DOI:
10.1039/C1PY00406A
(Paper)
Polym. Chem., 2012,
3, 172-181
Efficient synthesis of poly(2-hydroxypropyl methacrylate)-silica colloidal nanocomposite particles via aqueous dispersion polymerization†
Received
8th September 2011
, Accepted 4th November 2011
First published on 17th November 2011
Abstract
Colloidally stable poly(2-hydroxypropyl methacrylate)-silica nanocomposite particles can be efficiently prepared by aqueous dispersion polymerization at 60 °C using a binary mixture of an ultrafine aqueous silica sol and poly(N-vinylpyrrolidone) [PVP] as the stabilizer system. Optimization of the initial silica sol concentration allows relatively high silica incorporation efficiencies (close to 100%) to be achieved. The key to the formation of colloidally stable nanocomposite particles is the judicious choice of a suitable anionic initiator, PVP and a commercial 20 nm aqueous silica sol. In contrast, other formulations typically lead to either coagulation or flocculation. Such purified poly(2-hydroxypropyl methacrylate)-silica nanocomposite particles can have relatively narrow particle size distributions, with mean diameters typically varying from 480 to 900 nm as judged by dynamic light scattering (DLS), and with mean silica contents ranging from 10 to 22% by mass. The nanocomposite particles have a well-defined ‘core-shell’ morphology with a silica-rich surface, but a significant fraction of silica appears to be located within the interior of the nanocomposite particles.
Introduction
Over the past decade or so, colloidal vinyl polymer-silica nanocomposites have attracted substantial academic and industrial interest. Various synthetic routes to such nanocomposite particles have been extensively reviewed.1–4 These include in situpolymerization of various vinyl monomers in the presence of commercial ultrafine silica sols in either aqueous solution,5–13alcohol/water mixtures,14,15 or purely alcoholic media.16–18 Such colloidal nanocomposites have various potential applications, such as exterior architectural coatings,19 synthetic mimics for cosmic dust,20 and pH-responsive Pickering emulsifiers.21 However, often a significant problem for this in situpolymerization route is the relatively low silica incorporation efficiency that is typically observed. Excess silica sol can compromise the performance of these nanocomposite particles in some applications and also prevents the accurate determination of the mean silica content of the nanocomposite particles. Thus more efficient syntheses of such nanocomposite dispersions, preferably in aqueous media, are highly desirable.
One potential technical solution to this problem is mini-emulsion polymerization.22,23 This ensures that the silica sol is located exclusively on and/or within the monomer droplets, so that the silica incorporation efficiency is essentially 100%. However, such formulations usually require specialist equipment and are generally not widely used on an industrial scale. Recently, we reported that poly(2-vinylpyridine)-based [P2VP] colloidal nanocomposite particles could be efficiently prepared by emulsion polymerization at 60 °C using a commercial 20 nm aqueous silica sol as the sole stabilizing agent.9 The key to success here is simply the selection of a suitable cationic azo initiator. These P2VP-silica nanocomposite particles have a well-defined core-shell morphology with P2VP cores and silica shells; the mean particle diameter typically varied from 180 to 220 nm, and average silica contents ranged from 27 to 35% by mass. Very high silica incorporation efficiencies (88–99%) could be obtained for some formulations. Unfortunately, 2-vinylpyridine is relatively expensive and rather malodorous, and only high Tg nanocomposite particles could be obtained using this approach. Our groups has also reported the efficient preparation of vinyl polymer-silica nanocomposite particles by aqueous emulsion polymerization at 60 °C using a cationic azo initiator in combination with a commercially available glycerol-functionalized ultrafine aqueous silica sol in the absence of any surfactant, auxiliary comonomer, or non-aqueous co-solvent.10 For this formulation, various control experiments confirmed that the combination of a glycerol-functionalizedsilica sol with a cationic azo initiator is essential for the formation of colloidally stable nanocomposite particles. Silica mass contents are typically around 22–28%, with intensity-average particle diameters ranging between approximately 200 and 400 nm. Optimization of the initial silica sol concentration allows relatively high silica incorporation efficiencies (up to 95%) to be achieved. However, although it is commercially available, the glycerol-functionalized silica sol is significantly more expensive than non-functionalized silica sols. More recently, we have also explored the synthesis of core-shell nanocomposite particles via adsorption of small silica particles onto large sterically stabilized P2VP latex in aqueous solution.24 This approach can be optimized to ensure minimal excess silica, but it has only been conducted in dilute aqueous solution.
Purely aqueous dispersion polymerization syntheses are seldom reported in the literature. Presumably this is because relatively few water-miscible monomers are available that form water-insoluble polymers, which is a pre-requisite for such formulations. One of the few commodity vinyl monomers with this characteristic is 2-hydroxypropyl methacrylate (HPMA). Previously, we reported the preparation of HPMA-based colloidal nanocomposite particles using 4-vinylpyridine as an auxiliary comonomer to ensure a strong interaction between the organic and inorganic components.8 More recently, we reported the synthesis of sterically-stabilized poly(2-hydroxypropyl methacrylate) [PHPMA] latexes by aqueous dispersion polymerization at 60 °C using a PVP stabilizer.25 Another example of aqueous dispersion polymerization is the polymerization of aniline in the presence of either colloidal silica26 or water-soluble polymer such as PVP.27 In addition, Stejskal et al. reported the combination of high molecular weight polyacrylamide with a 22 nm silica sol to prepare polyaniline/silica nanocomposite particles.28 As far as we are aware, this latter work is the only study in which a binary stabilizer mixture of a colloidal sol and a soluble polymer has been evaluated. According to DLS studies, the final nanocomposite particle diameter ranged from 360 to 1190 nm (with larger values suggesting incipient flocculation) and the mean silica content could be varied from zero to 74%. Analysis confirmed that both the silica sol and the polyacrylamide co-stabilizer were incorporated into the nanocomposite particles, with the mean particle density being strongly dependent on the silica content as expected. In the present work we have extended Stejskal's ‘binary stabilizer’ concept to include vinyl polymerizations. More specifically, we have evaluated a binary mixture of PVP and a commercial 20 nm aqueous silica sol for the aqueous dispersion polymerization of HPMA, see Fig. 1. Given the relentless legislation-led drive towards zero volatile organic components (VOCs) in paint formulations, greater interest in the development of such wholly aqueous formulations based on commodity monomers is anticipated.
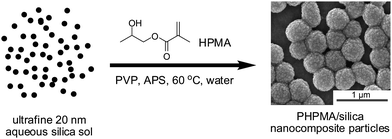 |
| | Fig. 1 Reaction scheme for the formation of poly(2-hydroxypropyl methacrylate)/silica nanocomposite particles by aqueous dispersion polymerization at 60 °C in the presence of both PVP and an ultrafine silica sol as the stabilizing agents. | |
Experimental section
Materials
2-Hydroxypropyl methacrylate (HPMA, 97%, Aldrich) was purified by passing through a column containing activated basic aluminium oxide and then stored at −20 °C prior to use. HPMA monomer is actually an isomeric mixture comprising approximately 75% of 2-hydroxypropyl methacrylate and 25% of 2-hydroxyisopropyl methacrylate.29 The aqueous silica sol (Bindzil 2040, Eka Chemicals, Bohus, Sweden) was supplied as a nominal 20 nm diameter at 40 w/w% solids. However, our in-house studies suggested that this sol actually had a solids content of around 42%. Both poly(N-vinylpyrrolidone) (PVP, K30, Mw = 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Acros) and ammonium persulfate (APS, Aldrich) were used as received. 2-(Methacryloyloxy)ethyl phosphorylcholine (MPC) was a gift from Biocompatibles Ltd; potassium 3-sulfopropyl methacrylate (KSPMA) and [2-(methacryloyloxy) ethyl]trimethyl ammonium chloride (QDMA) comonomers were both purchased from Aldrich; all the three comonomers were used as received. In certain control experiments, a commercial glycerol-modified aqueous silica sol (Bindzil CC40, 12 nm nominal diameter at 40 w/w %, also from Eka Chemicals), PVP with other molecular weights (K25, Mw = 24,000; K90, Mw = 360000, both from Aldrich), anionic initiator4,4′-azobis(4-cyanovaleric acid) (ACVA, Acros), cationic initiator2,2′-azobis(2-amidinopropane) dihydrochloride (AIBA, Aldrich), and a neutral initiator 2, 2′-azobisisobutyronitrile (AIBN, BDH) were used as received. All water used in these experiments was deionized using an Elgastat Option 3A water purifier.
000, Acros) and ammonium persulfate (APS, Aldrich) were used as received. 2-(Methacryloyloxy)ethyl phosphorylcholine (MPC) was a gift from Biocompatibles Ltd; potassium 3-sulfopropyl methacrylate (KSPMA) and [2-(methacryloyloxy) ethyl]trimethyl ammonium chloride (QDMA) comonomers were both purchased from Aldrich; all the three comonomers were used as received. In certain control experiments, a commercial glycerol-modified aqueous silica sol (Bindzil CC40, 12 nm nominal diameter at 40 w/w %, also from Eka Chemicals), PVP with other molecular weights (K25, Mw = 24,000; K90, Mw = 360000, both from Aldrich), anionic initiator4,4′-azobis(4-cyanovaleric acid) (ACVA, Acros), cationic initiator2,2′-azobis(2-amidinopropane) dihydrochloride (AIBA, Aldrich), and a neutral initiator 2, 2′-azobisisobutyronitrile (AIBN, BDH) were used as received. All water used in these experiments was deionized using an Elgastat Option 3A water purifier.
Particles synthesis
Most polymerizations were conducted using a Tornado IS6 Carousel Reaction Station (Radleys Discovery Technologies, UK) equipped with six 250 ml two-necked, round-bottomed glass flasks, six overhead Teflon stirrers, a water-cooled aluminium reflux condenser, a stainless steel central inlet/outlet for vacuum and inert gas, and a heating system. This Tornado unit conveniently allows up to six latex syntheses to be conducted simultaneously at a constant stirring rate.
A typical PHPMA/silica nanocomposite synthesis was carried out as follows: the appropriate amount of the aqueous silica sol (Bindzil 2040, 2.50 g of aqueous dispersion, equivalent to 1.05 g of dry silica), 0.50 g of PVP, 5.00 g of HPMA and 44.55 g of deionized water were degassed using a nitrogen purge for at least 30 min with continuous stirring at 20 °C. The reaction mixture was heated to 60 °C under a nitrogen atmosphere. The APSinitiator (50.0 mg; 1.0 wt % based on HPMA) was dissolved in degassed deionized water (4.0 g) and added to the reaction solution, giving a total water content of 50 g. Each polymerization was allowed to continue for 24 h at 200 rpm. Variation of these synthesis parameters enabled nanocomposite particles to be prepared over a wide particle size range with good control over the size distribution. Monomer conversions were typically greater than 95% at 60 °C. Control experiments were also conducted using the above protocol with the Bindzil CC40silica sol, either AIBA, ACVA or AIBNinitiator and in the presence or absence of the PVP stabilizer. Finally, HPMApolymerizations were conducted in the presence of the PVP stabilizer but in the absence of any silica sol.
The resulting milky-white colloidal dispersions were purified by repeated centrifugation redispersion cycles (3000–5000 rpm for 30 min.), with each successive supernatant being carefully decanted and replaced with deionized water. Excessive centrifugation rates (>8000 rpm) and times (>1 h) were avoided, since this results in the unwanted sedimentation of some of the excess silica sol and also makes redispersion of the sedimented nanocomposite particles much more difficult.
DLS was used to obtain intensity-average hydrodynamic particle diameters and polydispersities by analyzing the scattered light at 173°. Studies were conducted at 25 °C using a Malvern Zetasizer Nano ZS equipped with a 4 mW He–Ne solid-state laser operating at 633 nm. All measurements were performed in triplicate on highly dilute aqueous dispersions. Polydispersities are reported directly by the Malvern instrument software. According to the manual,30 cumulants analysis of dynamic light scattering data gives two parameters: a mean diameter and a width parameter known as the polydispersity (or polydispersity index, PDI). The cumulants analysis involves a polynomial fit to the log of the G1 correlation function, such that Ln[G1] = a + bt + ct2 + dt3 + et4 + …The value of b is known as the second-order cumulant, or the z-average diffusion coefficient. This is converted to a size using the known solution viscosity and several instrumental constants. Only the first three coefficients (a, b and c) are used in the standard analysis to avoid over-interpreting the data. The coefficient c of the squared term, when scaled as 2c/b2, is the polydispersity. Calculations for these parameters are defined in the ISO standard document 13321:1996 E.
Disk centrifuge photosedimentometry (DCP)
DCP (CPS Disk Centrifuge instrument, Model DC24000) was used to obtain the weight-average particle size distributions of the purified aqueous latexes. Solid-state particle densities were measured by helium pycnometry (Micromeritics AccuPyc 1330 instrument).
Thermogravimetric analyses (TGA)
TGA were conducted using a Perkin-Elmer Pyris 1 TGA instrument. Dried samples were heated in air to 800 °C at a heating rate of 10 °C min−1, and the observed mass loss was attributed to the quantitative degradation of the organic component, with the remaining incombustible residues being assumed to be that of pure silica (SiO2).
Silica incorporation efficiency10b
Monomer conversions were determined gravimetrically by measuring the solids content of the reaction solutions after drying to constant weight at 150 °C using an Ohaus M45 moisture analyzer. The monomer conversion was calculated as the difference between the experimental solids content after polymerization and the theoretical solids content at zero conversion normalized to the difference between the theoretical solids content at complete conversion and the theoretical solids content at zero conversion. The silica sol incorporation efficiency was determined using this monomer conversion together with the silica content of the purified nanocomposite particles obtained from carbon microanalyses and confirmed by TGA studies.
TEM samples were prepared by drying a drop of a dilute aqueous dispersion onto a carbon-coated copper grid, followed by analysis using a Philips CM100 instrument operating at 100 kV. SEM studies were performed on a Philips Inspect F scanning electron microscope. Samples were dried on adhesive carbon disks and sputter-coated with a thin layer of gold prior to examination.
Kinetic studies
Particle growth and monomer conversion curves were determined by periodic sampling of the mechanically-stirred polymerizing solution conducted on a larger scale. Efficient quenching of the HPMApolymerization was achieved by immersing in an ice-water mixture after exposure to air. Solids contents were determined gravimetrically with an Ohaus M45 moisture analyzer at a drying temperature of 150 °C, and DLS particle diameters were determined after dilution using deionized water.
Aqueous electrophoresis measurements were conducted using a Malvern Zetasizer NanoZS instrument. Measurements were conducted in the presence of 1.0 mM KCl as background electrolyte. Typically, the solution was initially adjusted to approximately pH 11 using KOH and then titrated to pH 2.0 using HCl.
Results and discussion
Effect of the presence of PVP, initiator type and silica sol type
Our preliminary experiments confirmed that the presence of PVP was crucial for the preparation of colloidally stable PHPMA/SiO2 nanocomposites. First, polymerizations were conducted using non-functionalized Bindzil 2040 silica sol with various initiators in the absence of PVP. Four types of initiator (anionic ACVA and APS, non-ionic AIBN, and cationic AIBA) were evaluated. In each case, there was no evidence for successful colloidal nanocomposite formation, with only macroscopic precipitation being observed. Polymerizations were also conducted using the glycerol-functionalized Bindzil CC40 silica sol with either anionic APS or cationic AIBAinitiators in the absence of any PVP stabilizer. It was found that this surface-modified CC40silica sol produced mainly colloidally stable nanocomposite particles, but some degree of coagulation (minor fraction) was also observed in both cases.
The importance of the judicious combination of anionic initiator and non-functionalized 2040 silica sol is also indicated in a comparative experiment. First, polymerizations were conducted using the Bindzil 2040 silica sol with various initiators in the presence of 1.50 g of PVP. For AIBN, there was no evidence for any nanocomposite formation. In contrast, colloidally stable nanocomposite particles were obtained in all cases when using the AIBA, ACVA and APSinitiators. However, those nanocomposites prepared using either AIBA or ACVAinitiators flocculated irreversibly on standing for several days. In contrast, the APS-synthesized nanocomposite particles remained stable with respect to aggregation on standing. Polymerizations were also conducted using Bindzil 2040 silica sol using a reduced amount of PVP (0.50 g) with either APS or AIBA. In this case the former initiator still produced stable nanocomposite particles, but the latter led to immediate coagulation. Moreover, some coagulation also occurred when HPMApolymerization was conducted using AIBA and Bindzil 2040 silica sol in the presence of 1.0 g of PVP. Thus it seems that APS is the most suitable initiator for this aqueous dispersion polymerization formulation. Finally, it is noteworthy that if colloidal nanocomposite syntheses were attempted using the glycerol-functionalized Bindzil CC40 silica sol with either AIBA or APS, complete coagulation was observed even in the presence of 1.50 g of PVP. Thus, unlike the observations made by Schmid et al.,10non-functionalizedsilica sols are definitely preferable for this particular HPMA-based formulation.
As both the APS-synthesized latex and the silica sol possess negative surface charge, this suggests that there is no electrostatic driving force for nanocomposite formation. A more likely explanation is the interaction between the PVP stabilizer chains and the silica surface. It is well known that PVP adsorbs onto (large) silica particles from aqueous solution,31,32 so it seems perfectly reasonable that small (in this case, 20 nm diameter) silica particles should adsorb onto large sterically-stabilized PHPMA latex (as in the present work). A similar explanation has already been invoked to explain the interaction between silicananoparticles and PEGMA-stabilized latexes.24 Thus, from the viewpoint of polymer thermodynamics, this favorable interaction provides a reasonable enthalpic driving force for silicananoparticles to be adsorbed onto PHPMA particles.
In summary, our preliminary experiments confirmed the importance of using an anionic persulfateinitiator, a non-functionalized aqueous silica sol and PVP as an additional stabilizer. This formulation proved to be robust: well-defined PHPMA/silica nanocomposite particles were obtained over a wide range of conditions. In contrast, surface-functionalized silica sol, and/or cationic azo initiator invariably led to either coagulation or flocculation, regardless of whether PVP was present or not.
Effect of varying the PVP molecular weight
Using the optimized formulation described above, the effect of varying the PVP molecular weight on the nanocomposite particle density, silica content, silica incorporation efficiency and nanocomposite particle diameter is summarized in Table 1. Three PVP molecular weights were evaluated: 24 K, 40 K and 360 K. SEM images are illustrated in Fig. 2 and the corresponding TEM images are shown in Fig. S1 (see ESI†). As the PVP molecular weight was increased, the mean particle diameter was reduced, as judged by DCP. However, DLS studies indicated a relatively broad size distribution for the nanocomposite dispersion prepared using the highest molecular weight of 360,000, which suggested that flocculation occurred in this case. This is consistent with the observation of a relatively low silica surface coverage of the nanocomposite particles, see Fig. 2c and Fig. S1c.† In contrast, SEM and TEM images of nanocomposite particles prepared using the two lower molecular weight PVP samples indicated much higher silica surface coverages, which is consistent with the improved colloidal stabilities observed for these aqueous dispersions. However, using the 40 K PVP led to a significantly higher silica incorporation efficiency (>99%). Hence this co-stabilizer was preferred for all subsequent nanocomposite syntheses.
Table 1 Effect of varying the PVP molecular weight on the synthesis of PHPMA/SiO2 colloidal nanocomposites prepared using 1.05 g of silica sol and 0.50 g of PVP
| Entry no. |
PVP type |
PVP Mw |
Particle density (g cm−3)a |
Silica content (wt %)b |
Silica incorporation efficiency (%) |
Hydrodynamic particle diameter (nm)c |
Weight-average particle diameter (nm)d |
|
As determined by helium pycnometry.
As determined by CHN microanalyses.
As measured by dynamic light scattering; polydispersities are given in brackets.
As measured by disk centrifuge photosedimentometry.
Incipient particle flocculation.
|
| 1 |
K25
|
24 K |
1.30 |
14 |
80 |
854 (0.03) |
681 ± 64 |
| 2 |
K30
|
40 K |
1.33 |
18 |
>99 |
652 (0.07) |
450 ± 57 |
| 3e |
K90
|
360 K |
1.32 |
18 |
>99 |
601 (0.26) |
275 ± 66 |
 |
| | Fig. 2
SEM images obtained for PHPMA/SiO2 particles prepared using a PVP stabilizer with a molecular weight of either 24 K, 40 K or 360 K combined with fixed amounts of silica (1.05 g) and PVP (0.50 g): (a) 24 K (entry 1 in Table 1); (b) 40 K (entry 2 in Table 1); (c) 360 K (entry 3 in Table 1). | |
Effect of varying the silica sol concentration
The initial silica sol concentration was systematically varied in order to improve its incorporation efficiency, as summarized in Table 2. Nanocomposite particles were first synthesized using 1.50 g of PVP. When the HPMApolymerization was conducted in the absence of any silica sol, substantial coagulation was observed and the size distribution of the purified latex was relatively broad as judged by both DLS and DCP. In the presence of 0.26 g of silica, polydisperse nanocomposite particles were obtained and DLS studies suggested incipient flocculation. At 0.53 g of silica, well-defined colloidal nanocomposite particles with a relatively low polydispersity were produced that exhibited good colloidal stability. This suggests that, in addition to the PVP co-stabilizer, the silica particles also play a significant stabilizer role in such formulations. Using the equation reported in our previous study,10b the silica incorporation efficiency was estimated to be close to 100% when either 0.26 g or 0.53 g of silica sol was used. The presence of the ultrafine silica at the surface of the nanocomposite particles was confirmed by SEM studies, which revealed a distinctive ‘raspberry’ morphology compared to the relatively smooth latex particles prepared in the absence of any silica (see Fig. 3). Higher silica surface coverages were observed on increasing the silica sol concentration (compare Fig. 3b to 3f), with a concomitant reduction in the mean particle diameter, as judged by both DLS and DCP. However, higher silica sol concentrations also led to significantly lower silica incorporation efficiencies. The mean silica contents of these PHPMA/SiO2 nanocomposite particles ranged from 10 to 17% by mass, which were broadly consistent with the particle density range of 1.27 to 1.32 g cm−3. It is perhaps noteworthy that the PHPMA latex particles and PHPMA-silica nanoparticles were not perfectly spherical in the electron micrographs. The Tg for rigorously dried PHPMA is 95 °C as judged by differential scanning calorimetry, but this value is lowered to 47 °C (i.e. below the polymerization temperature) by plasticization with water.33 This is presumably the case in the present work since the particles are synthesized by aqueous dispersion polymerization. Given a reaction temperature of 60 °C, the nanocomposite particles are most likely formed above their Tg, which hence makes them more susceptible to deformation. Alternatively, the observed deviation from a spherical morphology may be simply due to drying artifacts during preparation of electron microscopy samples.
Table 2 Effect of varying the silica sol concentration on the synthesis of PHPMA/SiO2 nanocomposite particles prepared using either 1.50 g or 0.50 g of PVP (40 K)a
| Entry no. |
Initial PVP mass (g) |
Initial silica mass (g) |
Particle density (g cm−3)b |
Silica content (wt %)c |
Silica incorporation efficiency (%) |
Hydrodynamic particle diameter (nm)d |
Weight-average particle diameter (nm)e |
|
Entry 13 in this Table is identical to entry 2 in Table 1.
As determined by helium pycnometry.
As determined by CHN elemental microanalyses.
As measured by dynamic light scattering; polydispersities are given in brackets.
As measured by disk centrifuge photosedimentometry.
Substantial coagulation.
Incipient particle flocculation.
Complete coagulation.
|
| 1f |
1.50 |
0.00 |
1.22 |
— |
— |
1088 (0.183) |
1153 ± 353 |
| 2g |
1.50 |
0.26 |
1.26 |
7 |
>99 |
887 (0.111) |
669 ± 207 |
| 3 |
1.50 |
0.53 |
1.27 |
10 |
>99 |
794 (0.062) |
650 ± 69 |
| 4 |
1.50 |
1.05 |
1.27 |
12 |
64 |
743 (0.020) |
545 ± 55 |
| 5 |
1.50 |
1.58 |
1.28 |
13 |
48 |
737 (0.077) |
526 ± 61 |
| 6 |
1.50 |
2.10 |
1.30 |
15 |
43 |
637 (0.051) |
410 ± 42 |
| 7 |
1.50 |
3.15 |
1.29 |
14 |
25 |
599 (0.015) |
334 ± 34 |
| 8 |
1.50 |
4.20 |
1.29 |
13 |
18 |
557 (0.040) |
— |
| 9 |
1.50 |
6.30 |
1.32 |
17 |
16 |
480 (0.017) |
— |
| 10h |
0.50 |
0.00 |
— |
— |
— |
— |
— |
| 11h |
0.50 |
0.26 |
— |
— |
— |
— |
— |
| 12 |
0.50 |
0.53 |
1.29 |
11 |
>99 |
902 (0.127) |
635 ± 75 |
| 13 |
0.50 |
1.05 |
1.33 |
18 |
>99 |
652 (0.074) |
450 ± 57 |
| 14 |
0.50 |
1.58 |
1.31 |
17 |
65 |
646 (0.037) |
448 ± 41 |
| 15 |
0.50 |
2.10 |
1.30 |
13 |
36 |
622 (0.058) |
413 ± 32 |
| 16 |
0.50 |
3.15 |
1.28 |
11 |
20 |
539 (0.045) |
372 ± 34 |
| 17 |
0.50 |
4.20 |
1.28 |
10 |
13 |
577 (0.054) |
392 ± 54 |
| 18 |
0.50 |
6.30 |
1.30 |
12 |
11 |
551 (0.055) |
370 ± 49 |
 |
| | Fig. 3 Representative SEM images obtained for PHPMA/SiO2 particles prepared using various SiO2 concentrations in the presence of 1.50 g of PVP: (a) 0 g of SiO2 (entry 1 in Table 2); (b) 0.26 g of SiO2 (entry 2 in Table 2); (c) 0.53 g of SiO2 (entry 3 in Table 2); (d) 1.05 g of SiO2 (entry 4 in Table 2); (e) 2.10 g of SiO2 (entry 6 in Table 2); (f) 6.30 g of SiO2 (entry 9 in Table 2). | |
In order to reduce the amount of PVP co-stabilizer used in these syntheses, the initial silica concentration was also systematically varied at a fixed 0.50 g of PVP 40 K (see Fig. S2). When either zero or 0.26 g of silica was utilized, complete coagulation occurred in both cases. In contrast, when using 1.50 g of PVP 40 K under the same conditions, the formulation was at least partially successful: some colloidal nanocomposite particles were obtained along with some coagulum. This difference further supports our hypothesis that both the PVP and the silica sol are required for the formation of well-defined PHPMA/SiO2 nanocomposite particles with reasonable colloidal stability. Increasing the initial silica mass from 0.53 to 3.15 g led to smaller mean nanocomposite particle diameters. However, on further increasing the initial silica mass from 3.15 to 6.30 g, the intensity-average particle diameter was almost unchanged, ranging from 539 to 577 nm. Like the series of nanocomposites prepared using 1.50 g of PVP, only partial surface coverage and relatively broad size distributions were achieved when 0.53 g of silica sol was used. Moreover, higher amounts of silica sol also led to significantly lower silica incorporation efficiencies, as confirmed by TEM studies (data not shown). Thus the optimum silica sol mass when using 0.50 g of PVP seems to be 1.05 g (see entry 13 in Table 2). Under these conditions, a relatively narrow particle size distribution was obtained, the silica incorporation efficiency was close to 100%, and a relatively high surface coverage of silica on the PHPMA/SiO2 particles was achieved (see Fig. 2b). The mean silica contents ranged from 10 to 18% by mass and the mean particle densities varied from 1.28 to 1.33 g cm−3. In summary, lower silica sol concentrations are desirable for achieving higher silica incorporation efficiencies.
Effect of varying the PVP concentration
The effect of varying the PVP concentration was studied using the optimized silica mass of 1.05 g. The results are summarized in Table 3, Fig. 4 and Fig. S3.† As indicated above, when HPMApolymerizations were conducted in the absence of any PVP, gross coagulation invariably occurred. Relatively large nanocomposite particles with rather broad size distributions were obtained when 0.25 g of PVP was used, which suggests some incipient flocculation. Increasing the PVP concentration from 0.50 to 4.0 g produced stable colloidal dispersions with reasonably narrow size distributions, with mean intensity-average particle diameters ranging from 652 to 762 nm. This suggests that the particle size of these PHPMA/SiO2 nanocomposites is surprisingly insensitive to the PVP co-stabilizer concentration. This differs from our earlier report of the synthesis of sterically-stabilized PHPMA latexes via aqueous dispersion polymerization,25 for which a higher PVP stabilizer concentration generally produced smaller particles.
Table 3 Effect of varying the PVP concentration on the synthesis of PHPMA/SiO2 nanocomposite particles in the presence of a constant amount of silica sol (1.05 g)a
| Entry no. |
Initial PVP mass (g) |
Particle density (g cm−3)b |
Silica content (wt %)c |
Silica incorporation efficiency (%) |
Hydrodynamic particle diameter (nm)d |
Weight-average particle diameter (nm)e |
|
In this Table, entry 3 is identical to entry 2 in Table 1 and entry 5 is identical to entry 4 in Table 1.
As determined by helium pycnometry.
As determined by CHN elemental microanalyses.
As measured by dynamic light scattering; polydispersities are given in brackets.
As measured by disk centrifuge photosedimentometry.
Complete coagulation.
Incipient particle flocculation.
|
| 1f |
0 |
— |
— |
— |
— |
— |
| 2g |
0.25 |
1.33 |
18 |
>99 |
859 (0.181) |
556 ± 273 |
| 3 |
0.50 |
1.33 |
18 |
>99 |
652 (0.074) |
450 ± 57 |
| 4 |
1.00 |
1.30 |
16 |
89 |
720 (0.040) |
488 ± 42 |
| 5 |
1.50 |
1.27 |
12 |
64 |
743 (0.020) |
545 ± 55 |
| 6 |
2.00 |
1.27 |
10 |
55 |
762 (0.037) |
571 ± 58 |
| 7 |
3.00 |
1.28 |
11 |
59 |
689 (0.027) |
467 ± 52 |
| 8 |
4.00 |
1.27 |
9 |
49 |
701 (0.055) |
— |
 |
| | Fig. 4
TEM images obtained for PHPMA/SiO2 nanocomposite particles prepared using a fixed mass (1.05 g) of silica and the following masses of PVP: (a) 0.25 g (entry 2 in Table 3); (b) 4.00 g (entry 8 in Table 3). | |
This observation also seems to be consistent with the study by Stejskal et al.,28 who found that the water-soluble polymeric stabilizer (in their case, polyacrylamide) was less efficiently incorporated into the final nanocomposite particles compared to the ultrafine silica sol. In other words, the silica acts as the primary stabilizer in both formulations. However, it seems that both the silica content of these nanocomposite particles and also the silica incorporation efficiency were reduced when using higher PVP concentrations. Moreover, there is an optimal PVP concentration for the production of PHPMA/SiO2 nanocomposite particles with relatively narrow size distributions. It is also noteworthy that at least some of these dispersions were weakly flocculated. Relatively large differences between the DLS diameter and the diameter estimated from electron microscopy are a good indication of significant flocculation.
Effect of varying the HPMA concentration
Having optimized the masses of PVP and silica sol in the formulation, the effect of varying the monomer concentration was investigated (see Table 4, Fig. 5 and Fig. S4†). More specially, nanocomposite particles were prepared using 0.50 g of PVP and 1.05 g of silica sol with 2.5 to 20.0 g of HPMA. It is known that the water-solubility of HPMA monomer is 13 w/w % at 25 °C,25 which is sufficient to allow an aqueous dispersion polymerization protocol to be formulated at reasonably high solids. In the present study, the HPMApolymerization was conducted at 60 °C. When the HPMA mass was increased from 2.50 to 17.5 g, nanocomposite particles were obtained with no macroscopic precipitation and the final solids content increased from 7.8 up to 29.8%. However, using 20.0 g of HPMA led to catastrophic coagulation. The mean DLS particle diameter increased monotonically from 535 to 1993 nm with increasing HPMA concentration, and the particle density was systematically reduced from 1.34 to 1.25 g cm−3. It is well-known that DLS is highly biased towards larger particles, thus there are significant differences between the mean DLS diameters and those estimated by electron microscopy studies, particularly for the rather broad size distributions obtained when using higher HPMA masses.7b,8 On the other hand, the DCP and DLS data shown in Table 4 may also indicate some degree of flocculation for nanocomposite particles prepared at higher solids (for example, see entries 6 and 7). Nevertheless, nanocomposite particles could be prepared with a relatively narrow size distribution and minimal flocculation at around 20% solids (see entry 5 in Table 4). We anticipate that even higher solids contents should be achievable using monomer-starved conditions, rather than our current one-pot batch protocol. Relatively high silica incorporation efficiencies were obtained when using 5.0 g or more of HPMA, although lower mean silica contents for these colloidal nanocomposites were also observed.
Table 4 Effect of varying the HPMA concentration for the synthesis of PHPMA/SiO2 nanocomposite particles at 60 °C using fixed masses of silica sol (1.05 g) and PVP co-stabilizer (0.50 g)a
| Entry no. |
Initial HPMA mass (g) |
Final solids content (%) |
Particle density (g cm−3)b |
Silica content (wt %)c |
Silica incorporation efficiency (%) |
Hydrodynamic particle diameter (nm)d |
Weight-average particle diameter (nm)e |
|
Entry 2 in this Table is identical to entry 2 in Table 1.
As determined by helium pycnometry.
As determined by CHN elemental microanalyses.
As measured by dynamic light scattering; polydispersities are given in brackets.
As measured by disk centrifuge photosedimentometry.
Incipient particle flocculation.
Complete coagulation.
|
| 1 |
2.5 |
7.8 |
1.34 |
22 |
66 |
535 (0.032) |
385 ± 55 |
| 2 |
5.0 |
12.4 |
1.33 |
18 |
>99 |
652 (0.074) |
450 ± 57 |
| 3 |
7.5 |
16.7 |
1.29 |
13 |
>99 |
773 (0.047) |
641 ± 62 |
| 4 |
10.0 |
19.8 |
1.27 |
11 |
>99 |
896 (0.083) |
713 ± 94 |
| 5 |
12.5 |
24.8 |
1.27 |
9 |
>99 |
1253 (0.297) |
944 ± 109 |
| 6f |
15.0 |
26.8 |
1.27 |
9 |
>99 |
1541 (0.544) |
— |
| 7f |
17.5 |
29.8 |
1.25 |
7 |
>99 |
1993 (0.908) |
— |
| 8g |
20.0 |
— |
— |
— |
— |
— |
— |
 |
| | Fig. 5
SEM images obtained for PHPMA/SiO2 nanocomposite particles prepared using 1.05 g of silica sol and 0.5 g of PVP 40 K co-stabilizer with the following masses of HPMA monomer: (a) 2.50 g (entry 1 in Table 4); (b) 10.0 g (entry 4 in Table 4); (c) 17.5 g (entry 7 in Table 4). | |
 |
| | Fig. 6
SEM image of the nanocomposite particles produced after statistical copolymerization of 5.0 g of HPMA with 0.50 g of MPC at 60 °C in the presence of 0.50 g of PVP and 1.05 g of silica sol. Note the unusual non-spherical particle morphology obtained in this case. | |
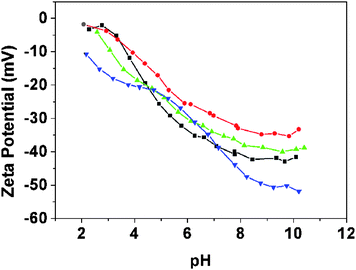 |
| | Fig. 7 Aqueous electrophoresis curves obtained for three PHPMA/SiO2 nanocomposite particles prepared using a PVP (40 K) mass of (■) 0.25 g, (▲) 0.50 g and (●) 4.00 g (see entries 2, 3 and 8 in Table 3) and also (▼) the pristine Bindzil 2040 silica sol as a reference. | |
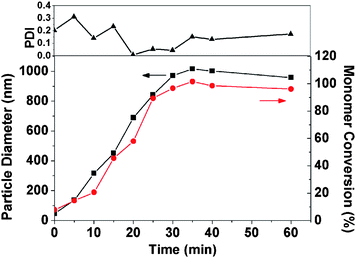 |
| | Fig. 8 Evolution of monomer conversion and particle growthvs. time for the synthesis of PHPMA/SiO2 nanocomposite particles at 60 °C. Conditions: 10.0 g of HPMA, 3.0 g of PVP (40 K) and 1.05 g of Bindzil 2040 silica sol in 100 g of water using 100 mg of APSinitiator and mechanically stirred at 200 rpm. | |
As shown in Fig. 8, after approximately 30 min, an intensity-average particle diameter of approximately 970 nm was attained at a monomer conversion of over 95%. Similar results were obtained for HPMApolymerizations conducted using alternative PVP and silica sol concentrations: in each case the minimum time period required for more than 95% HPMA conversion was around 35 min (see Fig. S6†). This is a significantly faster rate of polymerization than that observed during the synthesis of an 85:15 (HPMA-4VP)/SiO2 colloidal nanocomposite via aqueous dispersion polymerization at 25 °C, which required around 3 h for approximately 95% conversion.8 In this latter formulation, the lower conversion rate presumably results from the lower temperature, although it is also possible that the incorporation of the 4-vinylpyridine auxiliary comonomer may retard the overall rate of copolymerization. It is also noteworthy that the kinetics of HPMAhomopolymerization observed herein is much faster than that of styrene in the preparation of PS/SiO2 nanocomposites by aqueous emulsion polymerization,10b which required approximately 6–7 h at 60 °C to reach a styrene conversion of over 95%.
Particle formation mechanism
During the scaled-up synthesis described above, TEM images were obtained for the polymerizing reaction solution at various monomer conversions, see Fig. 9. Based on these observations, the following mechanism for nanocomposite particle formation is proposed, see Fig. 9. The nanocomposite particles are formed via three main steps. Initially, homogeneous nucleation occurs as the first PHPMA chains phase-separate from the aqueous solution and are subsequently coated by the silica sol and the PVP co-stabilizer that are present in the reaction medium. This leads to the formation of stable nascent nanocomposite nuclei. Then these nanocomposite nuclei become swollen with unreacted HPMA monomer. Further polymerization takes place within these growing particles. Presumably, there are relatively few free radicals per growing nanocomposite particle, which leads to minimal termination and a relatively fast rate of propagation. This may explain why some of the silicananoparticles appear to be incorporated within the interior of the nanocomposite particles. During this growth period, additional silica particles physically adsorb onto the growing particles to attain high surface coverage. Finally, at high HPMA conversion, the remaining silica is adsorbed and the particles reach their final dimensions.
 |
| | Fig. 9 (a) TEM images obtained for reaction solutions after the following polymerization times: 0 min, 10 min, 20 min, 30 min and 40 min. (b) Suggested scheme for the mechanism of formation of the PHPMA/SiO2 nanocomposite particles based on these TEM images. | |
Conclusions
In summary, an efficient route to colloidal vinyl (co)polymer-silica nanocomposite particles is reported. This new formulation involves aqueous dispersion polymerization of 2-hydroxypropyl methacrylate at 60 °C using a persulfateinitiator in combination with a non-functionalized ultrafine aqueous silica sol (Bindzil 2040) in the presence of a PVP co-stabilizer. Control experiments confirm the importance of the latter component, since colloidally stable nanocomposite particles are not obtained in its absence. Such nanocomposite particles usually have fairly uniform size distributions, mean particle diameters ranging from 480 to 900 nm, and mean silica contents ranging from 10 to 22%, depending on the precise synthesis conditions. SEM and TEM studies indicate well-defined ‘core-shell’ particle morphologies and silica-rich surfaces, which is consistent with aqueous electrophoresis measurements. However, a significant fraction of silica also appears to be located within the interior of the nanocomposite particles. Optimized concentrations of silica sol, PVP and HPMA monomer for a given set of conditions allow relatively high silica incorporation efficiencies to be obtained. Compared to previous studies, these poly(2-hydroxypropyl methacrylate)/silica nanocomposite particles are somewhat larger, but have lower silica contents. Finally, another interesting feature of this new formulation is the relatively fast rate of polymerization of the HPMA achieved at 60 °C.
Acknowledgements
We thank EPSRC for post-doctoral support of HZ (Platform grant EP/E012949/1). Dr P. Greenwood of Eka Chemicals (Bohus, Sweden) is thanked for donating the Bindzil 2040 and CC40 aqueous silica sols. Dr J. A. Balmer is acknowledged for obtaining the TEM images, Dr C. Shields is thanked for her assistance with the SEM studies, and Mr Lee Fielding is thanked for his help during the preparation of this manuscript.
References
- E. Bourgeat-Lami, J. Nanosci. Nanotechnol., 2002, 2, 1 CrossRef CAS.
- J. A. Balmer, A. Schmid and S. P. Armes, J. Mater. Chem., 2008, 18, 5722 RSC.
- H. Zou, S. S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893 CrossRef CAS.
- T. Wang and J. L. Keddie, Adv. Colloid Interface Sci., 2009, 147–148, 319 CrossRef CAS.
-
(a) M. Gill, J. Mykytiuk, S. P. Armes, J. L. Edwards, T. Yeates, P. J. Morel and C. Mollett, J. Chem. Soc., Chem. Commun., 1992, 108 RSC;
(b) S. Maeda and S. P. Armes, J. Colloid Interface Sci., 1993, 159, 257 CrossRef CAS;
(c) S. Maeda and S. P. Armes, J. Mater. Chem., 1994, 4, 935 RSC;
(d) M. G. Han and S. P. Armes, Langmuir, 2003, 19, 4523 CrossRef CAS.
-
(a) S. Maeda, M. Gill, S. P. Armes and I. W. Fletcher, Langmuir, 1995, 11, 1899 CrossRef CAS;
(b) M. D. Butterworth, S. Maeda, J. Johal, R. Corradi, S. F. Lascelles and S. P. Armes, J. Colloid Interface Sci., 1995, 174, 510 CrossRef CAS;
(c) S. Maeda and S. P. Armes, Synth. Met., 1995, 73, 151 CrossRef CAS.
-
(a) C. Barthet, A. J. Hickey, D. B. Cairns and S. P. Armes, Adv. Mater., 1999, 11, 408 CrossRef CAS;
(b) M. J. Percy, C. Barthet, J. C. Lobb, M. A. Khan, S. F. Lascelles, M. Vamvakaki and S. P. Armes, Langmuir, 2000, 16, 6913 CrossRef CAS;
(c) J. I. Amalvy, M. J. Percy, S. P. Armes and H. Wiese, Langmuir, 2001, 17, 4770 CrossRef CAS.
- M. J. Percy, V. Michailidou, S. P. Armes, C. Perruchot, S. J. Greaves and J. F. Watts, Langmuir, 2003, 19, 2072 CrossRef CAS.
- D. Dupin, A. Schmid, J. A. Balmer and S. P. Armes, Langmuir, 2007, 23, 11812 CrossRef CAS.
-
(a) A. Schmid, J. Tonnar and S. P. Armes, Adv. Mater., 2008, 20, 3331 CrossRef CAS;
(b) A. Schmid, S. P. Armes, C. A. P. Leite and F. Galembeck, Langmuir, 2009, 25, 2486 CrossRef CAS;
(c) A. Schmid, P. Scherl, S. P. Armes, C. A. P. Leite and F. Galembeck, Macromolecules, 2009, 42, 3721 CrossRef CAS;
(d) L. A. Fielding, J. Tonnar and S. P. Armes, Langmuir, 2011, 27, 11129 CrossRef CAS.
-
(a) J. L. Luna-Xavier, E. Bourgeat-Lami and A. Guyot, Colloid Polym. Sci., 2001, 279, 947 CrossRef CAS;
(b) J. L. Luna-Xavier, A. Guyot and E. Bourgeat-Lami, J. Colloid Interface Sci., 2002, 250, 82 CrossRef CAS;
(c) J. L. Luna-Xavier, A. Guyot and E. Bourgeat-Lami, Polym. Int., 2004, 53, 609 CrossRef CAS.
-
(a) S. Reculusa, C. Poncet-Legrand, S. Ravaine, C. Mingotaud, E. Duguet and E. Bourgeat-Lami, Chem. Mater., 2002, 14, 2354 CrossRef CAS;
(b) S. Reculusa, C. Mingotaud, E. Bourgeat-Lami, E. Duguet and S. Ravaine, Nano Lett., 2004, 4, 1677 CrossRef CAS;
(c) S. Reculusa, C. Poncet-Legrand, A. Perro, E. Duguet, E. Bourgeat- Lami, S. Mingotaud and S. Ravaine, Chem. Mater., 2005, 17, 3338 CrossRef CAS;
(d) A. Perro, S. Reculusa, E. Bourgeat-Lami, E. Duguet and S. Ravaine, Colloids Surf., A, 2006, 284–285, 78 CrossRef.
-
(a) M. Chen, L. M. Wu, S. X. Zhou and B. You, Macromolecules, 2004, 37, 9613 CrossRef CAS;
(b) M. Chen, S. X. Zhou and L. M. Wu, Macromolecules, 2005, 38, 6411 CrossRef CAS.
-
(a) M. J. Percy and S. P. Armes, Langmuir, 2002, 18, 4562 CrossRef CAS;
(b) M. J. Percy, J. I. Amalvy, S. P. Armes, S. J. Greaves and J. F. Watts, Langmuir, 2004, 20, 2184 CrossRef CAS.
-
(a) E. Bourgeat-Lami and J. Lang, J. Colloid Interface Sci., 1998, 197, 293 CrossRef CAS;
(b) E. Bourgeat-Lami and J. Lang, J. Colloid Interface Sci., 1999, 210, 281 CrossRef CAS;
(c) F. Corcos, E. Bourgeat-Lami, C. Novat and J. Lang, Colloid Polym. Sci., 1999, 277, 1142 CrossRef CAS;
(d) E. Bourgeat-Lami and J. Lang, Macromol. Symp., 2000, 151, 377 CrossRef CAS;
(e) S. Chalaye, E. Bourgeat-Lami, J. L. Putaux and J. Lang, Macromol. Symp., 2001, 169, 89 CrossRef CAS.
- K. Yoshinaga, T. Yokoyama, Y. Sugawa, H. Karakawa, N. Enomoto, H. Nishida and M. Komatsu, Polym. Bull., 1992, 28, 663 CrossRef CAS.
-
(a) A. Schmid, S. Fujii and S. P. Armes, Langmuir, 2005, 21, 8103 CrossRef CAS;
(b) A. Schmid, S. Fujii and S. P. Armes, Langmuir, 2006, 22, 4923 CrossRef CAS;
(c) A. Schmid, S. Fujii, S. P. Armes, C. A. P. Leite, F. Galembeck, H. Minami, N. Saito and M. Okubo, Chem. Mater., 2007, 19, 2435 CrossRef CAS.
- I. Sondi, T. H. Fedynyshyn, R. Sinta and E. Matijević, Langmuir, 2000, 16, 9031 CrossRef CAS.
-
(a) J. Leuninger, F. Tiarks, H. Wiese and B. Schuler, Farbe Lack, 2004, 110, 30 CAS;
(b)
Z. Xue and H. Wiese, International Patent WO0118081, 2001;
(c)
Z. Xue and H. Wiese, International Patent WO03000760, 2003.
- M. J. Burchell, M. Willis, S. P. Armes, M. A. Khan, M. J. Percy and C. Perruchot, Planet. Space Sci., 2002, 50, 1025 CrossRef CAS.
-
(a) S. Fujii, E. S. Read, S. P. Armes and B. P. Binks, Adv. Mater., 2005, 17, 1014 CrossRef CAS;
(b) S. Fujii, S. P. Armes, R. Murakami and B. P. Binks, Langmuir, 2006, 22, 6818 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 5775 CrossRef CAS.
- D. M. Qi, Y. Z. Bao, Z. X. Weng and Z. M. Huang, Polymer, 2006, 47, 4622 CrossRef CAS.
- J. A. Balmer, S. P. Armes, P. W. Fowler, T. Tarnai, Z. Gáspár, K. A. Murray and N. S. J. Williams, Langmuir, 2009, 25, 5339 CrossRef CAS.
- A. M. I. Ali, P. Pareek, L. Sewell, A. Schmid, S. Fujii, S. P. Armes and I. M. Shirley, Soft Matter, 2007, 3, 1003 RSC.
- J. Stejskal, P. Kratochvíl, S. P. Armes, S. F. Lascelles, A. Riede, M. Helmstedt, J. Prokeš and I. Křivka, Macromolecules, 1996, 29, 6814 CrossRef CAS.
- J. Stejskal, P. Kratochvíl and M. Helmstedt, Macromolecules, 1996, 12, 3389 CAS.
- J. Stejskal, T. Sulimenko, J. Prokeš and I. Sapurina, Colloid Polym. Sci., 2000, 278, 654 CAS.
- M. Save, J. V. M. Weaver, S. P. Armes and P. McKenna, Macromolecules, 2002, 35, 1152 CrossRef CAS.
- Malvern Instruments Ltd, Zetasizer Nano Series Manual, 2005 Search PubMed.
- T. Cosgrove, P. C. Griffiths and P. M. Lloyd, Langmuir, 1995, 11, 1457 CrossRef CAS.
- M. A. Cohen Stuart, G. J. Fleer and B. H. Bijsterbosch, J. Colloid Interface Sci., 1982, 90, 310 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2011, 133, 16581 CrossRef CAS.
- J. A. Balmer, O. O. Mykhaylyk, A. Schmid, S. P. Armes, J. P. A. Fairclough and A. J. Ryan, Langmuir, 2011, 27, 8075 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.