Enzymatically degradable nanogels by inverse miniemulsion copolymerization of acrylamide with dextran methacrylates as crosslinkers†
Daniel
Klinger
,
Eugen M.
Aschenbrenner
,
Clemens K.
Weiss
and
Katharina
Landfester
*
Max Planck Institute for Polymer Research, Ackermannweg 10, Mainz, Germany. E-mail: landfester@mpip-mainz.mpg.de; Fax: +49 6131 379-330; Tel: +49 6131 379-170
First published on 18th November 2011
Abstract
Nanogels consisting of polyacrylamide (PAAm), crosslinked with dextranmethacrylate (Dex-MA), were designed to be partially biodegradable by enzymatic cleavage of the methacryl-functionalized polysaccharide chains. Important properties of the described hydrogels such as a low initial degree of swelling and a high crosslinking efficiency—in order to ensure the morphological integrity during nanogel preparation, purification and storage—are highly favored. Primarily, those requirements strongly depend on the amount of crosslinking points, i.e.methacrylategroups in the network. This parameter was adjusted by either increasing the ratio of Dex-MA/AAm for a fixed degree of substitution (DS) of Dex-MA or increasing the DS of Dex-MA for a fixed ratio of Dex-MA/AAm. Secondly, the distribution of crosslinking points in the network is another crucial parameter which was optimized by lowering the molecular weight of the Dex-MA for analogous DS or by simultaneously increasing the Dex-MA/AAm ratio and decreasing the respective DS. This resulted in nanogels with a reduced initial degree of swelling and sol content, therefore indicating a more homogeneous distribution of the same amount of crosslinking points. Degradation of the hybrid gel nanoparticles was examined by turbidity and DLS measurements upon treatment with dextranase. It was found that the degradation behavior depends on the total amount of methacrylategroups in the network and the degree of substitution of the individual Dex-MA chains.
Introduction
Hydrogel nanoparticles (i.e. micro- or nanogels) consist of a chemically or physically crosslinked network structure and have gained considerable attention in recent years due to their unique features of merging the characteristics of macroscopic hydrogels with those of colloidal dispersions.1 The hydrophilic network structure allows the combination of structural integrity with the transport characteristics of a fluid for substances smaller than the mesh size of the gel.2 In addition to the good colloidal stability and the small sizes (<1 μm) of the particles, especially stimuli-responsive nanogels are of high interest for a broad variety of applications including the fields of drug delivery,3,4 chemical sensing,5 bioseparation,6 catalysis,7 optics,8 or colloidal crystals.9,10 A specific sensitivity can be achieved by the incorporation of functional groups into the gels, enabling a response of the network structure to external triggers. In this context two main mechanisms can be distinguished:(a) Induced by changes in the (physicochemical) environment, alteration of the physicochemical parameters of the network forming polymers results in a variation of the degree of swelling. The reversible volume phase transition is governed by an imbalance between repulsive and attractive forces (e.g.hydrogen bonds, electrostatic interactions) acting in the particles11 and can be influenced by different stimuli such as e.g. pH,12,13 temperature,10,14,15 ionic strength16 or combinations thereof.17
(b) Incorporation of labile crosslinking points into the gel enables the triggered (irreversible) disintegration of the network structure, yielding freely soluble polymer chains. Investigated approaches to realize the described concept of degradable microgels are based on e.g. pH-sensitive crosslinkers containing various acid labile moieties such as tertiary esters or acetals,18–20 light cleavable crosslinking molecules,21,22 or biodegradable polymers.23
Regarding degradable nanogels, complete particle disintegration offers the potential to release entrapped compounds upon the appliance of an external trigger. An important requirement is the prevention of diffusion of the embedded functional substance from the gel, until the respective stimulus is applied. This is achieved by e.g. initial mesh sizes of the gel, smaller than the hydrodynamic diameter of the active compound. Triggered cleavage of crosslinking points results in increased pore sizes thus enabling the release by diffusion.11,24
A highly interesting approach to degradable (micro-) gels is based on the utilization of dextrans as naturally occurring polysaccharides which can be cleaved upon incubation with dextranase.25 Covalent functionalization of dextran chains with either polymerizable vinyl groups26 or thermal initiators27 enables the formation of hydrogels by free radical (co)polymerization in aqueous media. The resulting networks can then be degraded by the addition of dextranase, inducing the release of embedded active compounds.23 Microgels based on dextran methacrylates have been investigated for the release of immunoglobulin G as a model protein.28
Investigated routes to enzymatically degradable microgels containing functionalized dextrans are so far based on precipitationpolymerizations from aqueous solutions. This preparation method is generally a widely used approach to microgel formation but is limited by some serious drawbacks. On one hand the microgels reported in the literature are in the size ranges of 4–10 μm28,29 and thus comparably large in their diameter. On the other hand, the introduction of a second comonomer is limited by its hydrophilicity.2 To ensure successful particle precipitation upon polymerization, highly hydrophilic comonomers can only be incorporated to a certain extent, thus restricting the versatility of this approach and these materials.
Compared to the described method of precipitationpolymerization, the inverse miniemulsion technique is highly advantageous. In this process, stable homogeneous droplets of an aqueous solution are dispersed in an organic solvent by applying high shear forces.30 The addition of an osmotic agent (an ultrahydrophilic compound) such as sodium chloride to the dispersed phase controls diffusion between the droplets by creating an osmotic pressure counteracting the Laplace pressure. Therefore, stable droplets of the same composition as the dispersed phase prior to emulsification are obtained and can be classified as “nanoreactors”.31 As a result, the composition of the polymeric particles after polymerization resembles the composition of the monomer phase, thus enabling the equal distribution of all different functionalities in each particle. Since the only main requirement for copolymerization of different monomers is their immiscibility with the continuous phase, this approach is highly tolerant to a broad variety of materials.30 So far, the inverse miniemulsion technique was mainly applied for capsule formation,32 sol–gel reactions,33 or hydrophobic particles preparation.34 However, this approach is very suitable for the synthesis of hydrogels in the droplets.35
A second highly advantageous point is the good control over the size of the particles obtained by inverse miniemulsion polymerizations. Observed particle sizes are generally in the sub-micron range and particle diameters can be adjusted by the choice and concentration of surfactant from roughly 100 to around 500 nm.31
The aim of this work is the preparation of enzymatically degradable poly(acrylamide-co-dextran methacrylate) nanogels by free radical copolymerization from aqueous solutions of acrylamide (AAm) and dextranmethacrylate (Dex-MA) in inverse miniemulsion. Here, the intention is to synthesize and characterize new materials by transferring the concept of enzymatically degradable dextran methacrylates as crosslinkers from hydrogels in the micrometre size range to the nanoscale by utilizing the inverse miniemulsion technique. Acrylamide as a highly water soluble monomer was chosen as a model compound to demonstrate the versatility of the inverse miniemulsion polymerization process towards the choice of monomers. In addition, the multiple methacrylation of dextran (DS > 0.05) leads to water soluble dextran methacrylates which are used as enzymatically degradable macromolecular crosslinking molecules.
Important properties of the resulting nanogels such as the initial degree of swelling and the sol content are assumed to be adjustable by systematically varying the amount of methacrylate units available for crosslinking. This can be achieved by either increasing the Dex-MA/AAm feed ratio for Dex-MAs of a fixed degree of substitution (DS) or by increasing the DS for fixed Dex-MA/AAm feed ratio. Systematic investigations on these parameters are part of this study and should give insight into the crosslinking performance of the Dex-MA molecules.
The enzymatic degradability of p(AAm-co-Dex-MA) nanogels represents an alternative approach to widely used acid degradable microgels, thus giving rise to potential release applications suitable for a broader range of pH values of the surrounding media. In this context, another important focus of the work presented is to optimize the synthetic parameters (i.e. the Dex-MA/AAm feed ratio) in order to yield nanogels which combine a high crosslinking density (i.e. small mesh sizes) and efficiency for ambient polymerization temperatures with a good enzymatic degradability. Nanogels fulfilling these criteria are assumed to be highly interesting materials for potential release applications. Here, a crucial prerequisite is the development of an effective synthetic pathway and a profound understanding of the stimuli-responsive behavior of these materials. Therefore, particular attention was paid on combining the design and characterization of the specific response mechanism at a molecular level with detailed investigations on the influence of these features on the overall sensitivity of the nanoscale polymeric hydrogel particles.
Experimental section
Materials
All chemicals were commercially available and used without further purification unless otherwise stated. Acrylamide was obtained from Fluka Chemicals, dextrans, potassium peroxodisulfate (KPS) and tetramethylethylenediamine (TEMED) from Sigma-Aldrich. Initiators (2,2′-azobis(2-methyl-butyronitrile) (V-59); 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70); and 2,2′-azobis(N,N′-dimethyleneisobutyramidine) dihydrochloride (VA-044)) were obtained from Wako Chemicals. Lubrizol U (poly(isobutylene-succinimide pentamine)) was kindly provided by Lubrizol, France.Instrumentation
1H-NMR (300 MHz) spectra were measured using a Bruker spectrometer. Particle sizes and particle size distributions were determined by dynamic light scattering (DLS) using a NICOMP zetasizer measuring at a fixed scattering angle of 90°. The measurements were carried out at 25 °C on diluted dispersions in the respective solvents. Particle sizes represent the number averages of the hydrodynamic diameters (dh). Polydispersities of the particles were determined as the full width at half maximum of the Gaussian fits of the particle size distributions and were depicted as error bars of the dh values in the respective diagrams. A Gemini 1530 SEM (Carl Zeiss AG, Oberkochen, Germany) with an InLens detector was used to acquire scanning electron micrographs (SEMs). Samples were prepared by drop-casting.Synthesis of dextran methacrylates
Dextran was functionalized using a modification of the protocol presented by Kim and Chu.36 In brief, the polysaccharide was dissolved in a 10 wt% LiCl solution of DMF at 90 °C. After cooling the reaction mixture to 60 °C, an equimolar amount of triethylamine (with respect to the anhydride) was added, followed by dropwise addition of a defined amount of methacrylic anhydride. The reaction was allowed to proceed for 2 h and then stopped by cooling to room temperature and precipitation in 2-propanol. The precipitate was separated by centrifugation (Sigma 3K30, 5 min @ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 min−1), dissolved in water, precipitated in 2-propanol and centrifuged again. Residual water soluble reactants were removed by dialysis (regenerated cellulose MWCO 3.5 k) against water for at least one day. Subsequently, the product was dried in a vacuum and analyzed by 1H-NMR spectroscopy in D2O. Used molar ratios of methacrylic anhydride to hydroxylgroups of dextran and obtained yields after purification are listed in Table 1.
000 min−1), dissolved in water, precipitated in 2-propanol and centrifuged again. Residual water soluble reactants were removed by dialysis (regenerated cellulose MWCO 3.5 k) against water for at least one day. Subsequently, the product was dried in a vacuum and analyzed by 1H-NMR spectroscopy in D2O. Used molar ratios of methacrylic anhydride to hydroxylgroups of dextran and obtained yields after purification are listed in Table 1.
| Sample | Dextran M w/g mol−1 | MA/Dex-OHa | DS | Yield (%) |
|---|---|---|---|---|
| a Molar ratio of methacrylic anhydride/hydroxylgroups of dextran. | ||||
| Dex-MA1 | 40000 |
0.05 | 0.12 | 90 |
| Dex-MA2 | 40000 |
0.10 | 0.17 | 82 |
| Dex-MA3 | 40000 |
0.15 | 0.33 | 89 |
| Dex-MA4 | 6000 | 0.05 | 0.09 | 45 |
| Dex-MA5 | 6000 | 0.10 | 0.19 | 39 |
| Dex-MA6 | 6000 | 0.15 | 0.29 | 31 |
| Dex-MA7 | 40000 |
0.03 | 0.05 | 78 |
The DS was evaluated from 1H-NMRspectra by calculating the ratio of the peak areas of the olefinic peak at 6.2 ppm relative to the anomeric proton of dextran assigned to the peak at 4.9 ppm by using the equation:
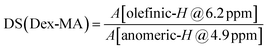 | (1) |
Synthesis of enzymatically degradable nanogels by inverse miniemulsion copolymerization
Dispersed phases were prepared by dissolving acrylamide (AAm) and dextranmethacrylate (Dex-MA) in an aqueous 0.5 M NaCl solution. While the total ratio of (AAm + Dex-MA)/solvent was kept constant with 1/1 w/v, the ratio of Dex-MA/AAm was varied. For initiation from the aqueous phase, the respective initiators (VA-044, KPS) were added to the dispersed phase. The solution was added under stirring to the continuous phase consisting of a solution of the non-ionic surfactant Lubrizol U (100 mg) in 10 g of cyclohexane. The inverse miniemulsion was formed by first stirring the mixture at 1750 rpm for 1.5 h and then homogenizing the obtained pre-emulsion with ultrasonication for 2 min at 90% intensity (Branson sonifier W450 Digital, 0.5′′ tip) at 0 °C. For initiation with oil-soluble initiators from the continuous phase, either V-59 or V-70 was added to the inverse miniemulsion after homogenizing. The polymerizations were carried out overnight in an oil bath set at a fixed temperature (see Table 2). For initiation with KPS, TEMED was added to the homogenized miniemulsion and the polymerization was carried out at room temperature overnight. After the polymerizations, coagulates were removed by filtration and the resulting dispersions were centrifuged at 5000 rpm for 30 min to collect the particles. The supernatant was removed and replaced by cyclohexane. Redispersion was carried out using a vortexer. In order to remove excess surfactant, the dispersions were further washed four times following the procedure described above.| Sample | Dex-MA type | M w Dex/g mol−1 | DS Dexa | Dex-MA contentb (wt%) | MA-unitsc (mol%) | Initiator | T polym/°C |
|---|---|---|---|---|---|---|---|
| a Calculated as DS = amount of functionalized glucopyranosylgroups/total amount of glucopyranosylgroups. b w.r.t. mass of Dex-MA + AAm. c w.r.t. monomer units of AAm + glucopyranosylgroups. d Contains non-functionalized dextran, not Dex-MA. | |||||||
| MG-1A | Dex-MA1 | 40000 |
0.12 | 5.0 | 0.24 | V-59 | 70 |
| MG-1B | Dex-MA1 | 40000 |
0.12 | 17.5 | 0.90 | V-59 | 70 |
| MG-1C | Dex-MA1 | 40000 |
0.12 | 30.0 | 1.69 | V-59 | 70 |
| MG-2A | Dex-MA2 | 40000 |
0.17 | 5.0 | 0.33 | V-59 | 70 |
| MG-2B | Dex-MA2 | 40000 |
0.17 | 17.5 | 1.25 | V-59 | 70 |
| MG-2C | Dex-MA2 | 40000 |
0.17 | 22.0 | 1.62 | V-59 | 70 |
| MG-2D | Dex-MA2 | 40000 |
0.17 | 30.0 | 2.34 | V-59 | 70 |
| MG-3A | Dex-MA3 | 40000 |
0.33 | 5.0 | 0.59 | V-59 | 70 |
| MG-3B | Dex-MA3 | 40000 |
0.33 | 13.0 | 1.63 | V-59 | 70 |
| MG-3C | Dex-MA3 | 40000 |
0.33 | 17.5 | 2.27 | V-59 | 70 |
| MG-3D | Dex-MA3 | 40000 |
0.33 | 30.0 | 4.28 | V-59 | 70 |
| MG-4A | Dex-MA4 | 6000 | 0.09 | 5.0 | 0.18 | V-59 | 70 |
| MG-5A | Dex-MA5 | 6000 | 0.19 | 5.0 | 0.36 | V-59 | 70 |
| MG-6A | Dex-MA6 | 6000 | 0.29 | 5.0 | 0.48 | V-59 | 70 |
| MG-1Ab | Dex-MA1 | 40000 |
0.12 | 30.0 | 1.69 | V-70 | 37 |
| MG-2Ab | Dex-MA2 | 40000 |
0.17 | 30.0 | 2.34 | V-70 | 37 |
| MG-3Ab | Dex-MA3 | 40000 |
0.33 | 30.0 | 4.28 | V-70 | 37 |
| MG-7Ab | Dex-MA7 | 40000 |
0.05 | 30.0 | 0.72 | V-70 | 37 |
| MG-1Db | Dex-MA1 | 40000 |
0.12 | 100.0 | 12.0 | V-70 | 37 |
| MG-8Aa | Dex | 40000 |
— | 30.0d | — | V-59 | 70 |
| MG-8Ab | Dex | 40000 |
— | 30.0d | — | V-70 | 37 |
| MG-8Ac | Dex | 40000 |
— | 30.0d | — | KPS/TEMED | 30 |
| MG-8Ad | Dex | 40000 |
— | 30.0d | — | VA-044 | 45 |
Purified dispersions in cyclohexane were examined with regard to the particle sizes and distributions by means of dynamic light scattering (DLS). Scanning electron microscopy (SEM) was used to investigate the particle morphologies. Freeze drying yielded the crosslinked gel particles as white powders. Transfer of the nanogels to the aqueous phase was achieved by simply swelling the dried powders overnight in water at 0.5% (w/v) at room temperature. Two additional washing steps by centrifugation at 14000 rpm for 90 min and redispersion in deionized water were performed to remove the sol content which consisted of unreacted monomer and crosslinker, soluble (non-crosslinked) polymers and oligomers. The purified particles were again freeze dried and redispersed in the respective medium by swelling at room temperature for 6 h at the desired concentration.
Degradation experiments of microgels in water
Enzymatic degradation of poly(AAm-co-Dex-MA) microgels was examined by adding 10 μL of dextranase solution (47 wt%) to 3 mL of a dispersion of the respective purified gel particles in water (0.0625 wt%). Particle degradation was monitored by turbidity measurements for 24 h at 37 °C and the final dispersions were investigated for their particles size distributions by DLS.Results and discussion
Hybrid nanogels consisting of polyacrylamide (PAAm), crosslinked with dextranmethacrylate (Dex-MA), were designed to be enzymatically degradable due to utilization of the methacryl-functionalized polysaccharide as crosslinker. Enzymatic cleavage of the dextran chains is proposed to lead to a disintegration of the network structure, thus enabling the release of potential embedded compounds from the gel. Scheme 1 depicts the described concept of nanogel formation by free radical copolymerization in inverse miniemulsion as well as the mechanism of network degradation upon dextranase-induced cleavage of Dex-MA crosslinkers.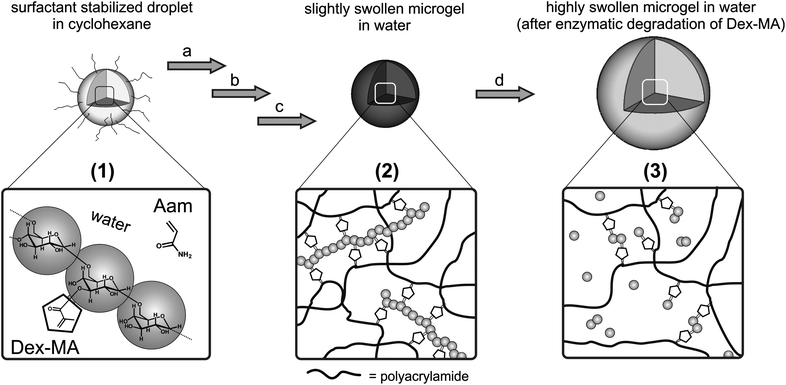 | ||
| Scheme 1 Schematic representation of the concept of enzymatically degradable poly(AAm-co-Dex-MA) microgels: synthesis and degradation. (1) Surfactant stabilized droplets in cyclohexane containing acrylamide and Dex-MA dissolved in water. Nanogel formation: (a) free radical copolymerization; (b) washing with cyclohexane and freeze drying; and (c) swelling in water, washing with water, freeze drying and transfer to water to obtain slightly swollen microgels (2). Enzymatic degradation: (d) addition of dextranase leads to highly swollen microgels (3) due to cleavage of Dex-MA. | ||
Important properties of the hydrogels, such as the initial swelling ratio and the crosslinking efficiency, are assumed to strongly depend on the amount of crosslinking points, i.e. the methacrylategroups in the network and their distribution therein. Those parameters can be adjusted by the variation of: (i) the ratio of Dex-MA/AAm for a fixed DS of Dex-MA; (ii) the DS of Dex-MA for a fixed ratio of Dex-MA/AAm and (iii) the molecular weight of the Dex-MA for analogous DS. Moreover, a fixed value of methacrylic groups in the network can be achieved by a combined variation of the Dex-MA/AAm ratio and the DS of the respective Dex-MA. Concerning the biodegradability of the nanogels, the enzymatic cleavability of the dextran based crosslinkers in the network depends on: (i) the total amount of methacrylic units for a fixed Dex-MA/AAm ratio or (ii) the DS of the Dex-MA for a fixed amount of methacrylic units.
Regarding potential release applications, a low initial degree of swelling preventing leakage of embedded compounds during nanogel preparation, purification and storage should be combined with a good biodegradability for the successful release of the payload upon enzymatic cleavage of dextrans. Systematic variation of the parameters described above and investigation of the resulting poly(AAm-co-Dex-MA) nanogels by means of the particle sizes, the initial degree of swelling, the relative crosslinking efficiency and the degradability were conducted.
Synthesis and characterization of p(AAm-co-Dex-MA) nanogels
Nanogel particle formation can be achieved by different approaches including precipitation (co)polymerization from aqueous solutions,1,2 crosslinking of preformed polymers in (mini)emulsions or copolymerisation of crosslinkers and monomers in inverse miniemulsion.35 The partially degradable hybrid nanogels were prepared by free radical copolymerization of acrylamide with dextranmethacrylate in inverse miniemulsion. The respective dispersed phase consisted of an aqueous solution of the monomer, the crosslinker and NaCl. In general, the inverse miniemulsion process is characterized by the presence of an osmotic pressure in the individual droplets counteracting the Laplace pressure. This effect is caused by the addition of NaCl as a highly hydrophilic compound (i.e. an osmotic agent or lipophobe) to the dispersed monomer phase and bears the advantage of the suppression of diffusion between the droplets, therefore rendering each droplet a nanoreactor.31 Compared to precipitationpolymerization based approaches, this method ensures an equal distribution of all monomers, crosslinkers and embedded compounds in every latex particle after polymerization. Especially regarding comparability among the microgels of different feed compositions, it is highly advantageous that the composition of the final polymeric particle resembles the composition of the dispersed phase before polymerization.30The continuous phase consisted of a 1% (w/v) solution of the nonionic surfactant Lubrizol U in cyclohexane. Dispersed phases contained mixtures of Dex-MA and acrylamide dissolved in aqueous 0.5 M NaCl solution with 50% (w/v) monomer + crosslinker. Polymerizations were initiated from the continuous phase by either using V-59 at 70 °C or V-70 at 37 °C. After polymerization (16 h) the resulting particles were repeatedly washed with cyclohexane to remove excess surfactant. The resulting dispersions in cyclohexane represent the nanogel particles in their collapsed (non-swollen) state. In order to investigate the influence of the amount of methacrylic (MA) groups available for crosslinking on the properties of the resulting nanogels, the DS of dextran and the weight ratio of Dex-MA/AAm were varied in the dispersed phase. Table 2 shows the respective compositions of the synthesized nanogels.
In the first attempt, Dex-MAs based on dextran of Mn = 40000 g mol−1 with different DS were used to form nanogels MG-1A to MG-3D. In comparison, gel particles MG-4A to MG-6A were synthesized in cyclohexane using functionalized dextrans of Mn = 6000 g mol−1 with DS and Dex-MA/AAm weight ratios analogous to MG-1A, MG-2A, and MG-3A. The as-synthesized microgels were washed with cyclohexane by centrifugation and redispersion. SEM was used to investigate the particle morphologies. Fig. 1 shows representative micrographs of nanogels MG-1A, MG-2A and MG-3A. The samples were prepared by drop casting from the purified dispersion in cyclohexane on silica wafers and the images depict the areas of high particle concentrations in the regions of the contact line during drying. In all cases, particular structures of well defined spherical morphologies and relatively narrow polydispersities were observed, thus confirming the successful preparation of nanoparticles. The non-swollen gel particles were further investigated in cyclohexane regarding their hydrodynamic diameters by DLS. Fig. 2 shows the obtained values in dependency on the amount of methacrylic groups.
 | ||
| Fig. 1 SEM images of nanogels MG-1A, MG-2A and MG-3A dropcast from cyclohexane dispersions. | ||
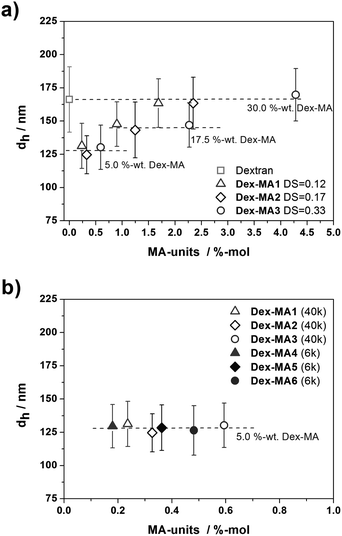 | ||
| Fig. 2 Hydrodynamic diameters in cyclohexane of p(AAm-co-Dex-MA) nanogels containing various amounts of functionalized dextrans of different DS: (a) Mn (Dex-MA) = 40000 g mol−1; and (b) Mn (Dex-MA) = 6000 g mol−1 in comparison to Mn (Dex-MA) = 40000 g mol−1 with similar DS. | ||
As shown in Fig. 2a, for fixed Dex-MA/AAm ratios, the variation of the total amount of MA units available for crosslinking is realized by using dextran methacrylates with different degrees of substitution. It becomes obvious that gel particles of analogous Dex-MA weight contents exhibit similar hydrodynamic diameters, independent of either the degree of substitution or the Dex-MA molecular weight (Fig. 2b). Furthermore, upon increasing the Dex-MA/AAm ratio, an increased particle size results. Sizes of nanogels containing 5 wt% Dex-MA are in the range of 130 nm (Fig. 2a and b), particles containing 17.5 wt% Dex-MA (Fig. 2a) exhibit average hydrodynamic diameters of around 145 nm and in the case of 30 wt% Dex-MA (Fig. 2a) average dh values of around 165 nm were detected. This effect was assigned to the increased viscosities (visual inspection) of the dispersed phases for increased Dex-MA contents prior to polymerization. Miniemulsification is achieved by applying high shear forces by ultrasonication, where a fission and fusion process results in the formation of stable droplets of a narrow size distribution. This process is influenced by the viscosity of the dispersed phase yielding bigger droplets for dispersed phases of higher viscosities.
All nanogels described above were synthesized by initiation with the hydrophobic V-59 from the continuous phase at 70 °C. Regarding the possibility of embedding active compounds in the gel networks during polymerization, mild conditions for the gel formation have to be applied in order to ensure the integrity of the compound to be delivered. Therefore, experiments to investigate different initiation and polymerization conditions were conducted. In a first set of experiments, non-crosslinked reference particles for the degradation studies were synthesized by inverse miniemulsion polymerization of acrylamide in the presence of non-functionalized dextrans of Mn = 40000 g mol−1. MG-8Aa was polymerized by initiation with V-59 at 70 °C from the continuous phase analogous to particles MG-1A to MG-3D. The resulting particles exhibited a similar hydrodynamic diameter of around 165 nm in cyclohexane. Particle formation reactions with unfunctionalized dextran at lower temperatures were carried out by using the oil soluble V-70 for initiation from the continuous phase at 37 °C (MG-8Ab), the water-soluble VA-044 for initiation from the dispersed phase at 45 °C (MG-8Ad) and the water-soluble KPS for initiation from the dispersed phase upon adding tetramethylethylenediamine (TEMED) to the continuous phase at room temperature (MG-8Ac). In the case of the initiation by KPS/TEMED no stable dispersions were obtained whereas utilization of either V-70 or VA-044 resulted without exception in stable dispersions of dextran-containing PAAm particles in cyclohexane. Resulting hydrodynamic diameters were in the same range (around 165 nm) as those of particles with 30 wt% of functionalized dextrans (see Fig. 3 for initiation with V-70, data for initiation with VA-044 are not shown).
 | ||
| Fig. 3 Hydrodynamic diameters in cyclohexane of p(AAm-co-Dex-MA) nanogels containing various amounts of functionalized dextranes (Mn = 40000 g mol−1) of different DS. Polymerizations were carried out at 37 °C using V-70 as an initiator. | ||
Since initiation with V-70 required the lowest polymerization temperatures, the respective conditions were used to synthesize MG-1Ab to MG-7Ab p(AAm-co-Dex-MA) microgels with weight contents of 30 wt% Dex-MA of different DS analogous to MG-1A to MG-3D particles. As shown in Fig. 3, resulting particles in cyclohexane exhibited hydrodynamic diameters of around 165 nm similar to the nanogels obtained from polymerizations at 70 °C.
Investigations on further increasing the amount of Dex-MA in the nanogels revealed that at very high Dex-MA contents (60 wt%), increasing polydispersity and ill-defined morphologies resulted (see Fig. S1 in the ESI†).
In this case, a dramatically increased viscosity (visual inspection) hinders homogenization of the pre-emulsion by ultrasonication. In addition, the high amount of the functionalized dextran is assumed to influence the stability of the droplets due to the interfacially active character of the amphiphilic Dex-MA. Since attempts to form nanogels from pure Dex-MA1 (MG-1Db) did not yield stable miniemulsions, the presence of a second comonomer seems to be a crucial requirement for the formation of enzymatically degradable gel particles by inverse miniemulsion polymerization.
Summarizing the preparation of p(AAm-co-Dex-MA) nanogels, it can be concluded that the free radical copolymerization in inverse miniemulsion allows for the formation of stable dispersions consisting of well defined spherical particles. The applied synthetic approach is highly versatile with respect to the polymerization conditions (i.e. initiating mechanism and temperature). Moreover, increasing the Dex-MA/AAm feed ratio was found to result in stable dispersions for Dex-MA contents up to 30 wt%. Resulting particle sizes were found to depend on the amount of Dex-MA in the dispersed phase prior to polymerization and were in the nanometre size range. The obtained nanogels were then further investigated with regard to their respective gel properties by transferring them to aqueous media.
Investigations on the relative crosslinking efficiency and the swelling behavior of p(AAm-co-Dex-MA) nanogels in water
Subsequent to particle purification by washing with cyclohexane, all dispersions were freeze-dried yielding the nanogels as white powders. In order to investigate the gel properties of the prepared p(AAm-co-Dex-MA) hybrid particles, dispersions in aqueous medium were obtained by simply dispersing and swelling the freeze-dried samples in water. Purification was achieved by repeated centrifugation and redispersion in water.The resulting dispersions of the swollen nanogels were stable without any additional surfactant. This effect is assumed to occur due to sterical stabilization by hydrated dangling chains of the swollen outer particle layer. Determination of the hydrodynamic diameters by DLS allowed calculation of the degrees of swelling (DGSs), or the swelling ratios, which were calculated as DGS = Vswollen/Vnon-swollen. The washed nanogel dispersions were assumed to consist solely of p(AAm-co-Dex-MA) networks, i.e. the gel content of the crosslinking copolymerization. The combined supernatants of the washing steps represent the sol content consisting of unreacted monomers, non-crosslinked polymer chains and non-incorporated oligomers. Freeze drying of both fractions was followed by gravimetric analysis and afforded the sol/gel content for every sample.
Generally, it is of high interest to determine the final composition of the purified nanogels in order to be able to specify the crosslinking efficiency and density as an important characteristic. However, the large variety of compounds (oligomers, monomers, crosslinkers, and surfactants) in the separated sol hindered an accurate quantification of the incorporated amount of crosslinker in the network by spectroscopic methods. While gravimetric investigations on the sol/gel content in combination with the measured degrees of swelling did not permit a quantitative evaluation of the crosslinking densities or an absolute conclusion regarding the inner morphologies as well, they allowed the expedient comparison of important properties, such as the initial swelling ratios and the relative crosslinking efficiencies. Those parameters strongly depend on the amount and distribution of crosslinking points—i.e.methacrylategroups—in the network and can be adjusted by the variation of: (i) the ratio of Dex-MA/AAm for a fixed DS of Dex-MA; (ii) the DS of Dex-MA for a fixed ratio of Dex-MA/AAm; and (iii) the molecular weight of the Dex-MA for analogous DS. Moreover, a fixed value of methacrylic groups in the network can be achieved by a combined variation of the Dex-MA/AAm ratios and the DS of the respective Dex-MA.
Fig. 4 shows the resulting DGSs and sol contents for nanogels polymerized at 70 °C with various amounts of Dex-MA1 (Fig. 4a), Dex-MA2 (Fig. 4b) and Dex-MA3 (Fig. 4c), thus representing three different classes of nanogels. Each class is characterized by a particular degree of substitution of the used Dex-MA. The amount of methacrylategroups was varied in every class by changing the ratio of Dex-MA/AAm. It becomes obvious that both DGS and sol content decrease with an increasing amount of Dex-MA, i.e. a higher number of methacrylategroups, thus indicating enhanced crosslinking efficiency.
 | ||
| Fig. 4 DGS and sol contents of p(AAm-co-Dex-MA) nanogels (prepared at 70 °C) in dependency on the amount of polymerizable MA units. (a) Nanogels polymerized with various amounts of Dex-MA1; (b) nanogels polymerized with various amounts of Dex-MA2; (c) nanogels polymerized with various amounts of Dex-MA3; and (d) comparison of DGS of nanogels polymerized with various amounts of Dex-MA1, Dex-MA2, and Dex-MA3. | ||
As shown in Fig. 4d, the same effect can be observed by comparing the DGSs of nanogels polymerized with the same Dex-MA/AAm ratio but containing Dex-MA crosslinkers with different degrees of substitution. Here as well, higher amounts of methacrylate units (realized by increased DS) result in lower initial degrees of swelling and consequently increased crosslinking efficiencies. Independent of the degree of substitution, all nanogels polymerized with 30 wt% of various types of Dex-MA exhibit DGS values in the same order of magnitude. It is concluded that the swelling behavior not only depends on the amount of polymerizable methacrylate units but also on the Dex-MA/AAm ratio. This suggests that also the amount of dextran chains in the network strongly influences the swelling behavior due to different interaction parameters of the hydrophobically functionalized dextran and polyacrylamide with water. The composition of the hybrid nanogels therefore represents an additional crucial parameter.
Fig. 5 shows the values for the DGS and sol contents for p(AAm-co-Dex-MA) nanogels containing 30 wt% Dex-MA of various DS (Dex-MA1, Dex-MA2, Dex-MA3 and Dex-MA4) polymerized at 37 °C initiated with V-70 from the oil phase.
 | ||
| Fig. 5 Characterization of the gel properties of p(AAm-co-Dex-MA) nanogels polymerized at 37 °C with 30 wt% of Dex-MAs with various degrees of substitution: DGS and sol contents in dependency on the amount of polymerizable MA units. | ||
Analogous to nanogels polymerized at 70 °C, 1.69 mol% of MA units correspond to the number of the MA moieties introduced with 30 wt% of Dex-MA1. Analogously 2.34 mol% correspond to Dex-MA2 and 4.28 mol% to Dex-MA3. In comparison to MG-1C, MG-2D, and MG-3D, all samples exhibit similar initial swelling ratios and only slightly increased sol contents. Therefore, an equally effective crosslinking polymerization (network formation) is assumed. Regarding a potential embedding of active substances into the gels, this versatility of polymerization conditions represents a big advantage. Whereas in the case of MG-7Ab microgels (0.72 mol% of MA-units by utilization of 30 wt% of Dex-MA7) a dramatic increase in the sol content is observed, the initial degree of swelling does not vary significantly from the values for the microgels containing similar amounts of dextran methacrylates of higher degrees of substitution. As mentioned above, the DGS of the particles is assumed to not only be affected by the crosslinking density but also depends on the Dex-MA/AAm ratio. It is assumed that the swelling behavior of MG-7Ab microgels is dominated by the high amount of Dex-MA rather than the comparatively low crosslinking efficiency.
In general, the total amount of crosslinking points in a p(AAm-co-Dex-MA) gel can be varied by either changing the Dex-MA/AAm ratio for a Dex-MA of a fixed DS or by changing the DS of the dextranmethacrylate for a fixed Dex-MA/AAm ratio. Another highly important factor which governs the properties of the hybrid microgels is the distribution of crosslinking points in the network. In order to investigate this parameter, microgels containing equal amounts of 1.5 mol% methacrylate units but varying in the Dex-MA/AAm ratio were prepared using dextran methacrylates of different degrees of substitution. Similar contents of MA-units were realized by increasing the Dex-MA/AAm ratio with decreasing DS of the Dex-MA used. The resulting microgels were investigated with respect to their initial degree of swelling and their sol content. Fig. 6 shows the obtained values of DGS in dependency on the Dex-MA/AAm ratio together with the dependency of the sol contents on the degree of substitution of the Dex-MA used.
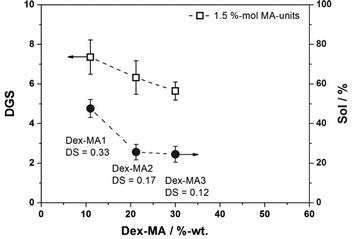 | ||
| Fig. 6 Investigations on the gel properties of p(AAm-co-Dex-MA) nanogels synthesized with a fixed amount of 1.5 mol% of methacryl units by changing the Dex-MA/AAm ratio and the DS of the Dex-MA: (a) DGS in dependency on the amount of different Dex-MAs; and (b) sol contents in dependency on the amount of different Dex-MAs. | ||
As can be seen, increasing the amount of Dex-MA simultaneously to decreasing the degree of substitution leads to reduced initial degrees of swelling and lower sol contents. A reduced sol content can be assigned to a better crosslinking efficiency which is assumed to be caused by a better distribution of the crosslinking points in the gel network. The DGSs exhibit a similar behavior (i.e. microgels polymerized with a higher amount of Dex-MA of a lower degree of substitution swell less). However, in this case the distribution of crosslinking points is not the single factor influencing the swellability of the hybrid gels. As mentioned above, the Dex-MA/AAm ratio also affects the swelling properties of the hybrid material due to different interactions of the modified dextran and polyacrylamide chains with water.
In order to further investigate the influence of the distribution of crosslinking points in the gel network, nanogel particles polymerized with dextran methacrylates based on dextran (6k) were compared to the samples containing Dex-MA (40k). In this experiment 6k Dex-MA crosslinkers were synthesized to exhibit similar degrees of substitution as the 40k analogues. Fig. 7 shows the DGS values and sol contents of the resulting microgels polymerized with each 5 wt% of different Dex-MAs of either 6k or 40k. As can be seen, both the initial degrees of swelling and the sol contents are reduced to some extent for the nanogels containing the respective low molecular weight Dex-MA crosslinkers, therefore indicating a slightly better distribution of crosslinks in the gel network.
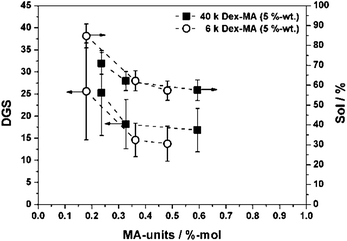 | ||
| Fig. 7 Investigations on the gel properties of p(AAm-co-Dex-MA) nanogels synthesized with 5 wt% of Dex-MAs of different DS: comparison of DGS and sol contents for 40k Dex-MAs with 6k Dex-MAs of similar degrees of substitution. | ||
In summary, free radical copolymerization of acrylamide with dextran methacrylates as biodegradable crosslinkers in inverse miniemulsion was found to be highly versatile with regard to the polymerization conditions (initiation mechanism and temperature) and the Dex-MA/AAm feed ratio. Increasing the amount of polymerizable methacrylate units available for crosslinking was carried out by either increasing the Dex-MA/AAm feed ratio for a fixed DS of Dex-MA or by increasing the DS of Dex-MA for a fixed Dex-MA/AAm feed ratio. As expected, a higher amount of MA units correlates with higher crosslinking efficiency as determined by low initial degrees of swelling and low sol contents.
Enzymatic degradation experiments
In order to investigate the behavior of the p(AAm-co-Dex-MA) nanogels in the presence of dextranase, aqueous particle dispersions were incubated with the enzymedextranase at 37 °C for 24 h. The dispersions contained 0.0625 wt% of purified gels after removal of the sol fraction. Particle degradation was monitored by turbidity measurements in transmission. In general, the turbidity τ(t) can be obtained by calculating the negative logarithm of the intensity ratio of transmitted light of the sample It to transmitted light of pure waterI0 as τ(t) = −ln (It/I0). The relative turbidity values were then calculated as the ratio of the time dependent turbidity after dextranase addition to the starting values, τ(t = 0), without added enzyme as τrel(t) = τ(t)/τ(t = 0) × 100 in percent.Keeping in mind potential applications of the microgels for release applications, a low initial degree of swelling represents a crucial parameter to prevent leakage of embedded compounds. Therefore, particles with high crosslinking efficiency and low sol content are of special interest. As discussed above, for particles containing 30 wt% of Dex-MA this criterion is fulfilled for all dextrans with different degrees of substitution. The relative turbidity curves shown in Fig. 8a thus enable the investigation of the degradability of microgels polymerized at 70 °C in dependency on the total amount of crosslinking points and the DS of the used Dex-MAs for a fixed Dex-MA/AAm ratio. The investigated nanogels MG-1C, MG-2D and MG-3D contained increasing numbers of crosslinking points due to an increasing DS of the Dex-MA used. Whereas the turbidity of MG-3D nanogels containing 4.48 mol% of MA-units (30 wt% Dex-MA3; DS = 0.33) was not influenced by the addition of dextranase, nanogels MG-1C with 1.72 mol% (30 wt% Dex-MA1; DS = 0.12) and MG-2D with 2.53 mol% (30 wt% Dex-MA2; DS = 0.17) of crosslinking points showed a pronounced decrease in turbidity upon 24 h incubation down to 47% and 65% respectively.
 | ||
| Fig. 8 Enzymatic degradation of p(AAm-co-Dex-MA) nanogels with various amounts of methacrylate units for a fixed Dex-MA content of 30 wt% (polymerized at 70 °C): (a) time dependent turbidity measurements; and (b) comparison of the degrees of swelling before and after the 24 h treatment with dextranase. | ||
This effect is based on the cleavage of dextran chains between the crosslinking points, covalently linking Dex-MA molecules to PAAm chains. The resulting decrease in crosslinking density causes the particle swelling and their partial dissolution in water which is assumed to follow a complex mechanism. In the early stages of the enzymatic treatment, the cleavage of dextran chains induces a swelling of the particles and the formation of hydrogel spheres with increased diameters. Further incubation possibly leads then to the diffusion of hydrophilic chains and/or smaller fragments out of the particles. Turbidity measurements were performed since they can take both phenomena into account. Regarding the increase of the swelling ratio, the corresponding loosened network structure is characterized by a decreased difference of the refractive indices of polymer and solvent. This is known to reduce the intensity of scattered light and therefore the relative turbidity.37,38 In addition, the diffusion of free hydrophilic chains out of the particles represents particle degradation yielding fragments of decreased sizes. As these smaller fragments are characterized by a decreased scattering intensity as well, the relative turbidity is decreased accordingly. Even though the performed turbidity measurements therefore allow convenient monitoring of the nanogel swelling and degradation and determination of the respective time scales, it has to be kept in mind that due to the superposition of the described two effects the correlation between degradation and turbidity is not linear. In order to get a more profound insight into the particle swelling, the described method was combined with measurements of particle sizes after enzymatic degradation. The latter yielded the DGSs which are shown in Fig. 8b in comparison to the non-degraded particles. As can be seen, a decrease in turbidity is accompanied by an increased degree of swelling of the gel particles thus confirming successful cleavage of crosslinkers. The performed measurements clearly exhibit a dependency of the degradation profile on the amount of methacrylate units and the degree of substitution of the used Dex-MA crosslinkers. While it becomes obvious that a higher amount of crosslinking points results in a less pronounced degradation (Fig. 8), it could not be determined whether this effect is based on a smaller mesh size of the network in general (the total amount of crosslinking points) or the distribution of the methacrylate units along the dextran chains (the degree of substitution of the respective Dex-MA). The first parameter is assumed to influence the degradation by a decreased accessibility of dextran chains in the network due to hindered diffusion of the enzyme molecules into the network. A higher DS of Dex-MA as the second parameter could interfere with a successful dextran chain cleavage by sterical hindrance due to the connected PAAm chains.
To test these assumptions, microgels with similar contents of methacrylate units but various amounts of dextran methacrylates of different degrees of substitution were investigated with respect to their degradation behavior. Fig. 9a shows the resulting turbidity curves and Fig. 9b the respective DGS obtained from particle sizes determined by DLS.
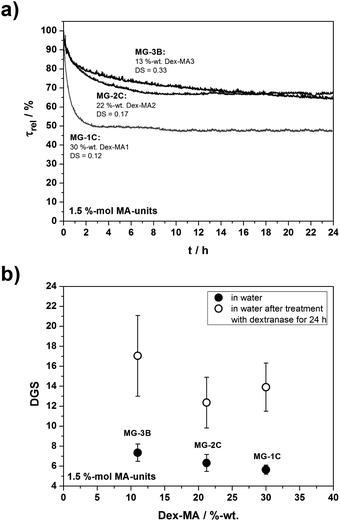 | ||
| Fig. 9 Enzymatic degradation of p(AAm-co-Dex-MA) nanogels with various contents of Dex-MAs with different DS for a fixed amount of methacrylate units of 1.5 mol%: (a) time-dependent turbidity measurements; and (b) comparison of the degrees of swelling before and after the 24 h treatment with dextranase. | ||
Turbidity measurements clearly demonstrate a decrease in the optical density for all samples which is assigned to network degradation upon cleavage of dextran chains. The DGS after enzymatic treatment indicates a pronounced increase of the swelling ratio for all degrees of substitution of Dex-MA.
Comparing the differences in the DGS after enzymatic treatment of MG-3B and MG-2C to the data obtained for microgels MG-3D and MG-2D (Fig. 8b) containing more Dex-MA with the same DS, it is observed that decreasing the amount of methacrylate units (i.e. the amount of the respective Dex-MA) results in an enhanced degradability. While for MG-3D containing 30 wt% of Dex-MA3 only a negligible increase in DGS after treatment with dextranase was observed, the DGS for MG-3B containing 12 wt% of Dex-MA3 changed from the initial value of DGS = 7.4 to DGS = 17.
A similar behavior was observed for microgels prepared with Dex-MA2. The measured increase of DGS = 6.3 to DGS = 12.3 after enzymatic cleavage for MG-2C (containing 22 wt% of Dex-MA2) is much higher than the difference of DGS = 5.5 to DGS = 8.5 obtained for MG-2D (containing 30 wt% of Dex-MA2). Therefore, it is assumed that one crucial parameter for successful microgel degradation is the total amount of crosslinking points (MA-units) in the gel. This observation can be interpreted by a reduced accessibility of the dextran chains for dextranase with increasing amounts of crosslinking points. By investigating the enzymatic degradation of crosslinked dextran methacrylates, Franssen et al. proposed that the degradability is reduced due to the presence of interpenetrating networks and the fact that the dextran chains are severely strained.26 Moreover, the diffusion of the enzyme in a more crosslinked network (i.e. more Dex-MA) is decreased as a result of a screening effect. A high density of crosslinking points correlates with small mesh sizes and would give access to the enzymatic degradation of the outer particle sphere due to an enhanced accessibility of the surface. In contrast, the relatively large size of the enzyme (44 kDa) is assumed to hinder its diffusion into the particle core due to comparably small mesh sizes thus resulting in incomplete particle degradation.
Besides the described screening effect, the interaction of the functionalized dextrans with the active sites of the enzyme can play an important role as well. As it is known from the literature,25dextranase is capable of hydrolyzing a glycosidic bond between a substituted and an unsubstituted glucopyranose unit of dextranmethacrylate in solution. In contrast, bonds between two adjacent functionalized residues cannot be cleaved. Regarding the synthesis of Dex-MA crosslinkers, the functionalization with methacrylategroups is assumed to result in a statistical distribution of the latter along the dextran backbone. Thus, the probability of obtaining chain segments with two substituted glucopyranose units next to each other increases with increasing DS. In the resulting hydrogel matrix those segments covalently connect PAAm chains forming the network. Since degradation of those structural elements is hindered, incomplete particle degradation occurs.
Another important observation is the dependency of the degradation rate of the microgels on the DS of the Dex-MAs (for a fixed amount of MA-units in the gel). As can be seen in Fig. 9a, the rate of particle disintegration (measured by turbidity) decreases for an increasing degree of substitution of dextran. This effect can be assigned to a changed interaction of the Dex-MA with the active sites of the enzyme. Investigations made in the group of Hennink proposed that dextran methacrylates with a low degree of substitution can easily be cleaved by dextranase yielding isomaltose and methacrylated isomaltotriose as the main degradation products.25,26 In this case, the long unsubstituted segments of the dextran chains between two functionalized glucopyranose units are assumed to exhibit enough conformational freedom to fold correctly in the binding site of dextranase. Thus degradation by even multiple scissions of these segments results in degradation profiles similar to native dextran. In contrast, increasing the DS results in shorter unsubstituted chain segments which are restricted in their conformational freedom due to adjacent methacrylated glucopyranose units. Even though these chain segments can still bind to the active sites of the enzyme, the affinity and the corresponding degradation rate is decreased. As the DS of the synthesized crosslinkers increases from Dex-MA1 to Dex-MA3, the corresponding decreases in the degradation rates are also assumed to be a result of the reduced affinity of Dex-MA to the binding sites of dextranase. This effect is even enhanced in the p(AAm-co-Dex-MA) nanogels since PAAm chains grafted from the dextran molecules cause an additional sterical hindrance with respect to the interaction with the active sites of the enzyme.
In another attempt, microgels MG-8Aa containing 30 wt% of unfunctionalized dextran were transferred to aqueous medium in order to investigate the dissolution behavior of non-crosslinked particles. Even though no crosslinking methacrylate functionalities were present in this sample, a turbidity of 13% relative to the initial turbidity of MG-1C microgels was observed. The detected value did not change upon 24 h of incubation with dextranase (see ESI†). Therefore it is assumed that, in addition to the above-discussed limitations of the enzymatic degradation, complete particle dissolution is also restricted by physical crosslinks due to entanglement of PAAm chains in the gels (visual inspection of pure PAAmnanoparticles).
Summarizing the statements made above, the observed degradation profiles are assumed to be based on a combination of five different parameters: (i) an increasing amount of crosslinking methacrylategroups reduces the accessibility of dextran chains for the enzyme due to comparably small mesh sizes of the network; (ii) hindered cleavage of two neighboring substituted glucopyranose residues results in incomplete particle degradation and increases with the DS; (iii) for increasing the DS the affinity of the Dex-MA to the binding site of the enzyme is reduced thus resulting in decreased degradation rates; (iv) PAAm chains grafted from the dextran backbone reduce the interaction of the polysaccharide with the enzyme due to sterical hindrance; and (v) physical crosslinks due to entanglement of PAAm chains hinder complete particle dissolution, independent of the enzymatic degradability. Nevertheless, the observed increase in particle volumes after treatment with dextranase holds great potential for the release of functional compounds from the network, triggered by increasing mesh sizes.
In the last experiment the influence of the polymerization conditions on the degradability of the microgels was investigated. As discussed above, potential release applications require mild polymerization conditions in order to prevent embedded functional compounds from damage. Microgels MG-1Ab to MG-3Ab were polymerized at ambient temperatures of 37 °C to facilitate a potential embedding of sensitive functional compounds.
As their swelling behavior was found to be similar to the gels polymerized at 70 °C it is assumed that also the degradation profiles do not vary significantly from those of the respective microgels MG-1C, MG-2D, and MG-3D. In Fig. 10 the time-dependent turbidity measurements of the enzymatic treatment are shown as well as the DGS before and after 24 h of incubation with dextranase.
 | ||
| Fig. 10 Enzymatic degradation of p(AAm-co-Dex-MA) nanogels with various amounts of methacrylate units for a fixed Dex-MA content of 30 wt% polymerized at 37 °C: (a) time-dependent turbidity measurements; and (b) comparison of the degrees of swelling before and after the 24 h treatment with dextranase. | ||
Compared to microgels of similar compositions polymerized at 70 °C, no significant deviations of the degradation profiles of the samples polymerized at 37 °C could be observed by turbidity measurements. In addition, the DGS after enzymatic treatment exhibits comparable increases in particle size upon dextran chain cleavage. Regarding potential release applications, MG-1Ab nanogels are the most promising candidates for a combination of reasons: (i) the low polymerization temperature gives rise to the potential embedding of sensitive compounds; (ii) the low initial degree of swelling and the low sol content point towards a high crosslinking efficiency and density, thus reducing leakage of potential embedded substances; and (iii) enzymatic degradation proceeds comparably fast and is characterized by a substantial increase in DGS, hence enabling the release of a payload upon the treatment with dextranase. As a result, p(AAm-co-Dex-MA) gel particles represent a highly interesting class of nano-scaled hybrid materials, which are characterized by their partial biodegradability and simple and versatile processing routes.
Conclusions
In conclusion, free radical copolymerization of acrylamide with dextran methacrylates as biodegradable crosslinkers in inverse miniemulsion represents a facile approach to p(AAm-co-Dex-MA) gel particles in the nanometre size range. The investigated preparation method was found to be highly versatile with regard to the polymerization conditions (initiation mechanism and temperature) and the Dex-MA/AAm feed ratio. Systematic investigations on increasing the amount of polymerizable methacrylate units available for crosslinking were carried out by either increasing the Dex-MA/AAm feed ratio for a fixed DS of Dex-MA or by increasing the DS of Dex-MA for a fixed Dex-MA/AAm feed ratio. As expected, a higher amount of MA-units correlates with a higher crosslinking efficiency as determined by low initial degrees of swelling and low sol contents. Enzymatic degradation profiles of the nanogels were examined by turbidity measurements and DLS. The investigations revealed a dependency of the degree and the rate of degradation on the crosslinking density (MA-units incorporated) of the gels and the DS of the used Dex-MAs.The described adjustment of the gel properties by variation of the synthetic parameters revealed p(AAm-co-Dex-MA) nanogels as highly interesting candidates for potential release systems in the nanometre size range. In this context, the combination of an ambient polymerization temperature with high crosslinking densities—as determined by low initial degrees of swelling and the low sol contents—gives rise to the potential embedding of sensitive compounds. Furthermore, enzymatic degradation proceeds comparably fast and is characterized by a substantial increase in DGS, hence enabling the release of a payload upon the treatment with dextranase.
As a result, p(AAm-co-Dex-MA) gel particles represent an extremely attractive class of nano-scaled hybrid materials, which are characterized by their partial biodegradability and simple and versatile processing routes. Moreover, their sensitivity towards dextranase is of great advantage for release applications since it is an orthogonal approach to widely used pH- or temperature-sensitive microgels.
Acknowledgements
D. K. acknowledges the International Max Planck Research School (IMPRS) for financial support.Notes and references
- M. Das, H. Zhang and E. Kumacheva, Annu. Rev. Mater. Res., 2006, 36, 117–142 CrossRef CAS.
- S. Nayak and L. A. Lyon, Angew. Chem., Int. Ed., 2005, 44(47), 7686–7708 CrossRef CAS.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33(4), 448–477 CrossRef CAS.
- N. A. Peppas and W. Leobandung, J. Biomater. Sci., Polym. Ed., 2004, 15(2), 125–144 CrossRef CAS.
- W. T. Wu, N. Mitra, E. C. Y. Yan and S. Q. Zhou, ACS Nano, 2010, 4(8), 4831–4839 CrossRef CAS.
- H. Kawaguchi and K. Fujimoto, Bioseparation, 1998, 7(4/5), 253–258 CrossRef CAS.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19(5), 1062–1069 CrossRef CAS.
- M. Karg, I. Pastoriza-Santos, J. Perez-Juste, T. Hellweg and L. M. Liz-Marzan, Small, 2007, 3, 1222–1229 CrossRef CAS.
- J. D. Debord and L. A. Lyon, J. Phys. Chem. B, 2000, 104(27), 6327–6331 CrossRef CAS.
- G. R. Hendrickson, M. H. Smith, A. B. South and L. A. Lyon, Adv. Funct. Mater., 2010, 20(11), 1697–1712 CrossRef CAS.
- C. C. Lin and A. T. Metters, Adv. Drug Delivery Rev., 2006, 58(12–13), 1379–1408 CrossRef CAS.
- S. R. Deka, A. Quarta, R. Di Corato, A. Falqui, L. Manna, R. Cingolani and T. Pellegrino, Langmuir, 2010, 26(12), 10315–10324 CrossRef CAS.
- G. M. Eichenbaum, P. F. Kiser, A. V. Dobrynin, S. A. Simon and D. Needham, Macromolecules, 1999, 32(15), 4867–4878 CrossRef CAS.
- C. Echeverria, D. Lopez and C. Mijangos, Macromolecules, 2009, 42(22), 9118–9123 CrossRef CAS.
- S. Nayak, H. Lee, J. Chmielewski and L. A. Lyon, J. Am. Chem. Soc., 2004, 126(33), 10258–10259 CrossRef CAS.
- G. M. Eichenbaum, P. F. Kiser, D. Shah, S. A. Simon and D. Needham, Macromolecules, 1999, 32(26), 8996–9006 CrossRef CAS.
- T. Hoare and R. Pelton, Macromolecules, 2004, 37(7), 2544–2550 CrossRef CAS.
- V. Bulmus, Y. Chan, Q. Nguyen and H. L. Tran, Macromol. Biosci., 2007, 7(4), 446–455 CrossRef CAS.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. J. Frechet, J. Am. Chem. Soc., 2002, 124(42), 12398–12399 CrossRef CAS.
- S. M. Standley, Y. J. Kwon, N. Murthy, J. Kunisawa, N. Shastri, S. J. Guillaudeu, L. Lau and J. M. J. Frechet, Bioconjugate Chem., 2004, 15(6), 1281–1288 CrossRef CAS.
- D. Klinger and K. Landfester, Soft Matter, 2011, 7(4), 1426–1440 RSC.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324(5923), 59–63 CrossRef CAS.
- O. Franssen, O. P. Vos and W. E. Hennink, J. Controlled Release, 1997, 44, 237–245 CrossRef CAS.
- N. A. Peppas, Y. Huang, M. Torres-Lugo, J. H. Ward and J. Zhang, Annu. Rev. Biomed. Eng., 2000, 2, 9–29 CrossRef CAS.
- O. Franssen, R. D. vanOoijen, D. deBoer, R. A. A. Maes, J. N. Herron and W. E. Hennink, Macromolecules, 1997, 30(24), 7408–7413 CrossRef CAS.
- O. Franssen, R. D. van Ooijen, D. de Boer, R. A. A. Maes and W. E. Hennink, Macromolecules, 1999, 32(9), 2896–2902 CrossRef CAS.
- S. Abdurrahmanoglu and Y. Firat, J. Appl. Polym. Sci., 2007, 106, 3565–3570 CrossRef CAS.
- O. Franssen, R. J. H. Stenekes and W. E. Hennink, J. Controlled Release, 1999, 59(2), 219–228 CrossRef CAS.
- O. Franssen, L. Vandervennet, P. Roders and W. E. Hennink, J. Controlled Release, 1999, 60(2–3), 211–221 CrossRef CAS.
- K. Landfester, M. Willert and M. Antonietti, Macromolecules, 2000, 33(7), 2370–2376 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48(25), 4488–4507 CrossRef CAS.
- D. Crespy, M. Stark, C. Hoffmann-Richter, U. Ziener and K. Landfester, Macromolecules, 2007, 40(9), 3122–3135 CrossRef CAS.
- R. Schiller, C. K. Weiss, J. Geserick, N. Husing and K. Landfester, Chem. Mater., 2009, 21(21), 5088–5098 CrossRef CAS.
- M. Maier, N. Kotman, C. Friedrichs, J. Andrieu, M. Wagner, R. Graf, W. Strauss, V. Mailänder, C. Weiss and K. Landfester, Macromolecules, 2011, 44, 6258–6267 CrossRef CAS.
- K. Landfester and A. Musyanovych, Chemical Design of Responsive Microgels, 2010, vol. 234, 39–63 Search PubMed.
- S.-H. Kim and C.-C. Chu, J. Biomed. Mater. Res., 2000, 49(4), 517–527 CrossRef CAS.
- M. D. Lecner, J. Serb. Chem. Soc., 2005, 70(3), 361–369 CrossRef.
- N. Al-Manasir, K. Z. Zhu, A. L. Kjoniksen, K. D. Knudsen, G. Karlsson and B. Nystrom, J. Phys. Chem. B, 2009, 113(32), 11115–11123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM investigations on p(AAm-co-Dex-MA) nanogels prepared with 60 wt% of Dex-MA1 and time dependent turbidity measurements of MG-8AaPAAm particles in comparison to MG-1C nanogels upon the treatment with dextranase. See DOI: 10.1039/c1py00415h |
| This journal is © The Royal Society of Chemistry 2012 |