Protein repellent hydrophilic grafts prepared by surface-initiated atom transfer radical polymerization from polypropylene
Charlotte Juel
Fristrup
a,
Katja
Jankova
a,
Rüya
Eskimergen
b,
Jens T.
Bukrinsky
b and
Søren
Hvilsted
*a
aDanish Polymer Centre, Department of Chemical and Biochemical Engineering, Technical University of Denmark, Building 227, DK-2800, Kgs. Lyngby, Denmark. E-mail: sh@kt.dtu.dk; Fax: +45 4588 2161; Tel: +45 4525 2965
bNovo Nordisk A/S, Novo Allé, DK-2880, Bagsværd, Denmark
First published on 14th November 2011
Abstract
Grafting of poly(ethylene glycol)methacrylate (PEGMA) and N,N-dimethylacrylamide (DMAAm) from UV-initiator modified polypropylene (PP) was performed by Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). The modification and hydrophilization of the PP substrates were confirmed with Attenuated Total Reflectance (ATR) Fourier Transform Infrared (FTIR) spectroscopy and Water Contact Angle (WCA) measurements. Confocal fluorescence microscopy of modified and unmodified substrates immersed in labelled insulin aspart showed superior repulsion of this protein for the poly(PEGMA) grafts, due to the achieved architecture.
Introduction
Non-specific fouling needs to be suppressed at interfaces between solid substrates and liquids for many applications within the biomedical field, e.g.pharmaceutical packaging, drug delivery systems, implantable devices, biosensors, and microarrays.1 For proteindrug formulations acceptable chemical and physical stability of the protein as well as a low level of non-toxic leachables are very important factors. In addition to that, inhibition of fouling is crucial in order for the drug to obtain compatibility with a substrate. Polypropylene (PP) is a substrate of interest for pharmaceutical packaging and delivery systems, due to its resistance to most chemicals, high fatigue strength, and good processability. However, PP is hydrophobic like most commercially available thermoplastics, which makes it less compatible with proteins compared with hydrophilic polymers.2 Therefore, the aim was to graft hydrophilic polymers from a PP substrate in order to improve the compatibility with a proteindrug formulation.Hydrophilic polymers can be grafted by Surface-Initiated Atom Transfer Radical Polymerization (SI-ATRP). This polymerization method can be performed on various polymeric, metallic or inorganic surfaces, and has proven to be a very robust and versatile grafting procedure with a controlled character.1,3 It has been greatly exploited and its use as a technique to develop biofunctional coatings is well documented and rapidly growing.1,4 The key step in grafting polymers through SI-ATRP is generation of initiating sites on the entire substrate. In principle, convenient procedures to transform different functional groups (OH, CHO, NH, NH2) on the surface to initiating groups for the chosen SIpolymerization method have been identified where the most exploited groups are surface hydroxyls, e.g., in cellulose.5 Recently poly(ether etherketone) (PEEK) has been immobilized with ATRP initiating moieties6,7 after transformation of the surface ketones to hydroxyls by a well established protocol.8 However, efficient ways for creating initiating sites for ATRP on inert surfaces are still challenging. One of the few reported methods, which has been successful, uses an UV-ATRP initiator, benzophenonyl bromoisobutyrate (BP-iBUBr). Firstly, it was proposed and used for PP,9 later it was also adapted for carbon nanotubes,10 and it was applied in the present investigations. When initiating groups on the surface are formed, SI-ATRP of a desired monomer can easily be performed in bulk, organic solvent or aqueous media. For biological applications mostly hydrophilic polymers are grafted, and the preferred medium is water or water/methanol. In this study it was decided to graft poly(ethylene glycol)methacrylate (PEGMA) and N,N-dimethylacrylamide (DMAAm) from PP. PEGMA was selected as coatings of poly(ethylene glycol) (PEG) are known to inhibit non-specific fouling due to their water solubility, hydrophilicity, and chain mobility.2,11 Poly(DMAAm) was of interest as it has been grafted from chromatographic packing material and prevented fouling during separation of proteins.12
In general, SI controlled polymerizations generate thin covalently linked polymer coatings on a substrate.3 The entire new surface layer on the substrate is different from the bulk and its properties. Thus new functionality to the substrate is added. Moreover, surface grafting influences the physical and chemical interactions of the substrate with the surrounding environment. This is essential for the application in question.
SI-ATRP was performed in a large scale on a substantial amount of test specimen in order to have sufficient modified substrates for a stability study to investigate the chemical and physical stability of insulin stored at three different temperatures.13 The modification was confirmed with Attenuated Total Reflectance (ATR) Fourier Transform Infrared (FTIR) and Water Contact Angle (WCA) measurements. Moreover, confocal fluorescence microscopy was applied to evaluate the tendency of the polymer materials to repel or adsorb proteins. For that purpose insulin aspart was labeled with a fluorophore.
Experimental
Materials
Dichloromethane (CH2Cl2, Sigma-Aldrich), N,N-dimethylacrylamide (DMAAm, Sigma-Aldrich), and triethylamine (TEA, Riedel-de Haën) were distilled over calcium hydride. 2,2′-Bipyridine (Bipy, 99%), 2-bromoisobutyrylbromide (Br-iBuBr, 98%), ethyl-2-bromoisobutyrate, copper(I) chloride (CuCl, 99%), 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA, 97%), 4-hydroxybenzophenone (98%), poly(ethylene glycol)methacrylate (PEGMA, Mn ≈ 360), and sodium bicarbonate (NaHCO3, 99.5%) were used as supplied by Sigma-Aldrich. Alexa Fluor® 488 (A488, Invitrogen), dimethylsulfoxide (DMSO, Fluka), ethanol (99.9 vol%, Kemetyl), and ultrapure water were used without further purification. Granulate of PP, Purell HM671T, was purchased from Basell. This PP was a gamma radiation resistant, high flow, and metallocenecatalyst based homopolymer. PP plates with the dimensions of 3.5 cm × 0.6 cm × 0.1 cm were injection-moulded at Danish Technological Institute. Freeze-dried insulin aspart known as NovoLog® or NovoRapid® was supplied by Novo Nordisk A/S.Modification of polypropylene surfaces
The UV-initiator, benzophenonyl bromoisobutyrate, was synthesized from 4-hydroxybenzophenone, according to the procedure of Huang et al.9 Anchoring of the initiator was made by immersion of the PP plates three times in a solution of the UV-initiator in toluene (10 mg mL−1). More than 200 plates were prepared. Subsequently, UV irradiation was performed for 30 min on both sides of the plates. The unreacted UV-initiator was removed from the plates by washing with CH2Cl2.
Polymerization of PEGMA from the 200 pieces of initiator-modified PP (PP-Br) plates was performed in aqueous media in one batch in a 1 L round bottom flask. 459 mL of ultrapure water and 276 mL of PEGMA were added to a reactor. Nitrogen was bubbled through the mixture for 60 min. 200 PP-Br plates were added and bubbling with nitrogen was proceeded for 40 min, followed by cooling on ice and bubbling with nitrogen for 50 min. The reactor was kept at 0 °C while CuCl (4.7932 g, 0.048 mol) and Bipy (12.804 g, 0.081 mol) were added. The bubbling with nitrogen was continued for 15 min. The polymerization was carried out at 30 °C for 45 min under stirring. Afterwards the PP-g-PPEGMA plates were immersed and stirred in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/methanol overnight and then in 5:1 water/ethanol for 1 hour.
:1 water/methanol overnight and then in 5:1 water/ethanol for 1 hour.
Grafting of poly(DMAAm) from 200 PP-Br plates was carried out in bulk in one batch in a 1 L glass reactor. 200 mL DMAAm was added to one flask and the other components i.e. 200 PP-Br plates, CuCl (2.1315 g, 0.021 mol), and HMTETA (21.5 mL, 0.077 mol) were added to another flask. After two freeze–pump–thaw cycles for the monomer and three cycles for the other components DMAAm was transferred by cannulation. The polymerization was performed at 90 °C for 23.5 hours under stirring. Purification of the PP-g-PDMAAm plates was carried out by immersion and sequentially stirring twice in water for 1 hour, once in 1:1 water/methanol for 1 hour and finally in 5:1 water/ethanol for 1 hour.
Polymerizations of either PEGMA or DMAAm with a sacrificial initiator were executed by the same protocol, but with only two PP-Br plates. As sacrificial initiator 0.06 mmol (11.7 μL) ethyl 2-bromobutyrate was used. The plates were taken out of the polymerization mixture, the latter was diluted, if necessary with THF, precipitated in heptane and after drying analyzed by SEC. The plates were further washed by stirring with copious amount of water, methanol, and THF.
Labelling of insulin
1 mg of A488 was dissolved in 100 μL of DMSO. 3.5 mg mL−1 of insulin aspart was prepared by transferring 8 mg of freeze-dried insulin aspart (containing water) to 1.5 mL of 0.1 M borate buffer, pH 9.5. The insulin solution was stirred for 30 min. UV-Vis spectroscopy was applied to confirm the concentration. The absorbance was 4.035 and it was determined by multiplying the dilution factor with the value from the UV-Vis spectrometer (15 × 0.269). The molar absorption coefficient, ε, at 280 nm was calculated from the formula ε(280) M−1 cm−1 = (#Trp)/(5500) + (#Tyr)(1490) + (#Cys)(125).14ε(280) = 4 × 1490 + 6 × 125 = 6710 L mol−1 cm−1. The molecular weight of insulin aspart was 5825.8 g mol−1 which resulted in a concentration of 3.5 mg mL−1. 1 mL of the insulin aspart solution was used for the labelling. 100 μL of A488 solution was added in three portions (33.33 μL at a time) in 1 hour (at 0, 20, and 40 min). The unreacted dye was removed by dialysis overnight.Adsorption tests
Four small circular pieces with a diameter of 4 mm were punched out for unmodified PP, PP-g-PPEGMA, and PP-g-PDMAAm. The four pieces were cleaned by immersion in ethanol for 10 min, followed by drying with compressed air. Three pieces were immersed in 50 μL of labelled insulin aspart in borate buffer at ambient temperature and they were taken out after 1, 4, and 24 hours. Each piece was cleaned by (1) rinsing with borate buffer pH 9.5 and (2) immersion in 1000 μL borate buffer, kept for 10 min on a blood mixer, and finally transferred to an Eppendorf tube with 1000 μL borate buffer and stored overnight at 5 °C. The fourth piece from the three different polymer materials was used to prepare a blank sample. The piece was immersed in a 1000 μL borate buffer and mixed for 10 min. Subsequently, it was transferred to another Eppendorf tube with 1000 μL borate buffer and stored under the same conditions as the samples.Methods
ATR FTIR spectra were obtained using a Spectrum One spectrometer from Perkin Elmer which was equipped with a universal ATR sample accessory. Thermogravimetric analysis (TGA) was performed on a TGA Q500 instrument (TA Instruments) from 25–800 °C with a heating rate of 20 °C min−1 under nitrogen flow. The investigated plates were cut in smaller pieces (a total of 25–35 mg) to fit into the platinum cups. WCA measurements were made on an OCA20 Contact Angle System from Dataphysics with a temperature controller. The temperature was set to 25 °C. The dynamic method called “sessile drop (needle in)” was used and the WCAs were computed using “Ellipse Fitting”. The measurements were carried out on three drops of deionised water at different spots and three values for both the advancing and receding angles were used to determine the average value. UV irradiation of the PP plates in order to anchor the initiator was performed with a UVA light source purchased from SolData Instruments. The light from the mercury vapour lamp had a broad maximum output at 365 nm. Intensity of the light was measured with a Sentry® UV detector which detected UV radiation from 280–400 nm. Typical measurements on the UV lamp were in the range of 7.2–7.9 mW cm−2. A Shimadzu UV1700, UV-Visspectrophotometer was applied to determine the concentration of insulin aspart. Measurements were carried out at 280 nm. The samples were diluted 15 times. Reset to zero was made on 0.1 M borate buffer, pH 9.5. Confocal fluorescence microscopy was performed on a Confocal Laser Scanning Microscope, CLSM 510 Meta from Zeiss with Zen710 software. Pinhole was set to 4.24 airy units. 2D-images were obtained with an objective of 10× magnification, and the following settings, tile scan 2 × 2 and 8 bit image, were applied.Size exclusion chromatography (SEC) of the polymers produced with the sacrificial initiator was performed in THF at room temperature at a flow rate of 1.0 mL min−1 on a set of PL guard and 2 PL gel mixed D columns (Polymer Laboratories), calibrated versuspolystyrene narrow molar mass standards. Additionally SEC in DMF was used to characterize the poly(DMAAm) free polymer. These SEC experiments were performed on an EcoSEC semi-microsystem from Polymer Standard Service (PSS) employing 100 Å and 300 Å PFG micro-columns with a bead size of 7 micrometre. The instrument was equipped with 2 detectors—a dual flow refractive index and a UV. DMF with 0.05 M LiCl was used as eluent at 50 °C. The molecular weights were estimated using the calibration curve constructed with poly(methyl methacrylate) (PMMA) standards and WinGPC Unity software from PSS.
Results and discussion
Initiating groups for Atom Transfer Radical Polymerization (ATRP) were first anchored on the PP surface before the graft polymerization could take place. Covalent C–C bonds should then be formed between the initiator and the PP when irradiated with UV light at 365 nm. Hydrophilic polymers of either poly(PEGMA) or poly(DMAAm) were grafted from the initiator modified PP surface using the conditions in Fig. 1.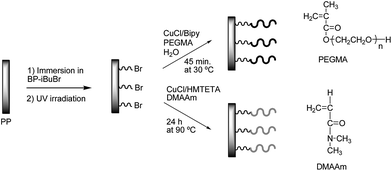 | ||
| Fig. 1 Grafting of poly(PEGMA) and poly(DMAAm) brushes on the surface. | ||
From the ATR FTIR spectra (Fig. 2) the presence of the poly(PEGMA) and poly(DMAAm) anchored to the PP substrates was observed. The carbonyl group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) from the ester in the initiator and poly(PEGMA) resulted in a stretching band at 1734 cm−1 whereas the CO absorption band from the amide in poly(DMAAm) appeared at 1634 cm−1. At 1256 cm−1 the C–O and C–N stretching bands were observed which were from the amide in poly(DMAAm) and the OC–O in initiator and poly(PEGMA). The absorption band at 1101 cm−1 was attributed to the characteristic absorption of the C–O in the ethylene oxide O–CH2–CH2–O–CH2– side chain. It was not possible to identify the initiating sites on the PP-Br plates with ATR FTIR, which other groups also have reported.5a,9,15 In principle, FT-IR was successfully used1 to recognize initiating groups on inorganic and metallic surfaces; however, it is incredibly difficult to apply a similar method to demonstrate the presence of initiating sites on polymeric surfaces. The first paper, which reports a successful detection ofATRP initiating groups on polymeric substrates, has applied PEEK films.6 The reason for this observation might be a more efficient chemical immobilization method than the UV initiator on PP. A reference sample was prepared in each of the polymerizations of PEGMA and DMAAm. The reference was a PP plate, which was subjected to the polymerization conditions, but it had no initiator immobilized. The ATR FTIR spectra of these 2 samples were identical with pure PP, and therefore not shown in Fig. 2.
O) from the ester in the initiator and poly(PEGMA) resulted in a stretching band at 1734 cm−1 whereas the CO absorption band from the amide in poly(DMAAm) appeared at 1634 cm−1. At 1256 cm−1 the C–O and C–N stretching bands were observed which were from the amide in poly(DMAAm) and the OC–O in initiator and poly(PEGMA). The absorption band at 1101 cm−1 was attributed to the characteristic absorption of the C–O in the ethylene oxide O–CH2–CH2–O–CH2– side chain. It was not possible to identify the initiating sites on the PP-Br plates with ATR FTIR, which other groups also have reported.5a,9,15 In principle, FT-IR was successfully used1 to recognize initiating groups on inorganic and metallic surfaces; however, it is incredibly difficult to apply a similar method to demonstrate the presence of initiating sites on polymeric surfaces. The first paper, which reports a successful detection ofATRP initiating groups on polymeric substrates, has applied PEEK films.6 The reason for this observation might be a more efficient chemical immobilization method than the UV initiator on PP. A reference sample was prepared in each of the polymerizations of PEGMA and DMAAm. The reference was a PP plate, which was subjected to the polymerization conditions, but it had no initiator immobilized. The ATR FTIR spectra of these 2 samples were identical with pure PP, and therefore not shown in Fig. 2.
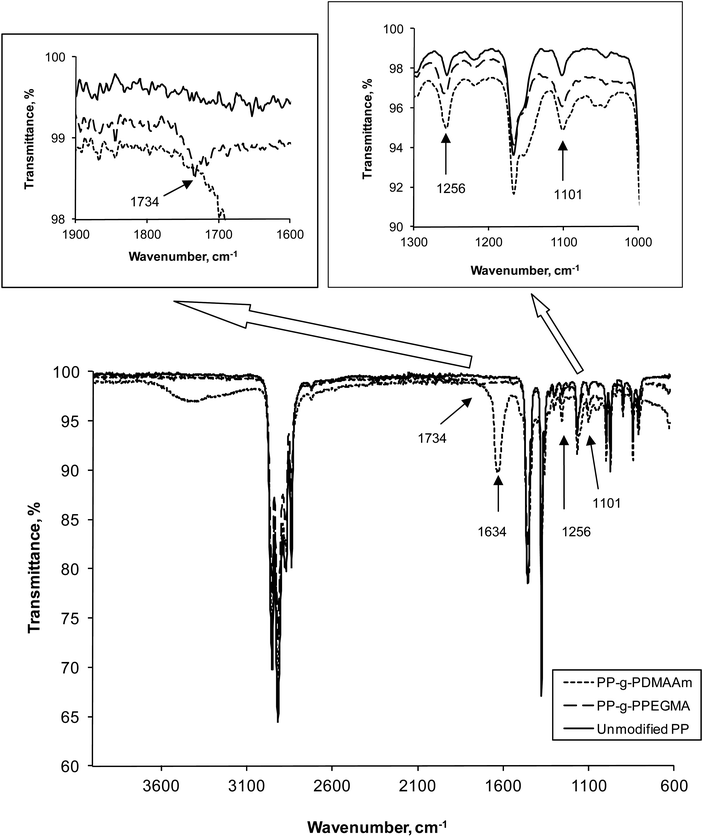 | ||
| Fig. 2 ATR FTIR spectra of the modified and unmodified PP. | ||
Meticulous gravimetric analyses performed on 15 washed and carefully dried un-modified PP and PP-g-PPEGMA plates revealed very small weight gains corresponding to only 0.13 wt% PPEGMA. Since 1 PP plate has a total surface area of 5 cm2 and a mass of 188 mg on average only 0.2 mg PPEGMA (0.04 mg cm−2) is grafted on one plate. However, the same analysis on the PP-g-PDMAAm plates revealed an average weight gain of 2.1 wt% corresponding to an average of 4 mg PDMAAm (0.8 mg cm−2) on one plate. In addition, TGA analyses showed that the PP-g-PDMAAm plates exhibited a weight loss upon heating of approximately 1.5% that could be assigned to PDMAAm. If one back calculates, this corresponds to 2.8 mg of PDMAAm grafted (0.56 mg cm−2) on the PP surface. Thus, the two different ways (simple gravimetry and TGA) of estimating the average amount of grafted PDMAAm on the PP plates are relatively in good agreement. A similar TGA analysis was not possible in the case of the PP-g-PPEGMA. Without the additional knowledge of grafting density estimation of the grafted chain-length is not possible.
WCAs were measured on unmodified PP and PP with the hydrophilic polymers of either poly(PEGMA) or poly(DMAAm). Both free polymers produced with the sacrificial initiator were water soluble, and therefore the WCAs of spin-coated free poly(PEGMA) or poly(DMAAm) could not be measured. When grafted, these polymers lowered the advancing and receding contact angles to some extent. The results in Table 1 show that in both cases the advancing and receding WCAs decreased. The grafting of poly(PEGMA) or poly(DMAAm) on the surface also increased the hysteresis due to the hydrophilicity of the grafts. Thus, the WCAs corroborate the changes in the grafted PP surface properties.
| Material | WCA (advancing)/° | WCA (receding)/° |
|---|---|---|
| PP | 112 ± 1 | 87 ± 4 |
| PP-g-PPEGMA | 83 ± 3 | 43 ± 2 |
| PP-g-PDMAAm | 104 ± 2 | 49 ± 3 |
When the applied ATRP conditions were used in solution it was revealed that some polymer chains have reacted with each other and formed crosslinks. The corresponding polymer coating will presumably also crosslink.6 Nevertheless, all polymer chains (crosslinked or non-crosslinked) were expected to inhibit protein adsorption when the modified substrates were immersed in a proteindrug formulation. The proteins should perceive the hydrophilic polymer chains as the aqueous media surrounding them. Thus the proteins were not supposed to denaturize or undergo conformational changes. Moreover, the proteins should not be physically adsorbed when the modified substrates were removed from the proteindrug formulation. Confocal fluorescence microscopy of modified and unmodified substrates immersed in labelled insulin showed that only PP-g-PPEGMA repelled the proteins (Fig. 3). Samples for fluorescence microscopy were taken out and analyzed after 1, 4, and 24 hours. The areas with adsorbed insulin increased over time on PP-g-PDMAAm whereas no visible difference was observed between 4 and 24 hours for unmodified PP.
 | ||
| Fig. 3 Overlay of images from transmission and confocal fluorescence microscopy of substrates immersed in labelled insulin. Unmodified PP after (a) 1 hour, (b) 4 hours, and (c) 24 hours; PP-g-PDMAAm after (d) 1 hour, (e) 4 hours, and (f) 24 hours; PP-g-PPEGMA after (g) 1 hour, (h) 4 hours, and (i) 24 hours. | ||
The efficiency of the crosslinked poly(PEGMA) grafts to repel labelled insulin seemed to be high and it is expected to be durable for a long time as the polymer chains are covalently attached to the surface. Adsorption of labelled insulin to PP-g-PDMAAm happened instantly and either the amount of denaturized insulin was increased or aggregates were formed. Factors like grafting density of the hydrophilic polymer and the size of the protein will influence the ability to inhibit fouling. Fouling has previously been observed for poly(DMAAm) grafts with low grafting density (in the mushroom regime).12 The grafting density of poly(DMAAm) and poly(PEGMA) ought to be comparable in this study. It might even be higher for poly(DMAAm) as PEGMA has side chains of about six ethylene oxide units. The crosslinked architecture of poly(PEGMA) must make the difference (Fig. 4).
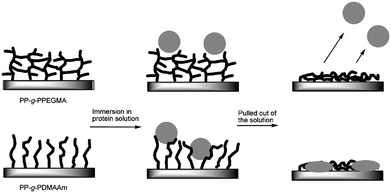 | ||
| Fig. 4 Crosslinked hydrophilic grafts of poly(PEGMA) were able to repel labelled insulin whereas insulin adsorbed onto the surface with poly(DMAAm) brushes when the substrates were pulled out of a protein solution as visualized. | ||
Polymerization in the presence of a sacrificial initiator is a widespread method to characterize polymer chains which are claimed to be comparable with the polymer grafts. Polymerization with a sacrificial initiator was conducted to try to improve the part about characterization of the polymer grafts.
PP plates with the attached hydrophilic poly(PEGMA) have a smooth surface, while the non-covalently attached poly(PEGMA) produced in solution from the sacrificial initiator appears to be crosslinked as well, thus determining the molecular weight characteristics was impossible. As seen in Fig. 5 the free polymer is even sticking onto the surface of PP. The polymerisation of PEGMA with the sacrificial initiator was also prepared in methanol. The free poly(PEGMA) from polymerization in methanol was soluble in alcohols, water and THF, and had a Mn of 6700 and a PDI of 1.18 (SEC in THF with PS calibration). However, this result does not imply that the polymer on the surface has the same characteristics when water was used as a solvent. Usually methanol is added to slow down the very fast polymerization of hydrophilic monomers in water.16
 | ||
| Fig. 5 PP plates with hydrophilic poly(PEGMA) grafted in the absence (left) and in the presence (right) of a sacrificial initiator. | ||
The same experiment when using DMAAm in the presence of a sacrificial initiator produced a free polymer with Mn = 2100 and PDI = 1.17 (SEC in DMF with PMMA standards). Additionally, low grafting density combined with short poly(DMAAm) chains may be the explanation for the absence of repellence of insulin from poly(DMAAm) modified surfaces by the conditions used.
Rejection of the relatively small size protein, insulin, is very dependent on sufficient grafting density or surface coverage in general. Groll et al.17 have compared linear and star PEG coated on silicon substrates with respect to the repulsion of insulin and the larger protein, lysozyme. In agreement with theoretical predictions they found that the branched structure of the star PEG and linear PEG with high grafting density could repel insulin. When the grafting density was lower only lysozyme was repelled. Therefore, the crosslinks between the poly(PEGMA) grafts must be able to compensate for a lower grafting density. Moreover, the antifouling properties of poly(DMAAm) might be more dependent on a high grafting density than other protein repellent hydrophilic grafts.
Conclusions
Poly(PEGMA) and poly(DMAAm) were grafted from PP using SI-ATRP. Lowering of the WCAs was observed after grafting of the polymers, thus hydrophilization of PP was achieved. ATR FTIR spectra confirmed the modifications of PP with poly(PEGMA) and poly(DMAAm), respectively. Poly(PEGMA) has presumably crosslinked as previously studied with homopolymerization of PEGMA in water formed a gel. The crosslinked poly(PEGMA) on the surface formed a very smooth film and showed excellent repulsion of labelled insulin aspart after 24 hours of exposure. Therefore, the poly(PEGMA) coating has the possibility of making polymer materials more usable for devices in contact with proteindrug formulations.Acknowledgements
CJF acknowledges the Technical University of Denmark, Novo Nordisk A/S, and the Danish Agency for Science Technology and Innovation for financial support. Furthermore, we thank Monika Butrimaité for the modification with PEGMA and Helle Markussen Nordholm from FeF Chemicals A/S for the assistance with UV-Vis spectroscopy and the confocal fluorescence microscopy. Irakli Javakhishvili is acknowledged for performing the SEC in DMF.References
- C. J. Fristrup, K. Jankova and S. Hvilsted, Soft Matter, 2009, 5, 4623–4634 RSC.
- (a) D. L. Elbert and J. A. Hubbell, Annu. Rev. Mater. Sci., 1996, 26, 365–394 CrossRef CAS; (b) J. H. Lee, H. B. Lee and J. D. Andrade, Prog. Polym. Sci., 1995, 20, 1043–1079 CrossRef CAS; (c) M. Malmsten, K. Emoto and J. M. Van Alstine, J. Colloid Interface Sci., 1998, 202, 507–517 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS.
- F. J. Xu, K. Neoh and E. T. Kang, Prog. Polym. Sci., 2009, 34, 719–761 CrossRef CAS.
- (a) A. Carlmark and E. Malmstrom, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS; (b) D. Plackett, K. Jankova, H. Egsgaard and S. Hvilsted, Biomacromolecules, 2005, 6, 2474–2484 CrossRef CAS; (c) D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- C. J. Fristrup, K. Jankova and S. Hvilsted, Polym. Chem., 2010, 1, 1696–1701 RSC.
- B. Yameen, M. Alvarez, O. Azzaroni, U. Jonas and W. Knoll, Langmuir, 2009, 25, 6214–6220 CrossRef CAS.
- (a) O. Noiset, Y. J. Schneider and J. Marchand-Brynaert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 3779–3790 CrossRef CAS; (b) O. Noiset, C. Henneuse, Y. J. Schneider and J. Marchand-Brynaert, Macromolecules, 1997, 30, 540–548 CrossRef CAS.
- J. Huang, H. Murata, R. R. Koepsel, A. J. Rusell and K. Matyjaszewski, Biomacromolecules, 2007, 8, 1396–1399 CrossRef CAS.
- A. E. Daugaard, K. Jankova, J. Bøgelund, J. K. Nielsen and S. Hvilsted, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4594–4601 CrossRef CAS.
- J. F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- B. R. Coad, B. M. Steels, J. N. Kizhakkedathu, D. E. Brooks and C. A. Haynes, Biotechnol. Bioeng., 2007, 97, 574–587 CrossRef CAS.
- C. J. Fristrup, K. Jankova, R. Eskimergen, J. T. Bukrinsky and S. Hvilsted, Eur. J. Pharm. Sci., submitted Search PubMed.
- C. N. Pace, F. Vajdos, L. Fee, G. Grimsley and T. Gray, Protein Sci., 1995, 4, 2411–2423 CrossRef CAS.
- N. Singh, J. Wang, M. Ulbricht, S. R. Wickramasinghe and S. M. Husson, J. Membr. Sci., 2008, 309, 64–72 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Chem. Rev., 2007, 107, 2270–2299 CrossRef CAS.
- J. Groll, Z. Ademovic, T. Ameringer, D. Klee and M. Moeller, Biomacromolecules, 2005, 6, 956–962 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |