (Kanto Chemical; purity >96%), tin(IV) chloride, anhydrous (Kanto Chemical; purity >98.0%), zinc chloride (Kanto Chemical; purity >98.0%), boron trifluoride diethyl ether complex (Wako Pure Chemical Industries), boron trifluoride phenol complex (Wako Pure Chemical Industries; BF3 about 30%), boron trifluoride acetic acid complex (Kanto Chemical; BF3 about 33%), and benzyl chloride (Wako Pure Chemical Industries; purity >99.0%) were used as received.Polymerization procedures
The polymerizations were carried out by the syringe technique under dry nitrogen in baked glass tubes equipped with a three-way stopcock. A typical example for St/Ip random copolymerization is given below. The copolymerization was initiated by adding solutions of premixed St/Ip mixture (St: 1.2 mol, 138 mL; Ip: 1.2 mol, 120 mL) at 3.3 mL min−1 by a syringe pump into a sec-butyllithium solution containing cyclohexane (840 mL), THF (6.0 mmol, 0.49 mL), and sec-butyllithium (2.0 mmol) at 40 °C. After adding all the St/Ip mixture, the polymerization mixture was kept stirred for 1 h at 40 °C. The polymerization was terminated with methanol (1 mL). The quenched solutions were washed with 300 mL of water three times to remove the initiator residues, dropped into methanol and acetone mixture (600/400 mL), decanted off any liquid, and vacuum dried to give the product styrene–isoprenerandom copolymers (r-SIR: random styrene–isoprene rubber) [yield = 93% (192 g), Mn = 119![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400, Mw/Mn = 1.03, St content = 48 mol%, Ip 3,4-unit content = 31 mol%]. The St and Ip 3,4-unit contents were determined by 1H NMR spectroscopy. St/Bd and St/Ip/Bdrandom copolymerizations were carried out similarly under dry nitrogen in an autoclave hastelloy reactor.
400, Mw/Mn = 1.03, St content = 48 mol%, Ip 3,4-unit content = 31 mol%]. The St and Ip 3,4-unit contents were determined by 1H NMR spectroscopy. St/Bd and St/Ip/Bdrandom copolymerizations were carried out similarly under dry nitrogen in an autoclave hastelloy reactor.
Cyclization procedures of r-SIR, r-SBR, r-SIBR, and r-pMSIR
The cyclizations were carried out by the syringe technique under dry nitrogen in baked glass tubes equipped with a three-way stopcock. A typical example for r-SIRcyclization is given below. The cyclization was initiated by adding CF3SO3H (4.4 mmol, 0.39 mL) into an r-SIR solution containing r-SIR (60 g) and cyclohexane (2.34 L) at 23 °C. After a predetermined period (60 min), the cyclization was terminated with 1% aqueous solution of Na2CO3 (500 mL). The quenched solutions were washed with 500 mL of water three times to remove CF3SO3H and Na2CO3 residues, dropped into methanol and acetone mixture (3/2 L), filtered and vacuum dried to give the product [yield = 97% (58 g), Mn = 76500, Mw/Mn = 1.55]. Several metal catalysts (AlCl3, ZnCl2, and FeCl3) were used as solids, which were not completely soluble in the reaction solution.
Measurements
The 1H NMR spectra were recorded on a JEOL JNM-LA400 (400 MHz). The number-average molecular weight (Mn) and polydispersity index (Mw/Mn) were determined by size-exclusion chromatography (SEC) in THF at 40 °C on three polystyrene gel columns [TSKgel GMH-M (7.8 mm i.d. × 30 cm) × 2 and TSKgel G2000H (7.8 mm i.d. × 30 cm); flow rate 1.0 mL min−1] connected to a Tosoh HLC-8020 system. The columns were calibrated against 9 standard polystyrene samples (GL Sciences; Mp = 580–600000; Mw/Mn = 1.02–1.12). Glass transition temperature (Tg) of the polymer was recorded on a DSC30differential scanning calorimeter (Mettler Toledo). Samples were first heated to 200 °C at 10 °C min−1, and cooled to −80 °C at 10 °C min−1 and then reheated to 200 °C at 10 °C min−1. All Tg values were obtained from the second scan, after removing the thermal history. Flexural properties were examined using an Autograph AG-5000B (Shimazu Co., Japan) compliant with the standard method for testing flexural properties of rigid plastics (ASTM D790). Impact strength was examined using a digital impact tester (Toyo Seiki Seisaku-sho, Ltd., Japan) compliant with the standard method for testing impact strength of rigid plastics (ASTM D256).
Results and discussion
Cationic cyclization of r-SIR with various catalysts
As previously reported,10,11 we prepared r-SIRvia living anionic polymerization using sec-butyllithium in cyclohexane as the initiator. The premixed styrene and isoprene monomers were incrementally and continuously added to the reaction mixture throughout the reaction, and a small amount of THF was employed as the randomizer to render the monomer sequence random and less blocky.11 Irrespective of the monomer-feed ratios, the molecular weights of the obtained copolymers were well-controlled and reached a relatively high value (Mn ≈ 105) with narrow MWDs (Mw/Mn ≤ 1.1; Table S1 in the ESI†). An 1H NMR analysis showed that the comonomer composition ratios of the obtained copolymers agreed well with the ratio in the initial feed ratio [styrene content (Fst) = 37–48%] and that the regiospecificities of the isoprene units were predominantly 1,4-rich (1,4-: 1,2-: 3,4- = 67–77: ∼1: 22–36), which agrees with the reported values.11The cationic cyclizations of the obtained r-SIR were then examined in cyclohexane or toluene at several temperatures with moderate polymer concentrations, using various Brönsted acids whose Hammett acidity functions (H0) were −14.1 (CF3SO3H), −13.8 (ClSO3H), −12 (H2SO4), −7.86 (CH3SO3H), −2.7 (CF3COOH), and less than −2.7 (CCl3COOH) (Table 1).12 Consistent with previous work, trifluoromethanesulfonic acid (CF3SO3H) efficiently induced the reaction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond to afford the cyclized products, which were completely soluble in THF without gelation, indicating that the Friedel–Crafts reaction predominately occurred between the CC double bond of the isoprene unit and the phenyl group of the adjacent styrene units (entries 1–8 in Table 1). The consumption of the olefinic protons was evaluated based on a decrease in the peak intensity ratios of the CH protons compared to all of the polymer peaks for r-SIR. The high CF3SO3H concentration or reaction temperature resulted in more CC double bonds being consumed. Under these conditions, the SEC curves exhibited broader MWDs with small peaks in the higher-molecular-weight regions, indicating that a small amount of product was formed by the intermolecular linking reaction. In addition, weaker Brönsted acids were inactive or less active in cyclohexane, even at higher concentrations. However, when the reaction was performed in toluene, which has a dielectric constant that is slightly higher than that of cyclohexane, ClSO3H and H2SO4 induced cyclization, but with less CC bond consumption. Although toluene might participate in the Friedel–Crafts reaction, almost no effects were observed on the 1H NMR spectra as well as the properties of the products most probably due to the predominant intramolecular cyclization. These results indicate that a high acidity (H0 < −12) is necessary for the efficient cationic cyclization of r-SIR, and that superacids with H0 < −14.1 are effective even in non-polar solvents, such as cyclohexane.
C double bond to afford the cyclized products, which were completely soluble in THF without gelation, indicating that the Friedel–Crafts reaction predominately occurred between the CC double bond of the isoprene unit and the phenyl group of the adjacent styrene units (entries 1–8 in Table 1). The consumption of the olefinic protons was evaluated based on a decrease in the peak intensity ratios of the CH protons compared to all of the polymer peaks for r-SIR. The high CF3SO3H concentration or reaction temperature resulted in more CC double bonds being consumed. Under these conditions, the SEC curves exhibited broader MWDs with small peaks in the higher-molecular-weight regions, indicating that a small amount of product was formed by the intermolecular linking reaction. In addition, weaker Brönsted acids were inactive or less active in cyclohexane, even at higher concentrations. However, when the reaction was performed in toluene, which has a dielectric constant that is slightly higher than that of cyclohexane, ClSO3H and H2SO4 induced cyclization, but with less CC bond consumption. Although toluene might participate in the Friedel–Crafts reaction, almost no effects were observed on the 1H NMR spectra as well as the properties of the products most probably due to the predominant intramolecular cyclization. These results indicate that a high acidity (H0 < −12) is necessary for the efficient cationic cyclization of r-SIR, and that superacids with H0 < −14.1 are effective even in non-polar solvents, such as cyclohexane.
| Entry | Catalyst | [Catalyst]0/mM | Solv.b | Temp./°C | CC consumptionc (%) |
M n | M w/Mnd | Intermolecular-linkinge (%) | T g /°C |
|---|---|---|---|---|---|---|---|---|---|
|
a The cyclization was carried out for 1 h (except for entry 4: reaction time = 0.5 h): [r-SIR]0 = 3.2 wt%. The r-SIRcopolymers were prepared by anionic living copolymerization; r-SIR-1 (Fst = 48%, Mn = 119400, Mw/Mn = 1.03, Tg = 16 °C) for entries 1, 2, and 5, r-SIR-2 (Fst = 48%, Mn = 112400, Mw/Mn = 1.04, Tg = 20 °C) for entries 3, 4, and 9–14, and r-SIR-3 (Fst = 37%, Mn = 119200, Mw/Mn = 1.10, Tg = −1 °C) for entries 6–8.
b
CHx and Tol represent cyclohexane and toluene, respectively.
c Determined from the CH ratio in the 1H NMR spectra.
d The number-average molecular weight (Mn) and distribution (Mw/Mn) were determined by size-exclusion chromatography (SEC) against PSt standards.
e Determined by the peak intensity ratio of the higher molecular weight region in the SEC curves.
f The glass transition temperature (Tg) was determined by differential scanning calorimetry.
|
|||||||||
| 1 | CF3SO3H | 0.4 | CHx | 23 | 38 | 104700 |
1.10 | 2 | 47 |
| 2 | CF3SO3H | 1.9 | CHx | 23 | 91 | 66300 |
1.60 | 19 | 130 |
| 3 | CF3SO3H | 1.9 | CHx | 23 | 92 | 76500 |
1.55 | 19 | 129 |
| 4 | CF3SO3H | 1.9 | Tol | 23 | 90 | 79700 |
1.29 | 15 | 129 |
| 5 | CF3SO3H | 19 | CHx | 23 | 99 | 25300 |
2.18 | >20 | 152 |
| 6 | CF3SO3H | 0.4 | CHx | 23 | 72 | 82500 |
1.32 | 7 | 81 |
| 7 | CF3SO3H | 0.4 | CHx | 40 | 80 | 79200 |
1.40 | 10 | 102 |
| 8 | CF3SO3H | 0.4 | CHx | 70 | 87 | 63900 |
1.76 | 12 | 120 |
| 9 | ClSO3H | 8.3 | Tol | 40 | 10 | 36300 |
2.20 | 3 | 42 |
| 10 | ClSO3H | 8.3 | CHx | 40 | ∼0 | 112400 |
1.04 | — | 20 |
| 11 | H2SO4 | 8.3 | Tol | 40 | 19 | 55000 |
1.66 | 11 | 43 |
| 12 | CH3SO3H | 8.3 | Tol | 40 | ∼0 | 112400 |
1.04 | — | 20 |
| 13 | CF3COOH | 8.3 | Tol | 40 | ∼0 | 112400 |
1.04 | — | 20 |
| 14 | CCl3COOH | 8.3 | Tol | 40 | ∼0 | 112400 |
1.04 | — | 20 |
The thermal properties of the obtained products were also evaluated by DSC, which showed that all of the cyclized products exhibited a single glass transition peak, suggesting the formation of random copolymers. As the consumption of the CC bonds increased, the Tg's of the copolymers increased from the ambient temperature (16–20 °C) to above 100 °C (Table 1). The highest concentration of CF3SO3H led to the highest Tg for the cyclized r-SIR at 152 °C (entry 5), which is predominantly due to the intramolecular cyclization between the CC bond in the isoprene units and adjacent the aromatic ring of the styrene units, resulting in the formation of a bicyclic tetrahydronaphthyl unit in the backbone chain.
In general, Lewis acids are also used for both Friedel–Crafts reactions and cationic polymerizations.9,13 A series of Lewis acids, including AlCl3, TiCl4, FeCl3, SnCl4, ZnCl2, and BF3 complexes, was also used as the catalyst for the cyclization of r-SIR (Table 2). However, most of the Lewis acid catalysts were not effective in cyclohexane, due to the non-polar solvent and the low protogen content of anhydrous cyclohexane, which generally contains approximately 5 ppm of water (entries 1–6). In contrast, BF3 complexes with protic molecules, such as phenol (BF3·2PhOH), induced CC bond consumption, yielding cyclized r-SIR with a relatively high Tgvia Friedel–Crafts alkylation (entry 7 and 8). In contrast to BF3·2PhOH, which was effective in cyclohexane, the acetic acid complex (BF3·2CH3COOH) was ineffective for the cyclization of r-SIR in cyclohexane but was effective in toluene. In addition, when coupled to benzyl chloride (BzCl), cyclization occurred more effectively with the boron complexes (entries 9, 12, 13, and 14) because BzCl acts as a cationogen in the presence of Lewis acids to generate the benzyl cation.6 In the BF3/BzCl system, the cyclization of r-SIR proceeded well and yielded cyclized r-SIR with less content from intermolecular-linking reactions and a relatively narrow MWD, even under high CC bond conversion conditions. This result is due to the residual carbocationic species in the protonated isoprene units being capped with the chloride anions derived from BzCl to suppress the intermolecular reaction.
| Entry | Catalyst | [Catalyst]0/mM | Solv.b | Temp./°C | CC consumptionc (%) |
M n | M w/Mnd | Intermolecular-linkinge (%) | T g /°C |
|---|---|---|---|---|---|---|---|---|---|
|
a The cyclization was carried out for 1 h: [r-SIR]0 = 3.2 wt%. The r-SIR copolymers were prepared by anionic living copolymerization; r-SIR-2 (Fst = 48%, Mn = 112400, Mw/Mn = 1.04, Tg = 20 °C) for entries 4–8, 10, and 12–14, and r-SIR-4 (Fst = 47%, Mn = 128400, Mw/Mn = 1.05, Tg = 19 °C) for entries 1–3, 9, and 11.
b
CHx and Tol represent cyclohexane and toluene, respectively.
c Determined from the CH ratio in the 1H NMR spectra.
d The number-average molecular weight (Mn) and distribution (Mw/Mn) were determined by size-exclusion chromatography against PSt standards.
e Determined by the peak area ratio of the higher molecular weight region in the SEC curves.
f The glass transition temperature (Tg) was determined by differential scanning calorimetry.
g [BF3 complex]0/[benzyl chloride (BzCl)]0 = 3/1.
|
|||||||||
| 1 | AlCl3 | 1.9 | CHx | 23 | ∼0 | 128400 |
1.05 | — | 19 |
| 2 | TiCl4 | 1.9 | CHx | 23 | ∼0 | 128400 |
1.05 | — | 19 |
| 3 | FeCl3 | 1.9 | CHx | 23 | ∼0 | 128400 |
1.05 | — | 19 |
| 4 | SnCl4 | 12 | CHx | 50 | ∼0 | 112400 |
1.04 | — | 20 |
| 5 | ZnCl2 | 10 | CHx | 50 | ∼0 | 112400 |
1.04 | — | 20 |
| 6 | BF3·Et2O | 22 | CHx | 50 | ∼0 | 112400 |
1.04 | — | 20 |
| 7 | BF3·2PhOH | 3.3 | CHx | 50 | 41 | 98000 |
1.11 | 7 | 58 |
| 8 | BF3·2PhOH | 7.5 | CHx | 50 | 74 | 82100 |
1.30 | 16 | 105 |
| 9 | BF3·2PhOH/BzClg | 7.5 | CHx | 50 | 83 | 94500 |
1.33 | 10 | 112 |
| 10 | BF3·2AcOH | 7.5 | CHx | 40 | ∼0 | 112400 |
1.04 | — | 20 |
| 11 | BF3·2AcOH | 6.2 | Tol | 40 | 71 | 85700 |
1.33 | 7 | 83 |
| 12 | BF3·2AcOH/BzClg | 7.5 | CHx | 40 | ∼0 | 112400 |
1.04 | — | 20 |
| 13 | BF3·2AcOH/BzClg | 6.2 | Tol | 40 | 79 | 86900 |
1.14 | 8 | 117 |
| 14 | BF3·2AcOH/BzClg | 12.5 | Tol | 40 | 81 | 78600 |
1.26 | 10 | 120 |
Fig. 1 shows the Tg values of the cyclized copolymers obtained with various catalysts as a function of the consumption of the CC double bonds in the r-SIR. In all of the cases, the Tg of the copolymers gradually increased from 20 °C to 150 °C as the CC double bonds were consumed. In the BF3/BzCl system, the potential production of cyclized r-SIR containing benzyl units may affect the thermal properties of the resulting polymers. In the BF3/BzCl system, the cyclization was triggered by generating benzyl cation to result in the cyclized r-SIR contained benzyl units, which was also confirmed by 1H NMR (Fig. S1 in the ESI†). However, there are no significant differences in Tg values of the resulting polymers, suggesting that the initial loading of BzCl (1 wt % with respect to r-SIR) was too small to affect these properties. Thus, the intramolecular cyclization of r-SIR was efficiently catalyzed by both superacidic Brönsted acids and Lewis acids to yield soluble cycloolefincopolymer analogues with high glass transition temperatures (Tg's) containing bicyclic tetrahydronaphthyl units in the backbone chain.
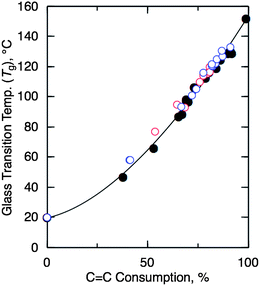 | ||
| Fig. 1 Glass transition temperature (Tg) of cyclized r-SIR obtained with various acidic catalysts as the function of CC bond consumption in the original r-SIRcopolymers as in the same experiments in Tables 1 and 2. Catalyst: CF3SO3H (filled circles), BF3·2PhOH (blue circles), and BF3·2CH3COOH/BzCl (red circles). | ||
Cationic cyclization of various r-SIR analogues
To clarify the effects of the molecular structure on its reactivity during cationic cyclization and the properties of the cyclized products, we prepared a series of random styrene–diene copolymers (including r-SIR) with various microstructures in the isoprene units and copolymers that contained other styrene and diene monomer units, including 1,3-butadiene and p-methylstyrene, as summarized in Table 3.| Entry | Prepolymer (before cyclization)a | After cyclizationb | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Copolymers | F st (mol%) | F Ip (mol%) | F Bd (mol%) | M n | M w/Mnd | Microstructure in dienec (%) | CC consump.c (%) |
M n | M w/Mnd | Intermol. linkingd (%) | T g /°C | |||
| 1,4 | 1,2 | 3,4 | ||||||||||||
|
a Anionic living copolymerization was performed with sec-butyllithium in the presence of THF in cyclohexane at 40 °C; see also the Experimental section.
b
Cyclization was carried out with CF3SO3H in cyclohexane for 1 h at 23 °C: [prepolymer]0 = 3.2 wt%, [CF3SO3H]0 = 1.9 mM.
c Determined from the CH ratio in the 1H NMR spectra.
d The number-average molecular weight (Mn) and distribution (Mw/Mn) were determined by size-exclusion chromatography against PSt standards.
e The glass transition temperature (Tg) was determined by differential scanning calorimetry.
f
CF3SO3H was used as a CH2Cl2 solution to avoid gelation.
g
[CF3SO3H]0 = 19 mM.
h Calculated from the initial feed ratio of monomers.
|
||||||||||||||
| 1 | r-SIR50-1 | 48 | 52 | — | 112400 |
1.04 | 67 | 1 | 32 | 92 | 76500 |
1.55 | 19 | 129 |
| 2 | r-SIR50-2 | 48 | 52 | — | 111100 |
1.05 | 86 | 1 | 13 | 81 | 84000 |
1.68 | 30 | 113 |
| 3 | r-SIR50-3 | 48 | 52 | — | 131400 |
1.04 | 46 | 6 | 48 | 77 | 102100 |
1.34 | 19 | 106 |
| 4 | r-SBR50 | 48 | — | 52 | 113400 |
1.04 | 76 | 24 | — | 14f | 111900 |
1.11 | 7 | 23 |
| 5 | r-SBR50 | 48 | — | 52 | 113400 |
1.04 | 76 | 24 | — | 100f,g | 71800 |
2.22 | 41 | 134 |
| 6 | r-SBR70 | 69 | — | 31 | 117500 |
1.03 | 90 | 10 | — | 100f,g | 98500 |
1.24 | 15 | 113 |
| 7 | r-SIBR50 | 46h | 42h | 12h | 145400 |
1.04 | n.d. | n.d. | n.d. | 54 | 110100 |
1.43 | 22 | 91 |
| 8 | r-pMSIR50 | 52 | 48 | — | 107100 |
1.05 | 68 | 2 | 30 | 91 | 55500 |
1.52 | 12 | 130 |
The randomness of the sequence of isoprene and styrene and the microstructures of the isoprene units were controlled by changing the loading concentration of THF during anionic copolymerization (entries 1–3). The obtained r-SIRcopolymers exhibited various microstructures with 1,4-regiospecificities in the isoprene units that decreased with increasing THF concentrations (1,4-: 1,2-: 3,4- = 46–86: 1–6: 13–48). In all of the cases, the obtained copolymers had relatively high and controlled molecular weights (Mn > 105) and narrow MWDs (Mw/Mn < 1.1), irrespective of the differences in the microstructures. Furthermore, the cyclization of the obtained r-SIR copolymers proceeded well with CF3SO3H as the catalyst to afford the cyclized copolymers with relatively high Tg independent of the microstructure. This result is most likely due to the adjacent units of isoprene and styrene producing a bicyclic structure, irrespective of the regiostructure of the isoprene unit, resulting in products with high Tg's, as shown in Scheme 1 and Fig. S2 in the ESI†.
Using a similar procedure, r-SBR, r-SIBR, and r-pMSIR were also prepared via living anionic polymerization (entries 4–8). In all of the cases, copolymerizationvia the incremental and continuous addition of the monomer solutions in the presence of THF afforded the copolymers with well-controlled molecular weights and MWDs, although the microstructure of the r-SIBR terpolymers could not be determined due to the complexity of the 1H NMR spectra. All of the obtained copolymers exhibited relatively low Tg's ranging from −10 to 20 °C, which are consistent with the values reported in the literature. Next, the cyclization of the obtained copolymers was examined using CF3SO3H in cyclohexane at an ambient temperature. The reactions of r-SBR and r-SIBR proceeded more slowly than that of r-SIR under the same conditions (Table 3, entries 1 and 4), whereas r-pMSIR exhibited almost the same reactivity as r-SIR (entry 8). Furthermore, r-SBR exhibited a greater tendency to undergo intermolecular linking-induced gelation compared to r-SIR, and a dilute solution of CF3SO3H in CH2Cl2 was required to avoid gelation. Consequently, the reaction of r-SBR yielded a cyclized copolymervia a CH2Cl2 solution with a higher CF3SO3H content (entry 5). These results indicate that the protonation of the diene units plays a pivotal role in cationic cyclization. The CC double bonds in the isoprene units, which generate tertiary carbocationic species, induce effective cyclizationvia an intramolecular Friedel–Crafts reaction, while the CC double bonds in the 1,3-butadiene units, which generate more reactive secondary cations, are less reactive but can induce intermolecular linking reactions with lower selectivity under relatively rigorous conditions. Similar efficient intramolecular cyclizations of low molecular weight compounds via tertiary carbocationic species were also reported in Friedel–Crafts chemistry.14 The methyl substituent in the isoprene unit would also contribute to more effective cyclization due to the formation of sterically favored 6-endo-trig-like conformation.
Fig. 2 shows the SEC curves of the copolymers of r-SIR, r-SBR, and r-pMSIR, which have comonomer compositions of approximately 1:1 (styrene:isoprene, styrene:1,3-butadiene, and p-methylstyrene:isoprene = 48:52, 48:52, and 52:48, respectively). Fig. 2 also shows their cyclized products that are formed using CF3SO3H. In all of the cases, the main peaks shifted toward lower molecular weights after the cyclization reactions due to the reduction in the hydrodynamic volume that occurs upon cyclization. This result was confirmed by the fact that the absolute molecular weight of the main peak, which is obtained by light scattering, did not change during cyclization in our previous work.7 In addition, the small peaks in the higher-molecular-weight region resulted from some intermolecular linking and/or cross-linking reactions, which were observed for r-SIR and r-SBR. In particular, the SEC curves for the cyclization of r-SBR exhibited a higher ratio for the intermolecular linking reaction compared to the cyclization of r-SIR. In addition, r-pMSIR underwent less intermolecular linking because the intermolecular Friedel–Crafts alkylation between the different polymer chains barely occurred on the phenyl group of the p-substituted styrene unit. Although the intermolecular cross-linking reaction might also increase the Tg of the products, such effects were not observed in our case,7 at least for the soluble products with low cross-linking contents. The peaks in the lower-molecular-weight region were most likely caused by long-range intramolecular Friedel–Crafts reaction to form macrocyclic chain and/or cleavage of the main chain via β-scission between the adjacent styrene and diene units to form a styryl cation and a terminal CC double bond (see also Fig. S2 in the ESI†).
![Size-exclusion chromatograms for intramolecular Friedel–Crafts cationic cyclization of (A) r-SIR (entry 1 in Table 3), (B) r-SBR (entry 5 in Table 3), and r-pMSIR (entry 8 in Table 3) with CF3SO3H in cyclohexane at 23 °C: [copolymer]0 = 3.2 wt%, [CF3SO3H]0 = 1.9 (for r-SIR and r-pMSIR) or 19 mM (for r-SBR).](/image/article/2012/PY/c1py00433f/c1py00433f-f2.gif) | ||
| Fig. 2 Size-exclusion chromatograms for intramolecular Friedel–Crafts cationic cyclization of (A) r-SIR (entry 1 in Table 3), (B) r-SBR (entry 5 in Table 3), and r-pMSIR (entry 8 in Table 3) with CF3SO3H in cyclohexane at 23 °C: [copolymer]0 = 3.2 wt%, [CF3SO3H]0 = 1.9 (for r-SIR and r-pMSIR) or 19 mM (for r-SBR). | ||
Fig. 3 shows the 1H NMR spectra of the various random copolymers and their cyclized products from the post-polymerization reactions. Before the cyclization of the copolymers, their spectra showed the typical signals for the random styrene–isoprene, styrene–1,3-butadiene, and p-methylstyrene–isoprene copolymers, in which the signals at 3.8–5.7 ppm were assigned to the olefinic protons of the isoprene and 1,3-butadiene units. After the cyclization reactions, the double-bond signals dramatically decreased, and the peaks of the aromatic protons at 6.0–7.5 ppm became broader in all of the cases. These results indicate that cyclization most likely proceeded between the CC double bond of the diene unit and the phenyl group of the adjacent styrene unit via the Friedel–Crafts reaction and independent of the monomer structures.
![1H NMR spectra (CDCl3, 25 °C) of r-SIR (entry 1 in Table 3) before (A) and after cyclization (B), r-SBR (entry 5 in Table 3) before (C) and after cyclization (D), and r-pMSIR (entry 8 in Table 3) before (E) and after cyclization (F) using CF3SO3H in cyclohexane at 23 °C: [copolymer]0 = 3.2 wt%, [CF3SO3H]0 = 1.9 (for r-SIR and r-pMSIR) or 19 mM (for r-SBR).](/image/article/2012/PY/c1py00433f/c1py00433f-f3.gif) | ||
| Fig. 3 1H NMR spectra (CDCl3, 25 °C) of r-SIR (entry 1 in Table 3) before (A) and after cyclization (B), r-SBR (entry 5 in Table 3) before (C) and after cyclization (D), and r-pMSIR (entry 8 in Table 3) before (E) and after cyclization (F) using CF3SO3H in cyclohexane at 23 °C: [copolymer]0 = 3.2 wt%, [CF3SO3H]0 = 1.9 (for r-SIR and r-pMSIR) or 19 mM (for r-SBR). | ||
Thermal and mechanical properties of various cyclized copolymers
The thermal and mechanical properties of the cyclized copolymers obtained from the various random copolymers were evaluated. Fig. 4 shows the Tg values of the cyclized copolymers as a function of the consumption of the CC double bonds during cyclization. In all of the cases, the Tg's of the cyclized copolymers increased as the reaction proceeded. They reached higher values (Tg > 130 °C) than the values (Tg ≈ 100 °C) obtained for the corresponding homopolymers of styrene or the cyclized homopolymers of dienes. For the r-SIRcopolymers, all of the Tg values were plotted on a single line despite the differences in the original microstructures. This result indicates that similar bicyclic structures can be generated from the various regiostructures of the isoprene units via intramolecular cyclization, as shown in Scheme 1. In addition, the Tg's of the cyclized r-pMSIR were similar to those of the cyclized r-SIR, whereas those of the cyclized r-SBR exhibited slightly lower values. These results are consistent with the original Tg values of the corresponding homopolymers, as follows: poly(p-methylstyrene) (Tg = 109 °C), polystyrene (Tg = 105 °C), polyisoprene (Tg = −65 °C), and poly(1,3-butadiene) (Tg = −90 °C).15,16
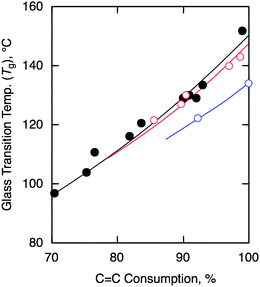 | ||
| Fig. 4 Glass transition temperature (Tg) of cyclized r-SIR (filled circles), r-SBR (blue circles), and r-pMSIR (red circles) obtained with CF3SO3H as CC bond consumption in the original copolymers. | ||
The mechanical properties of the obtained cyclized copolymers were also evaluated via the flexural testing of the hot-press-molded samples (Table 4). Both the cyclized r-SIR and the cyclized r-SBR exhibited relatively high flexural moduli and greater strengths compared to the polystyrenehomopolymer. In particular, the cyclized r-SBR exhibited superior impact strength as determined by the IZOD impact test, which is most likely due to the flexible butadiene units. Thus, the mechanical properties of the cyclized copolymers were comparable to those of the polystyrenehomopolymer, but the cyclized copolymers exhibited superior thermal properties.
| Polymer | M n | M w/Mnd | CC consumptione (%) |
T g /°C | Flexural modulusg/MPa | Flexural strengthg/MPa | IZOD impact strength (unnotched)h/kJ m−2 |
|---|---|---|---|---|---|---|---|
|
a Cyclized r-SIR (entry 2 in Table 1).
b Cyclized r-SBR (entry 5 in Table 3).
c Prepared by free radical polymerization of styrene.
d The number-average molecular weight (Mn) and distribution (Mw/Mn) were determined by size-exclusion chromatography against PSt standards.
e Determined from the CH ratio in the 1H NMR spectra.
f The glass transition temperature (Tg) was determined by differential scanning calorimetry.
g According to ASTM D790.
h According to ASTM D256.
|
|||||||
| Cyclized r-SIRa | 66300 |
1.60 | 91 | 130 | 3200 | 89 | 9 |
| Cyclized r-SBRb | 71800 |
2.22 | 100 | 134 | 2900 | 100 | 14 |
| Polystyrene c | 83800 |
2.23 | — | 100 | 3400 | 72 | 10 |
Conclusions
Cationic cyclizationvia intramolecular Friedel–Crafts alkylation to form a bicyclic structure was performed on the random copolymers of various styrene and diene derivatives. These cyclizations were catalyzed by either strong Brönsted acids or Lewis acids with cationic sources. In particular, CF3SO3H and BF3·2AcOH/benzyl chloride proved to be efficient catalytic systems with respect to their activities and controllabilities. Similar to the styrene–isoprenecopolymer, the random styrene–butadiene and 4-methylstyrene–isoprene copolymers also underwent cationic cyclization with CF3SO3H to produce polymers with relatively high glass-transition temperatures. The cyclized copolymers exhibited high Tg's, relatively robust mechanical properties, and a robust impact strength. These results suggest that cyclized copolymers based on styrene and dienes are promising candidates to use as thermoplastics with relatively high heat resistances.Acknowledgements
We thank Mr Masatsuka Shirakawa and Dr Toshiya Uozumi for technical support and useful suggestions. This work was supported in part by the Global COE Program “Elucidation and Design of Materials and Molecular Functions”.References
- G. B. Butler, Cyclopolymerization and Cyclocopolymerization, M. Dekker, Inc., New York, 1992 Search PubMed.
- (a) G. J. Van Veersen, J. Polym. Sci., 1951, 6, 29–32 CrossRef CAS; (b) D. J. Patterson and J. L. Koenig, Makromol. Chem., 1987, 188, 2325–2337 CrossRef CAS; (c) S. Riyajan, D.-J. Liaw, Y. Tanaka and J. T. Sakdapipanich, J. Appl. Polym. Sci., 2007, 105, 664–672 CrossRef CAS.
- (a) M. A. Golub and J. Heller, Can. J. Chem., 1963, 41, 937–953 CrossRef CAS; (b) R. K. Agnihotri, D. Falcon and E. C. Fredericks, J. Polym. Sci., Part A-1: Polym. Chem., 1972, 10, 1839–1850 CrossRef CAS; (c) G. Kaszas, J. E. Puskas and J. P. Kennedy, J. Appl. Polym. Sci., 1990, 39, 119–144 CrossRef CAS; (d) J. Lal, Polymer, 1998, 39, 6183–6186 CrossRef CAS.
- (a) Y. Tanaka, H. Sato and I. G. Gonzalez, J. Polym. Sci., Polym. Chem. Ed., 1979, 17, 3027–3029 CrossRef CAS; (b) A. Priola, M. Bruzzone, F. Mistrali and S. Cesca, Angew. Makromol. Chem., 1980, 18, 21–35 CrossRef; (c) E. A. Abdel-Razik, Polymer, 1988, 29, 1704–1708 CrossRef CAS; (d) C. Wang, Mater. Chem. Phys., 2005, 89, 116–121 CrossRef CAS.
- Y. Shangxian, L. Lin, G. Jiangnan, S. Cuihua and W. Jiliang, J. Photopolym. Sci. Technol., 1993, 6, 7–14 CrossRef.
- C. Wang, X. Huang and J. Yang, Eur. Polym. J., 2001, 37, 1895–1899 CrossRef CAS.
- A. Nakahara, K. Satoh and M. Kamigaito, Macromolecules, 2009, 42, 620–625 CrossRef CAS.
- (a) G. A. Olah, Friedel–Crafts and Related Reactions, Interscience publishers a division of John Wiley & Sons Inc., New York-London-Sydney, 1964 Search PubMed; (b) D. P. N. Satchell and R. S. Satchell, Chem. Rev., 1969, 69, 251–278 CrossRef CAS.
- (a) J. P. Kennedy, Cationic Polymerization of Olefins: a Critical Inventory, Wiley-Interscience, New York, 1975 Search PubMed; (b) Cationic Polymerizations, ed. K. Matyjaszewski, Marcel Dekker, New York, 1996 Search PubMed.
- (a) A. F. Johnson and D. J. Worsfold, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 449–455 CrossRef CAS; (b) H. L. Hsieh, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 153–161 CrossRef CAS; (c) H. L. Hsieh, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 163–172 CrossRef CAS.
- (a) D. J. Worsfold and S. Bywater, Can. J. Chem., 1964, 2, 2884–2892 CrossRef; (b) M. Morton, N. Calderon, L. J. Fetters and J. F. Meier, Polym. Eng. Sci., 1962, 2, 106–107 CrossRef; (c) M. Morton, Anionic Polymerization, Principles and Practice, Academic Press, Inc., New York, 1983 Search PubMed; (d) H. L. Hsieh and R. P. Quirk, Anionic Polymerization: Principles and Practical Applications (Plastics Engineering Series), M. Dekker, Inc., New York, 1996 Search PubMed.
- (a) G. A. Olah, G. K. S. Prakash and J. Sommer, Superacids, John Wiley & Sons Inc., New York, 1985 Search PubMed; (b) R. A. Cox and K. Yates, Can. J. Chem., 1983, 61, 2225–2243 CrossRef CAS; (c) B. L. Booth and T. A. El-Fekky, J. Chem. Soc., Perkin Trans. 1, 1979, 2441–2446 RSC; (d) S. Saito, S. Saito, T. Ohwada and K. Shudo, Chem. Pharm. Bull., 1991, 39, 2718–2720 CAS.
- H. Imai, T. Saegusa and J. Furukawa, Makromol. Chem., 1965, 81, 92–99 CrossRef CAS.
- A. A. Khalaf and R. M. Roberts, J. Org. Chem., 1969, 34, 3571–3574 CrossRef CAS.
- (a) J. M. G. Cowie and I. J. McEwen, Macromolecules, 1977, 10, 1124–1128 CrossRef CAS; (b) J. M. G. Cowie, Eur. Polym. J., 1975, 11, 297–300 CrossRef CAS; (c) R. F. Boyer, Macromolecules, 1992, 25, 5326–5330 CrossRef CAS.
- (a) A. Brunacci, J. M. G. Cowie, R. Ferguson, J. L. Gómez Ribelles and A. Vidaurre Garayo, Macromolecules, 1996, 29, 7976–7988 CrossRef CAS; (b) Van Krevelen, Properties of Polymers, 3rd edn, Elsevier Science Publishers, 1990, p. 144 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00433f |
| This journal is © The Royal Society of Chemistry 2012 |