Synthesis of 3,3,3-trifluoropropene telomers and their modification into fluorosurfactants
Frédéric
Boschet
*a,
Georgi
Kostov
a,
Bruno
Ameduri
*a,
Andrew
Jackson
b and
Bernard
Boutevin
a
aInstitut Charles Gerhardt, Ingénierie et Architectures Macromoléculaires, UMR CNRS 5253, Ecole Nationale Supérieure de Chimie de Montpellier, 8 Rue de l'Ecole Normale, 34296, Montpellier, France. E-mail: bruno.ameduri@enscm.fr; frederic.boschet@enscm.fr
bChemtura, 1801 U.S. Highway 52 West, West Lafayette, Indiana 47906-2200, USA
First published on 17th November 2011
Abstract
Original fluorinated surfactants based on 3,3,3-trifluoropropene (TFP) as alternatives to perfluorooctanoic acid (PFOA) were synthesized in three to five straightforward steps in good overall yields. First, the radical or thermal telomerization of TFP in the presence of perfluoroisopropyl iodide as the chain transfer agent led to various TFP telomers of different molecular weights. They were further chemically modified into various ionic and non-ionic surfactants. To obtain anionic surfactants, TFP telomers bearing iodine end-group was converted into allylic group on which thioglycolic acid was added under a radical or photochemical process. Non-ionic surfactants were obtained by esterification of the anionic acid with poly(ethylene glycol)monomethylether. Cationic surfactants were obtained by ethylene end-capping of the TFP telomers followed by a nucleophilic substitution by either triethylamine or pyridine. All surfactants showed good inertness to bases and acids, and satisfactory surface properties. They exhibit interesting critical micellar concentration values comparable to that of PFOA (0.06, 4.10, and 3.20 versus 3.00 g L−1).
Introduction
Fluorinated polymers exhibit exceptional properties leading to high tech applications.1 Most of these specialty polymers are prepared by radical (co)polymerization of fluorinated monomers in aqueous media (suspension or emulsion polymerizations).1–3 However, these hydrophobic monomers require the use of a surfactant (preferentially fluorinated) to favor the incorporation of these unsaturated derivatives into the micelles, and to enable the synthesis of fluoropolymers. It is well known that most efficient surfactants involved in such processes are amphiphilic,4 and are composed of a polar head and one or more perfluorinated chains,5,6 mainly C7F15 or C8F17,7–12 such as perfluorooctanoic acid C7F15CO2H (PFOA) or the perfluorooctane sulfonic acid, C8F17SO3H (PFOS). These fluorosurfactants are involved in more than 200 applications including detergents, firefighting foams, coatings, cosmetics, emulsifiers, adhesives, electroplating, antifogging, and antistatic agents…13 However, recent studies directed by the U.S. Environmental Protection Agency (EPA)14 showed that these surfactants are bioaccumulable13,15,16 (due to the perfluorinated chain that is too stable, for example perfluorooctane sulfonic acid's half-life is ca. 3.26 years in human blood17,18), toxic, and persistent,19,20 and that decreasing their production by as much as 95% was scheduled by the end of 2010, followed by definitive stop of their production by 2015. Numerous analyses13,21 across the planet have demonstrated that these compounds are still present in many rivers,22 seas,23 and even on the ice cap21 (moreover, traces were found in the livers of polar bears), and as an example, according to a German study, the Bavarian Environmental Agency15 has shown that both these perfluorinated surfactants can be found at concentrations of 22.3 mg L−1 (for PFOS) and 6.8 mg L−1 (for PFOA) in the plasma of the studied population.At that point, the main companies (Arkema, Asahi Glass, Ciba, Clariant, Daikin, 3M/Dyneon, Dupont and Solvay Solexis) involved in the production of derivatives and fluorinated polymers have been joining efforts since 2005 in a consortium called the “Stewardship Program”24 that aims to reflect the future, and to seek for new alternatives to PFOA and PFOS. Indeed, these PFOA or PFOS have been synthesized from tetrafluoroethylene (TFE) telomers.1,8 Although short perfluorinated chains (C4)25 or oligo(hexafluoropropylene oxide)26–29-based surfactants have already led to original non-bioaccumulable alternatives to PFOA, it was of interest to find new derivatives.
The purpose of this article is to deal with the synthesis of cationic, anionic and non-ionic surfactants by simple and efficient modifications of fluorinated telomers obtained by iodine transfer polymerization. The concept is illustrated using the radical telomerization of 3,3,3-trifluoropropene and the chemical modifications of the resulting TFP telomers in 2–4 steps to obtain the various types of surfactants.
The synthesis of 3,3,3-trifluoropropene (TFP) was pioneered by Haszeldine43 in the early 1950s from dehydroiodination of CF3–CH2–CH2I and is now commercially available (from Great Lakes-Chemtura now DuPont). TFP has been used in telomerization with several telogens (Table 1) using various type of initiation: redox, UV, γ-radiation, thermal, or peroxides. Results show that perfluoroalkyl iodides RFI are the best telogens.1
| Telogen | Initiation | Product obtained | Ref. |
|---|---|---|---|
| a BPO: dibenzoyl peroxide, DTBP: di-tert-butyl peroxide. | |||
| H3COCOC(CH3)2H | Peroxides | H3COCOC(CH3)2, [CH2CH(CF3)]nH | 30 |
| (CH3)2C(OH)H | γ-rays (T < 90 °C) | (CH3)2C(OH), [CH2CH(CF3)]nH (n = 1, 2) | 31–34 |
| C6H5CH2–Cl | Fe(CO)5 | C6H5CHX [CH2CH(CF3)]nY (n = 1, 2; X = Cl, H; Y = H, Cl) | 35 |
| CCl4 | Fe(CO)5 | Cl3C[CH2CH(CF3)]nCl (n = 1–3) | 36 |
| CCl4 | CuCl2/ICl | Cl3C[CH2CH(CF3)]nCl (n = 1–7) | 37 |
| C6H5CH2–Br | Fe(CO)5 | C6H5CHX [CH2CH(CF3)]nY (n = 1, 2; X = Br, H; Y = H, Br) | 38,39 |
| Br2CH–Br | Fe(CO)5 | Br2CH[CH2CH(CF3)]nBr (n = 1–3) | 40 |
| CF3CBrHCH2CBr2[CH2CH(CF3)]2H | |||
| Br2CH–Br | Peroxides | XCBr2[CH2CH(CF3)]nY (Y = X = H or Br; n = 1, 2) | 40 |
| Br2CH–Br | Fe(CO)5 | BrCH2[CH2CH(CF3)]nBr (n = 1, 2) | 40 |
| CBr4 | BPO | Br3C[CH2CH(CF3)]nBr (n = 1–3) | 41 |
| CF3I | UV, 5 days | CF3[CH2CH(CF3)]nI (n = 1, 2) | 42 |
| CF3I | 225 °C, 36 h | CF3[CH2CH(CF3)]nI (n = 1–3) | 42 |
| CF3I | UV, various T | Normal and reverse monoadducts, few amounts of diadducts | 43 |
| C6F13I | Δ, UV, Fe3+/benzoin or peroxides | C6F13[CH2CH(CH3)]nI (n = 1, 2) | 44 |
| (CF3)2CFI | DTBP | (CF3)2CF[CH2CH(CH3)]nI (n = 1, 2) | 44 |
| Cl3SiH | UV | Cl3Si[CH2CH(CF3)]nH (n = 1, 2) | 45 |
| (C2H5O)2P(O)H | DTBP, 130 °C | (C2H5O)2P(O)(TFP)nH (n = 1–5) | 46–48 |
| THF | DTBP, 140 °C | Monoadduct (48%) | 49 |
| 2-Me-1,3-dioxolane | Fe(CO)5 | 2,4-bis(TFP)-2-Me-dioxolane | 50 |
| CH3SSCH3 | UV | CH3SCH2CH(CF3)SCH3 | 51 |
| HBr | UV | CF3CH2CH2Br | 42 |
| cyclopentadiene | 180 °C, 72 h |
exo![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :endo = 62:38 :endo = 62:38 |
52 |
Experimental section
Materials
All reagents were used as received unless stated.
3,3,3-Trifluoropropene (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–CF3, TFP) and 2-iodoperfluoropropane ((CF3)2CF–I), and perfluorooctanoic acid (C7F15–COOH, PFOA) were kindly supplied by Great Lakes Chemical Corporation, now Chemtura (West Lafayette, USA). 2-Iodoperfluoropropane was treated with sodium thiosulfate and then distilled prior to use to remove impurities and molecular iodine. 1,1,1,3,3-Pentafluorobutane (Solkane® 365mfc, CF3–CH2–CF2–CH3) was kindly supplied by Solvay S.A. (Tavaux, France). tert-Butyl peroxypivalate in solution of isododecane (Trigonox® 25-C75, tBuOOC(O)tBu, TBPPi) (purity 75 wt.%), di-tert-butyl peroxide (Trigonox® B, DTBP) (purity 99%), and azobisisobutyronitrile (AIBN) (purity 99%) were gifts from Akzo Nobel (Châlons-sur-Marne, France). Ethylene (CH2CH2, E) was purchased from Air Liquide (France).
CH–CF3, TFP) and 2-iodoperfluoropropane ((CF3)2CF–I), and perfluorooctanoic acid (C7F15–COOH, PFOA) were kindly supplied by Great Lakes Chemical Corporation, now Chemtura (West Lafayette, USA). 2-Iodoperfluoropropane was treated with sodium thiosulfate and then distilled prior to use to remove impurities and molecular iodine. 1,1,1,3,3-Pentafluorobutane (Solkane® 365mfc, CF3–CH2–CF2–CH3) was kindly supplied by Solvay S.A. (Tavaux, France). tert-Butyl peroxypivalate in solution of isododecane (Trigonox® 25-C75, tBuOOC(O)tBu, TBPPi) (purity 75 wt.%), di-tert-butyl peroxide (Trigonox® B, DTBP) (purity 99%), and azobisisobutyronitrile (AIBN) (purity 99%) were gifts from Akzo Nobel (Châlons-sur-Marne, France). Ethylene (CH2CH2, E) was purchased from Air Liquide (France).
Allyl acetate (CH2CH–CH2–O–CO–CH3), triethylamine (N(C2H5)3), pyridine, zinc dust, polyethylene glycol monomethylether (n = 13), thioglycolic acid (HSCH2CO2H), magnesium sulfate (MgSO4), hydrochloric acid, and sodium thiosulfate (Na2S2O3) were purchased from Sigma-Aldrich (Saint Quentin-Fallavier, France). Tert-butanol, 2-butanone, dichloromethane, pentane, all of analytical grade, were purchased from SDS (France). Deuterated acetone d6 was purchased from Euroiso-top (Grenoble, France) (purity >99.8%).
Characterization
Synthesis
The adducts were already characterized by Kostov et al.44
Diadduct: (CF3)2CF(C3H3F3)2-I; 2-iodo-2H,3H,3H,4H,5H,5H-4,6-bis(trifluoromethyl)perfluoroheptane (normal adduct), 1-iodo-1H,1H,2H,3H,4H,4H-2,3,5-tris(trifluoromethyl) perfluorohexane (reverse adduct). b.p. = 68–71 °C/20 mm Hg.
19F NMR (CDCl3): −69.6, −70.9, −71.5, −72.9 (assigned to CF3 of both TFP (normal and reverse adducts), 6 F); −77.3, −78.5, −78.8 (t, (CF3)2, 6 F); −185.5, −187.2 (dt, C(F), 1 F).
1H NMR (CDCl3): 1.9–2.3 (m, *C–CH2–C*, 2 H); 2.3–2.7 (m, RF–CH2, 2 H); 2.6–3.2 (m, *CH(CF3)), 25% of one diastereoisomer overlapping with RF–CH2protons and 75% in the range of 2.75–3.2; negligible reverse adduct (absence of signal at 3.5, 1 H)); 4.15–4.45 (m, *CH(CF3)I, 1 H).
13C NMR (CDCl3): 130.8 (130.6), 128.0 (127.8), 125.2 (125.0), 122.4 (122.3) (q of d (coupling of two diastereoisomers), CF3 of TFP between two CH2groups in normal adduct, 1JCF = 279.7, 1 C); 128.4 (128.16), 125.66 (125.42), 122.92 (122.68), 120.18 (119.94) (q of d, CF3 adjacent to I in normal adduct, 1JCF = 276.7, 1 C); 125.04 (124.94), 122.27 (122.10), 119.39 (119.26), 116.55 (116.43), 124.79 (124.66), 121.95 (121.82), 119.11 (118.99), 116.27 (116.15) (two q of d, CF3, 1JCF = 285.7, 2JCF = 28.17, 6 C); 92.63–89.02 (d of sept, C(F), 1JCF = 207.3, 2JCF = 32.2, 1 C); 38.10, 37.8, 37.6, 37.3, 37.0, 36.7 (sext, C*H(CF3) of TFP between two CH2groups, 2JCF = 27.17, 1 C); 35.56, 33.29 (d, CH2 of TFP-I, 1 C); 27.70, 27.51, 27.29, 27.10 (two d, CH2, 2JCF = 19.1, 1 C); 19.53–18.55, 17.23–16.30 (d of q (tr/er), *CH(CF3)I, 2JCF = 29.2, 1 C).
Synthesis of RF(C3H3F3)nCH2CHICH2OC(O)CH3. The TFP telomer (n = 1 or 2, 0.18 mol) and a 1.2-fold excess of allyl acetate (0.215 mol) were introduced to a 250-ml three-neck round-bottom flask equipped with a double condenser and a thermometer. Then the mixture was heated up to 82 °C and was stirred. When the temperature reached 80 °C, AIBN (C0 = 0.015–0.050) was introduced stepwise over the 10 h reaction time. No exotherm was observed. After 10 h, the reaction was stopped; the crude product was cooled to room temperature, filtered and analyzed by GC. The reaction mixture was distilled to purify the iodoacetate (yield = 75–80%).
(CF3)2CF(C3H3F3)2CH2CH(I)CH2OCOCH3; 8,9,9,9-tetrafluoro-2-iodo-4,6,8-tris(trifluoromethyl)nonyl acetate. b.p. = 78–80 °C/0.1 mm Hg
19F NMR (CDCl3): no signal at −67 (no reverse product); −70.5 to −72.77 (m, 2 × CF3 of TFP, 6 F); −77.2 to −78.75 (m, (CF3)2, 6 F); −186.4, −187.14, −187.73 (t, C(F), 1 F)
1H NMR (CDCl3): 1.6–1.9 (m, *C–CH2–*C, 2 H); 2.02–2.25 (CH3OCO, 3 H); 2.25–2.4 (CH2–CHI, 2 H); 2.3–2.5 (RF–CH2, 2 H); 2.6–3.0 (*CH(CF3), 1 H); 4.2 (m, 1 H of CH(I) + 1 H of *CH(CF3)); 4.4 (m, ester CH2OCOCH3, 2 H).
Synthesis of RF(C3H3F3)nCH2CH
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) CH2.
Zn dust (2 equivalents about iodoacetate) was activated with 1.2 g of a mixture of acetic acid/acetic anhydride (1:1). Then, methanol (40 mL) was added, and the mixture was introduced into 250-ml two-neck round-bottom flask with a reverse condenser and a magnetic stirrer. The temperature was increased to 65 °C while stirring. The iodoacetate (0.12 mol) from the previous step dissolved in methanol (30 mL) was added dropwise within 3 h at reflux to a zinc slurry with vigorous stirring. Upon complete addition, the stirring was maintained for additional 2 h and a colorless product was obtained. The zinc complex was filtered off, the filtrate was diluted with dichloromethane (1:1 vol./vol.), washed with 10% hydrochloric acid aqueous solution (100 ml) and washed again with distilled water. The organic phase was added dropwise to anhydrous magnesium sulfate under stirring to eliminate traces of water, filtered and twice distilled (quantitative conversion, yield after distillation 75%). The product obtained is a colorless liquid.
CH2.
Zn dust (2 equivalents about iodoacetate) was activated with 1.2 g of a mixture of acetic acid/acetic anhydride (1:1). Then, methanol (40 mL) was added, and the mixture was introduced into 250-ml two-neck round-bottom flask with a reverse condenser and a magnetic stirrer. The temperature was increased to 65 °C while stirring. The iodoacetate (0.12 mol) from the previous step dissolved in methanol (30 mL) was added dropwise within 3 h at reflux to a zinc slurry with vigorous stirring. Upon complete addition, the stirring was maintained for additional 2 h and a colorless product was obtained. The zinc complex was filtered off, the filtrate was diluted with dichloromethane (1:1 vol./vol.), washed with 10% hydrochloric acid aqueous solution (100 ml) and washed again with distilled water. The organic phase was added dropwise to anhydrous magnesium sulfate under stirring to eliminate traces of water, filtered and twice distilled (quantitative conversion, yield after distillation 75%). The product obtained is a colorless liquid.
(CF3)2CFCH2CH(CF3)CH2CH(CF3)CH2CHCH2; 8,9,9,9-tetrafluoro-4,6,8-tris(trifluoromethyl)non-1-ene. b.p. = 62–65 °C/20 mmHg.
19F NMR (CDCl3): −71.7, −72.2, −72.5 (t, 2 × CF3 in TFP, 6 F); −77.7 to −78.9 (dt, (CF3)2, 6 F); −186.9, −187.4 (m, C(F), 1 F).
1H NMR (CDCl3): 1.55–2.2 (m, CH2 from *C–CH2–C*, 2 H); 2.0–2.2 (m, allylCH2, 2 H); 2.3–2.5 (m, 2 H of (CF3)2CF–CH2 + 25% of diastereoisomer *CH(CF3) from the side of (CF3)2CF–); 2.9 (m, 1 H of *CH(CF3) from the side of the allyl); 5.20 (m, CH2, 2 H); 5.7 (m, –CH, 1 H).
13C NMR (CDCl3): 133.21 (132.89) (d, CH2CH-, 1 C); 131.76 (131.61), 128.98 (128.83), 126.20 (126.05), 123.43 (123.28) (q, CF3 of TFP on the side of (CF3)2CF–, 1JCF = 279.7, 1 C); 131.18 (131.00), 128.40 (128.22), 125.62 (125.45), 122.84 (122.67) (q, CF3 of TFP adjacent to allyl, 1JCF = 279.7, 1 C); 125.10 (125.06), 124.82 (124.79), 122.26 (122.23), 121.98 (121.95), 116.59 (116.55), 116.30 (116.26) (two qd, (CF3)2, 1JCF = 285.7, 2JCF = 28.2, 2 C); 188.29 (118.24) (d, -CHCH2, 1 C); 92.74–88.77 (d of sept, (CF3)2CF, 1JCF = 206.25, 2JCF = 32.2, 1 C); 40.37–39.38 (qn, *CH(CF3) adjacent to (CF3)2CFCH2–, 2JCF = 25.15, 1 C); 35.85–34.68 (qn of d, *CH(CF3) adjacent to allyl, 2JCF = 27.16, 1 C); 32.78–32.75 (d, CH2 adjacent to vinyl, 1 C); 32.10, 32.08, 31.91 (t, CH2 between 2*C, 1 C); 28.94, 27.70 (two d, CH2-CF(CF3)2, 2JCF = 19.12, 1 C).
Synthesis of RF(C3H3F3)n(CH2)3SCH2COOH. The allylic TFP telomer (0.05 mol) and thioglycolic acid (0.06 mol) in acetonitrile (50 mL) were introduced to a 250-ml three-neck round-bottom flask equipped with a condenser and a thermometer. Then the mixture was heated up to 75 °C while stirring. At that temperature, tert-butyl peroxypivalate was introduced (3 mol% with respect to allylic TFP telomer). After 3 h, the reaction was stopped. The crude product was cooled to room temperature, and the solvent rotavaped under vacuum. RF(TFP)2(CH2)3SCH2COOH was obtained in 73% yield. The same procedure was also carried out at room temperature without any radical initiator but photoinitiated (P = 50 W, wide spectral range of wavelength of the UV lamp located at 15 cm from the reaction medium). The obtained yield was slightly higher (81%) than that noted above.
Synthesis of RF(C3H3F3)n(CH2)3SCH2COO(CH2CH2O)nCH3. In a 100 mL two-neck round-bottom flask equipped with a Dean–Stark apparatus and a condenser, polyethylene glycol monomethylether (HO(CH2CH2O)13CH3) (17.6 × 10−3 mol) was dissolved in toluene (50 mL) at 140 °C, and the solution of the previous product (17.6 × 10−3 mol) was added. Methane sulfonic acid (CH3SO3H) was used as the catalyst (5 mol% with respect to PEGMe). Water generated by the esterification was eliminated by azeoptropic distillation, and the progress of the reaction was monitored by FTIR (vanishing of the OH band at 3500 cm−1). After evaporating the toluene under vacuum, surfactant A was obtained in 74% yield. NMR (Fig. 1 and 2); FTIR (Fig. 3).
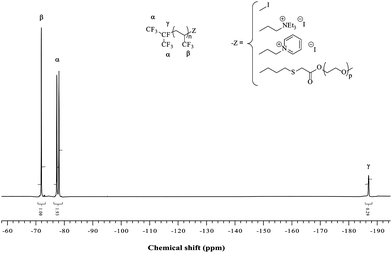 | ||
| Fig. 1 19F NMR spectrum of RF-TFP-I telomer and its derivatives (A, B, and C surfactants) recorded in CDCl3. | ||
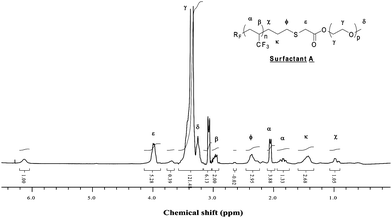 | ||
| Fig. 2 1H NMR spectrum of (CF3)2CF(C3H3F3)n(CH2)3SCH2COO(CH2CH2O)nCH3Surfactant A recorded in CDCl3. | ||
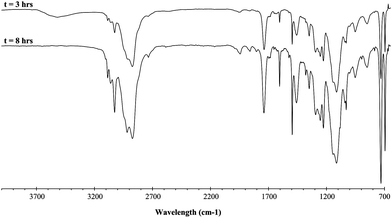 | ||
| Fig. 3 FTIR spectra during the esterification reaction of (CF3)2CF(C3H3F3)n(CH2)3SCH2COOH with PEO–OH leading to (CF3)2CF(C3H3F3)n(CH2)3SCH2COO(CH2CH2O)nCH3 after 3 h (upper spectrum) and 8 h (lower spectrum). | ||
Synthesis of RF(C3H3F3)n(CH2)2I. A 160 or 300 mL Hastelloy Paar autoclave equipped with a manometer, a magnetic stirrer, and safety valve (3000 psi), was pressurized with 30 bars of nitrogen to check for leaks. The autoclave was then conditioned for the reaction with several nitrogen/vacuum cycles (10−2 mbar) to remove any trace of oxygen. TFP telomer, tert-butanol, and tert-buyl peroxypivalate (TBPPi 0.1 mol % with respect to ethylene) were then introduced in the autoclave. Ethylene was introduced by double weighing (1.2
:1.0 compared to TFP telomer). The ethylenation was carried out at 75 °C for 6 h. A drop of pressure was observed. After the reaction stopped, the autoclave was cooled and placed in an ice bath to purge the unreacted ethylene. After distillation, the ethylenation yielded 75–80% of iC3F7(TFP)2CH2CH2I telomer as a colourless liquid.
iC3F7(TFP)2CH2CH2I b.p. = 44–46 °C/0.2 mm Hg
1H NMR (acetone d6, ppm): 3.6–3.4 (m, central –CH2C*H(CF3)–, 1H); 3.4–3.1(m, –CH2CH2I, 2H); 2.8–2.7 (m, –CH2C*H(CF3)–, 1H); 2.3–2.1, centre 2.2, (m, HA, HB in AB system RF–CH2C*H–, 2H), –(C*H–CH2C*H–, 2H) and (–CH2CH2I, 2H).
19F NMR (acetone d6, ppm; Fig. 1) δ: −69.2 and −71.6 [2 × CF3 in –CH2C*H(CF3), 6F]; −78.5 (2 × CF3 end-groups, 6F); −187 (m, ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CF–, 1F).
CF–, 1F).
13C NMR (acetone d6, ppm) d: 130.8 (130.6); 128.0 (127.8); 125.2 (125.0); 122.4 (122.3); 1JCF = 265 Hz; q (coupling of two diastereoisomers) 2 × 1C (CF3 of TFP between two CH2groups); 128.4; 125.7; 122.9; 120.1; 125.0; 122.3; 119.4; 116.3; two q of d (1JCF = 275.7 Hz; 2JCF = 28.2 Hz; 2 × 1C from terminal (CF3)2– groups); 92.6–89.0 d of hept., 1C [C(F)], 1JCF = 207.3 Hz; 2JCF = 32.2 Hz; 40.8; 40.7; 40.5; 40.3; 35.2; 34.9, 34.3; 34.1; d of q, 2JCF = 27.2 Hz, 2×1C [C*H(CF3) of TFP between two CH2groups]; 32.5; 31.8, d, 1C [–C(F)–CH2– of TFP, 2JCF = 27.1 Hz]; 29.0; 28.9; 28.7; 28.5; q, 1C (–CH2–CH2I of E); 27.0; 26.8; 26.6; t, 1C (C*–CH2–C*) 2JCF = 19.1 Hz; 0.8; 0.0; s, 1C (–CH2–I, 2 diastereoisomers).
Synthesis of RF(C3H3F3)n(CH2)2NR+I−. (NR stands for pyridinium or triethyl ammonium.)
The ethylenated TFP telomer (0.05 mol) was introduced to a 250-ml three-necked round-bottom flask equipped with a condenser and a thermometer. Pyridine or triethylamine (0.1 mol) was added dropwise in DMF or MeOH, respectively (50 mL). Then, the mixture was purged with argon for 15 min and heated up to 40 °C (or room temperature for NEt3) under stirring. After 10 h, the reaction was stopped. The crude product was cooled, and the solvent rotavaped under vacuum and then the product was precipitated from ether. RF(C3H3F3)n(CH2)2NR+I− (where NR represents a pyridinium or triethyl ammonium function) was obtained in 40–50% yield. 19F NMR spectra were similar to those of the ethylenated TFP telomers.
Results and discussion
To our knowledge only surfactants based on 3,3,3-trifluoropropene (TFP) and vinylidene fluoride (VDF) has been prepared53 and it was worth investigating the synthesis of cationic surfactants which bear weak points which are able to be metabolized or enzymatically degraded.54 In contrast to perfluorinated materials, hydrogenofluorinated compounds can be degraded by first, elimination of HF, yielding a double bond that can be oxidized and cleaved.20 This degradation can be advantageously achieved by the methylene and methynegroup brought by each TFP unit.54The objective of the present article consists in synthesizing original TFP-based surfactants by simple chemical modifications of TFP telomers,44,55,56 achieved by radical telomerization of TFP with isoperfluoropropyl iodide.44
The overall paths for the syntheses of these surfactants is described in Scheme 1. From the radical telomerization of 3,3,3-trifluoropropene (TFP), several surfactants can be obtained (anionic, cationic and non-ionic) in satisfactory yields (50% overall yields from iC3F7I). The initial step is the telomerization of 3,3,3-trifluoropropene (TFP) in the presence of perfluoroalkyl iodides ((CF3)2CFI) that was reported in patents from Great Lakes55,56 and by Kostov et al.44 The telomerization in bulk as that initiated by organic peroxides such as di-tert-butylperoxide led to TFP telomers in high yields (>80%). TFPhomopolymerization was reported to lead to poly(TFP) homopolymer but the presence of (CF3)2CFI as an efficient chain transfer agent drastically lowered DPn to achieve either monoadduct or diadduct or triadduct for a high amount of TFP in the feed. The degree of polymerization (DPn = n in (CF3)2CF–(TFP)n-I) could be tuned by the initial molar ratio [TFP]0/[((CF3)2CFI]0. The 19F NMR spectra (Fig. 1) enabled to assess DPn value from the ratio between trifluoromethyl groups of TFP units and those of the perfluoroisopropylgroup. Fig. 1 exhibits the signals of the perfluoroisopropyl end-group (two CF3 and CF centered at −77.7, −78.5, and −188.9 ppm, respectively). It also displays the signal at −72 ppm assigned to the CF3group of TFP corresponding to the normal addition of (CF3)2CF• onto TFP [(CF3)2CF–CH2CH(CF3)–] while that assigned to the reverse addition [(CF3)2CF–CH(CF3)CH2–] expected at −67 ppm was never observed, indicating a selective normal addition, at least for the first adducts.
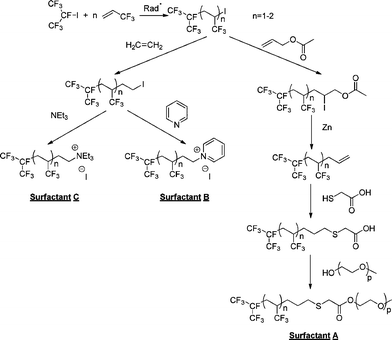 | ||
| Scheme 1 Straightforward strategies for the preparation of 3,3,3-trifluoropropene-based cationic and non-ionic surfactants. | ||
The synthesis of the non-ionic surfactants was carried out in three steps. First, the preparation of RF–(TFP)x–CH2–CHCH2 allylic derivative was reported,44 and consists in the direct radical addition of RF-(TFP)x-I onto allyl acetate (Yield = 75–80%) followed by the deiododeacetalization in the presence of zinc and methanol. As for tetrafluoroethylene telomers,57 the yields were high showing a suitable electron-withdrawing effect of CHCF3group to easily generate (CF3)2CF–CH2–CH(CF3)• radical. The conversion of iodoacetate was quantitative while allylic TFP telomers were obtained 75% yield after distillation. Second, the allylic derivative was further reacted with thioglycolic acid in acetonitrile to obtain a carboxylic ω-functionalized compound (as an anionic surfactant, PFOA type). That reaction occurred either photochemically (81% yield) or in the presence of a source of radicals arising from tert-butyl peroxypivalate (73% yield). 1H NMR enabled that reaction to be monitored by the vanishing of both allyl double bond and mercaptogroups centered at 5.0–6.5 and 1.5 ppm, respectively. 19F NMR spectrum was similar to that of the precursor. Finally, the esterification with an ω-hydroxy poly(ethylene oxide) oligomer (PEO-OH) led to the formation of an original RF-(TFP)x–CH2–CH2–CH2–S–CH2–CO2–PEO non-ionic surfactant. The 1H NMR spectrum (Fig. 2) exhibits the characteristic signals centered at 4.0, 3.4, 3.3, and 3.0 assigned to the –SCH2CO2– group, ethylene oxide units, methyl end-group, and CHCF3, respectively. To ensure a complete conversion of acid into ester, the reaction was carried out in toluene and the water-toluene azeotrope was continuously distilled using a Dean–Stark apparatus at 95 °C (b.p. of water and toluene are 100 and 110 °C, respectively). The reaction was monitored by FTIR spectroscopy (Fig. 3). The vanishing of OHgroup frequency was observed at 3600 cm−1 while that at 1750 cm−1, assigned to the carbonyl of the ester, increased. The former frequency disappeared after 8 h reaction, meaning that the esterification was complete, and was confirmed visually by the absence of water-toluene azeotrope.
To obtain the series of cationic surfactants, first ethylene end-capping of the TFP telomers was achieved. This reaction, initiated by an organic peroxide, quantitatively led to RF-(TFP)x–CH2–CH2–I in 89% yield with a quantitative conversion of (CF3)2CF(TFP)nI. This successful ethylenation was evidenced by 1H, 13C and 19F NMR spectroscopy. The chemical shifts of ethylene end-groups were centered at about 29.0, 28.9, 28.7 and 28.5 ppm for –CH2–CH2–I, 8.0 and 0.0 for –CH2–I on the 13C NMR spectrum, and 2 ppm (–CH2–CH2–I) in the 1H NMR spectrum (details are given in the experimental section). The nucleophilic substitution of RF–(TFP)x–CH2–CH2–I by either triethylamine (NEt3) or pyridine (C5H5N) at room temperature or at 40 °C, respectively, led to cationic surfactants bearing ammoniumgroups (Scheme 1) in satisfactory yield (>75%) with a slight dehydroiodination (Fig. 4 and 5). These mild experimental conditions avoid any dehydroiodination.
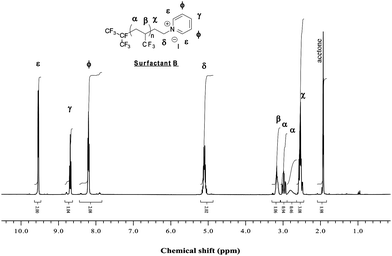 | ||
| Fig. 4 1H NMR spectrum of (CF3)2CF(TFP)n(CH2)2NC5H5+I− (Surfactant B) recorded in acetone d6. | ||
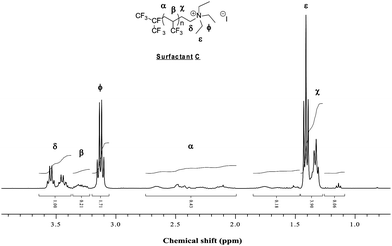 | ||
| Fig. 5 Expansion of the 1H NMR spectrum (0.8 to 3.9 ppm) of (CF3)2CF(TFP)n(CH2)2N(CH2CH3)3+I− (Surfactant C) recorded in CDCl3. | ||
The structures of both cationic surfactants were proved by 1H NMR spectroscopy. In the 1H NMR spectrum of surfactant B (Fig. 4) the group of signals present in the 8.1 to 9.6 ppm range (with an integral ratio of 1: 0.5: 1) was assigned to the pyridinium group. In the case of surfactant C, the 1H NMR spectrum (Fig. 5) obviously does not display any signal in the 7–9 ppm range while the signal of the ethyl group of the triethylammonium are located at 1.4 (CH3) and 3.15 ppm (CH2).
Physicochemical properties of surfactants are often searched since they can be used under hostile conditions (e.g. oil wells, acidic buffers for emulsion polymerization of tetrafluoroethylene). These surfactants exhibit good overall inertness to acids and bases especially iC3F7(TFP)(CH2)3SCH2CO2(PEO)CH3, and a satisfactory solubility in water and methanol but a poor solubility in apolar solvents, and acetone (Table 2).
| Media | T/°C | Time (days) | Surfactant | ||
|---|---|---|---|---|---|
| A | B | C | |||
| (1) Base-acid resistance | Weight losses (%) | ||||
| 98% H2SO4 | 25 | 7 | 0.0 | <1.2 | 0.0 |
| 60% HNO3 | 25 | 7 | - | >40.0 | <20 |
| 37% HCl | 25 | 7 | 0.0 | >15.0 | 0.0 |
| 40% NaOH | 25 | 7 | 0.0 | 0.0 | 0.0 |
| (2) Solubility in selected solvents | Solubility (g/100mL) | ||||
| Water | 25 | 3 | >10 | >10 | >10 |
| Methanol | 25 | 3 | >10 | >10 | >10 |
| Diethyl ether | 25 | 3 | <1 | <1 | <1 |
| Benzene | 25 | 3 | <2 | <2 | <2 |
| Acetone | 25 | 3 | <10 | <10 | <10 |
| A: |

|
||||
| B: |

|
||||
| C: |

|
||||
Surface tensions of the various surfactants were assessed at different concentrations to determine the critical micellar concentration (CMC) of the surfactants in water at 25 °C. The results are displayed in Fig. 6, and the CMC values were found to be 0.06, 4.10, 3.20, and 3.00 g L−1 for surfactants A, B, C, and PFOA, respectively. It can be seen that both cationic surfactants behave like PFOA while the non ionic surfactant (A) has a higher surface tension but a lower CMC.
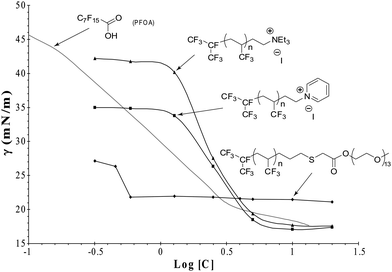 | ||
| Fig. 6 Surface tension versus the concentration of TFP-based surfactants compared to that of PFOA. | ||
Conclusions
Original 3,3,3-trifluoropropene telomers were synthesized under radical conditions and were modified into hydrogenofluorosurfactants by simple chemistry in high overall yields. Their physicochemical properties showed good inertness to bases and acids. Their surface tensions were assessed and show interesting CMC values similar to that of PFOA. Nonionic surfactant A should behaves as a very interesting surfactant, because its CMC is quite low (0.06 g L−1). Further work on the biodegradation of these surfactants is under progress. Also surfactants based on vinylidene fluoride with alternating C–H and C–F bonds (which may act as degradation sites), and also containing both TFP and VDF units appear as excellent alternatives to perfluorinated chains. Such compounds are currently under investigation.Acknowledgements
The authors would like to thank Great Lakes/Chemtura company (Dr S. Brandstater, and V. Sharma) for financial support.Notes and references
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier, Amsterdam, 2004 Search PubMed.
- J. Scheirs, Modern Fluoropolymers, John Wiley and Sons Ltd., New York, 1997 Search PubMed.
- G. Hougham, P. E. Cassidy, K. Johns and T. Davidson, Fluoropolymers, Kluwer/Plenum, New York, 1999 Search PubMed.
- L. Nordstierna, I. Furó and P. Stilbs, J. Am. Chem. Soc., 2006, 128, 6704–6712 CrossRef CAS.
- Y. Kondo, H. Miyazawa, H. Sakai, M. Abe and N. Yoshino, J. Am. Chem. Soc., 2002, 124, 6516–6517 CrossRef CAS.
- M.-P. Krafft and J. G. Riess, Chem. Rev., 2009, 109, 1714–1792 CrossRef CAS.
- S. Ishikawa, S. Masumoto and H. Eguchi (Tosoh Corp), Japanese Patent 2001/219969.
- E. Kissa, Fluorinated Surfactants: Synthesis, Properties and Applications, CRC Press, New York, 2001 Search PubMed.
- Surfactants in Tribology, ed. G. Biresaw and K. L. Mittal, CRC Press, Boca Raton, 2008 Search PubMed.
- G. M. Nicolle and A. E. Merbach, Chem. Commun., 2004,(7), 854–855 RSC.
- J. Gan, M. El Bakkari, C. Belin, C. le Margottin, P. Godard, J.-L. Pozzo and J.-M. Vincent, Chem. Commun., 2009,(34), 5133–5134 RSC.
- H. V. Berlepsch, C. Böttcher, K. Skrabania and A. Laschewsky, Chem. Commun., 2009,(17), 2290–2292 RSC.
- J. Kovarova and Z. Svobodova, Neuroendocrinol. Lett., 2008, 29, 599–608 CAS.
- U.S. Environmental Protection Agency, Preliminary Risk Assessment: Perfluorooctanic Acid (PFOA) and Fluorinated Telomers, April 2003 Search PubMed.
- H. Fromme, O. Midasch, D. Twardella, J. Angerer, S. Boehmer and B. Liebl, Int. Arch. Occup. Environ. Health, 2007, 80, 313–319 CrossRef CAS.
- S. K. Ritter, Chem. Eng. News, 2010, 88, 12–17 Search PubMed.
- E. Brede, M. Wilhelm, T. Göen, J. Müller, K. Rauchfuss, M. Kraft and J. Hölzer, Int. J. Hyg. Environ. Health, 2010, 213, 217–223 CrossRef.
- K. Steenland, T. Fletcher and D. A. Savitz, Environ. Health Perspect., 2010, 118, 1100–1108 CrossRef CAS.
- H. Hori, E. Hayakaiva, H. Einaga, S. Kutsuna, K. Koike, T. Ibusuki, H. Kiatagawa and R. Arakawa, Environ. Sci. Technol., 2004, 38, 6118–6124 CrossRef CAS.
- J. R. Parsons, M. Saez, J. Dolfing and P. de Voogt, in Reviews of Environmental Contamination and Toxicology, Springer, New York, 2008, vol. 196, pp. 53–71 Search PubMed.
- J. W. Martin, M. M. Smithwick, B. Braune, P. F. Hoekstra, D. C. G. Muir and S. A. Mabury, Environ. Sci. Technol., 2004, 38, 373–380 CrossRef CAS.
- E. Sinclair, D. Mayack, K. Roblee, N. Yamashita and K. Kannan, Arch. Environ. Contam. Toxicol., 2005, 50, 398–410 CrossRef.
- N. Saito, K. Harada, K. Inoue, K. Sasaki, T. Yoshinaga and A. Koizumi, J. Occup. Health, 2004, 46, 49–59 CrossRef CAS.
- PFOA stewardship Program, http://www.epa.gov/oppt/pfoa/pubs/pfoastewardship.htm, Accessed February 5th, 2009.
- J. Guo, P. Resnick, K. Efimenko, J. Genzer and J. M. DeSimone, Ind. Eng. Chem. Res., 2008, 47, 502–508 CrossRef CAS.
- H. Sawada, T. Kawase, Y. Ikematsu, Y. Ishii, M. Oue and Y. Hayakawa, Chem. Commun., 1996,(2), 179–180 RSC.
- H. Sawada, T. Kawase, K. Yamashitau and Y. Hayakawa, Chem. Commun., 1996,(7), 827–828 RSC.
- J. L. Panza, A. J. Russell and E. J. Beckman, Chem. Commun., 2002,(9), 928–929 RSC.
- W. Schwertfeger, K. Hintzer and E. Obermaier (3M), Eur. Patent 2006/093885 A1.
- N. S. Ikonnikov, N. I. Lamova and A. B. Terent'ev, Izv. Akad. Nauk SSSR, Ser. Khim., 1988, 117–121 CAS.
- R. A. Zamyslov, A. G. Shostenko, I. V. Dobrov and V. E. Myshkin, Zh. Org. Khim., 1980, 16, 897–901 CAS.
- A. G. Shostenko, I. V. Dobrov and A. V. Chertorizhskii, Khim. Prom-st., 1983, 339–340 CAS.
- R. A. Zamyslov, Zh. Vses. Khim. O-va. im. D. I. Mendeleeva, 1986, 31, 589–591 CAS.
- R. A. Zamyslov, A. G. Shostenko, I. V. Dobrov and N. P. Tarasova, Kinet. Katal., 1987, 28, 977–979 CAS.
- A. B. Terent'ev and T. T. Vasil'eva, Ind. Chem. Libr., 1995, 7, 180–201 CrossRef CAS.
- T. T. Vasil'eva, I. A. Fokina, S. V. Vitt and V. I. Dostovalova, Izv. Akad. Nauk SSSR, Ser. Khim., 1990, 8, 1807–1811 Search PubMed.
- W. Keim, G. H. Raffeis and D. Kurth, J. Fluorine Chem., 1990, 48, 229–237 CrossRef CAS.
- R. G. Gasanov, T. T. Vasil'eva and S. I. Gapusenko, Kinet. Katal., 1991, 32, 1466–1470 CAS.
- T. T. Vasil'eva, I. A. Fokina and S. V. Vitt, Izv. Akad. Nauk SSSR, Ser. Khim., 1991, 1384–1388 CAS.
- T. T. Vasil'eva, V. A. Kochetkova, V. I. Dostovalova, B. V. Nelyubin and R. K. Freidlina, Izv. Akad. Nauk SSSR, Ser. Khim., 1989, 2558–2562 CAS.
- T. T. Vasil'eva, V. A. Kochetkova, B. V. Nelyubin, V. I. Dostovalova and R. K. Freidlina, Izv. Akad. Nauk SSSR, Ser. Khim., 1987, 808–811 CAS.
- R. N. Haszeldine, J. Chem. Soc., Abstr., 1952, 2504–2513 CAS.
- R. N. Haszeldine, J. Chem. Soc., Abstr., 1951, 2495–2504 CAS.
- G. K. Kostov, B. Ameduri and S. M. Brandstadter, Collect. Czech. Chem. Commun., 2008, 73, 1747–1763 CrossRef CAS.
- R. N. Haszeldine, M. J. Newland and J. B. Plumb, J. Chem. Soc., 1965, 2101–2107 RSC.
- R. N. Haszeldine, D. L. Hobson and D. R. Taylor, J. Fluorine Chem., 1976, 8, 115–124 CrossRef CAS.
- H. D. Block (Bayer), DE Patent 1976/2,514,640.
- G. Kostov, B. Ameduri and S. Brandstadter, J. Fluorine Chem., 2007, 128, 910–918 CrossRef CAS.
- J. Chen, Y.-F. Zhang, X. Zheng, A. Vij, D. Wingate, D. Meng, K. White, R. L. Kirchmeier and J. M. Shreeve, Inorg. Chem., 1996, 35, 1590–1601 CrossRef CAS.
- A. B. Terent'ev, E. V. Pastushenko, D. E. Kruglov and T. A. Rybininia, Izv. Akad. Nauk SSSR, Ser. Khim., 1992, 2768–2772 CAS.
- G. Haran and D. W. A. Sharp, J. Chem. Soc., Perkin Trans. 1, 1972, 34–38 RSC.
- W. J. Feast, M. Gimeno and E. Koshravi, Polymer, 2003, 44, 6111–6121 CrossRef CAS.
- G. K. Kostov, F. Boschet, J. Buller, L. Badache, S. Brandstadter and B. Ameduri, Macromolecules, 2011, 44, 1841–1855 CrossRef CAS.
- G. Kostov, F. Boschet and B. Ameduri, J. Fluorine Chem., 2009, 130, 1192–1199 CrossRef CAS.
- A. Jackson, V. Sharma, B. E. Edwards, J. Boggs, V. Hedrick, S. Brandstadter, J. Chien, E. Norman, R. Kaufman, B. Ameduri, G. Kostov and G. Leman (Chemtura), US Patent 2007/027349.
- B. Ameduri, S. M. Brandstadter and G. K. Kostov (Great Lakes Chemical Corporation, USA), WO Patent 2008/019068 A2.
- B. Ameduri, B. Boutevin, M. Nouiri and M. Talbi, J. Fluorine Chem., 1995, 74, 191–197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |