DOI:
10.1039/C1PY00381J
(Paper)
Polym. Chem., 2012,
3, 162-171
Hierarchical self-assembly and secondary structures of linear polypeptides graft onto POSS in the side chain through click chemistry†
Received
26th August 2011
, Accepted 20th October 2011
First published on 15th November 2011
Abstract
In this study, we used click chemistry to synthesize a new macromolecular self-assembling building block, linear polypeptide-g-polyhedral oligomeric silsesquioxane (POSS) copolymers, from a mono-azido-functionalized POSS (N3-POSS) and several poly(γ-propargyl-L-glutamate) (PPLG) oligomers. The incorporation of the POSS unit on the side chain of the PPLG moiety enhanced the α-helical conformation in the solid state, as determined by Fourier transform infrared spectroscopy, solid state nuclear magnetic resonance and wide-angle X-ray diffraction analysis. PPLG-g-POSS underwent hierarchical self-assembly, which was characterized using small-angle X-ray scattering and wide-angle X-ray diffraction analyses, to form a hexagonal cylinder packing nanostructure featuring α-helical conformations and POSS aggregates. The attachment of POSS onto the side chain of PPLG could stabilize the α-helical secondary structure with an increase in temperature, compared with pure PPLG by temperature-dependent FTIR analyses.
Introduction
Polypeptides, which are polymers, have received much interest because of their close relationship to proteins, their flexibility in functionality, and their molecular recognition properties. It is well-known that the secondary structure of a peptide chain plays a crucial role in the formation of a well-defined protein tertiary structure.1Polypeptides form hierarchically ordered structures, such as α-helices, which can be regarded as a rigid rod stabilized by intramolecular hydrogen bonding interactions, and β-sheets, a secondary structure stabilized by intermolecular hydrogen bonding interactions.2 Conformational studies of model polypeptides are an important step toward mimicking the biological activity of more complex proteins.3 For several decades, most methods for synthesizing poly(peptide-b-nonpeptide) (rod/coil) block copolymers, with potential applications in tissue engineering and drug delivery, have followed nature's strategies for producing these supramolecular bioactive assemblies.4–15 The nonpeptide blocks have often been used as macroinitiators; for example, polystyrene,11 poly(ethylene oxide),16 poly(dimethylsiloxane),9–17 poly(ε-caprolactone),18poly(N-isopropylacrylamide),19 poly(butadiene),20 poly(isoprene),21 poly(2-ethyl-2-oxazoline),22 and polyfluorene23 have been used. Klok et al. reported that the Fourier transform infrared (FTIR) spectra of poly(styrene-b-γ-benzyl-L-glutamate) (PS-b-PBLG) copolymers revealed significant stabilization of the α-helical secondary structure relative to those of the corresponding PBLG oligomers.11 We have previously combined the well-defined macromolecular architectures of polyhedral oligomeric silsesquioxane (POSS) and PBLG to generate polymeric building blocks (POSS-b-PBLG). We proposed that the incorporated POSS moieties on the chain ends of PBLG units would enhance the latter's α-helical conformations in the solid state.24–26 In those studies, the POSS moieties are only located on the chain end, and the amount of POSS in the polypeptide is extremely low. As a result, we linked the POSS moiety on the side chain of the polypeptide in this study and discussed how the self-assembly and secondary structures change from the polypeptide graft to the POSS nanoparticle.
Click chemistry has been widely used for the modification of polypeptides because of their high efficiency, specificity and tolerance of functional groups.27–30 For example, Hammond et al. proposed the high efficiency of grafting onto polypeptide side chains, while maintaining α-helical conformation of the poly(γ-propargyl-L-glutamate) (PPLG) polypeptide backbone with azide functional poly(ethylene oxide) (PEO-N3) using click chemistry.27 Chen et al. presented the preparation of glycopolypeptides with highly effective glycosylation by a click reaction of PPLG with sugar azide.28 In this study, we first synthesized the different degree of polymerizations through ring-opening polymerization (ROP) of γ-propargyl-L-glutamate N-carboxyanhydride using butylamine as an initiator. Secondly, the PPLG-g-POSS copolymers were synthesized from azido functionalized POSS (N3-POSS) and PPLGvia a click reaction as shown in Scheme 1. In this study, the secondary structures of PPLG and PPLG-g-POSS were characterized by Fourier transform infrared spectroscopy (FTIR), solid state nuclear magnetic resonance (NMR) and wide-angle X-ray diffraction (WAXD) analyses.
Experimental
Materials
Isobutyltrisilanol-POSS was obtained from Hybrid Plastics. Propargyl alcohol and L-glutamic acid were purchased from Tokyo Kasei Kogyo. Copper(I) bromide (CuBr) was purified by washing with glacial acetic acid overnight, followed by washing with absolute ethyl ether and then drying under vacuum. N,N-Dimethylformamide (DMF), sodium azide (NaN3), triethylamine (TEA), trichloro[4-(chloromethyl)phenyl]silane, N,N,N,N,N-pentamethyldiethylenetriamine (PMDETA, 99%), and trifluoroacetic acid-d (d-TFA) were purchased from Aldrich. All solvents were distilled prior to use. Mono-benzyl azide POSS (N3-POSS) was prepared according to our previous literature procedure.26
L-glutamic acid was cooled to lower than room temperature under vacuum, and excess propargyl alcohol was added to the reactor using a syringe. Following the addition of tert-butanol, the mixture became a white powder suspension. As in the procedure above, concentrated sulfuric acid was added slowly dropwise with a syringe while the reaction was being cooled on an ice bath. The solution gradually became clear, and the reaction was over within 24 h. The solution was quenched with TEA. Ethyl ether was then added, and the solution became a suspension. The suspension solution was put into a beaker, and an alcohol solution was carefully added until the white powder appeared. The resulting suspension was filtered to extract the residual white solid. The solid was washed three times with ethyl ether in a beaker, and the purified compound was collected by filtration.
Synthesis of N-carboxyanhydride from γ-propargyl-L-glutamates27,28
To a degassed mixture of γ-propargyl-L-glutamates, ethyl acetate was added under vacuum at lower than room temperature. As the above procedure was completed, N2 gas filled the reactor, and it was then heated to reflux. The solid triphosgene (1/3 mole ratio of γ-propargyl-L-glutamates) was added. The γ-Propargyl-L-glutamates were gradually dissolved to become a clear solution. The solution was stirred for 2–3 h at room temperature. The solution was filtered to collect the filtrate and extracted with a cold 0.5% sodium bicarbonate solution. The organic layer was separated and dried over anhydrous MgSO4 at 0 °C. Filtration and evaporation resulted in clear oil. The clear oil was dissolved in ethyl acetate and was passed through aluminium oxide. The solvent was removed under vacuum to yield a pure oily liquid.
The γ-propargyl-L-glutamate N-carboxyanhydride (PLG-NCA) was frozen using liquid nitrogen under vacuum conditions. DMF was added as a solventvia a syringe, and the solution was warmed to 0 °C. N2 gas then filled the reactor. Then, the initiator containing the aminegroup compounds was added and stirred for 48 h at a lower temperature. The residual polypeptide was collected by removing the solvent under vacuum. The solid residue was dissolved in DMF and precipitated with cold diethyl ether or methanol. The pure polymer products were collected by filtration, and the solid was dried under vacuum at room temperature.
Synthesis of PPLG-g-POSS polymersvia click chemistry
N3-POSS (2.27 g, 2.395 mmol), PPLG (0.20 g, 1.198 mmol), and CuBr (0.17 g, 1.180 mmol) were dissolved in DMF (25 mL) in a flask equipped with a magnetic stir bar. After one brief freeze/thaw/pump cycle, PMDETA (24.64 μL, 1.180 mmol) was added. The reaction mixture was then carefully degassed through three freeze/thaw/pump cycles, placed in an oil bath thermostatted at 60 °C, and stirred for 24 h. After evaporating all of the solvent under reduced pressure, the residue was dissolved in tetrahydrofuran (THF) and passed through a neutral alumina column to remove coppercatalysts. The concentrated THFsolvent was evaporated, and the product was precipitated by methanol. The resulting polymer products were filtered off and dried under vacuum at room temperature; they yielded the linear PPLG-g-POSS as a white powder.
Characterization
1H NMR spectra were recorded at room temperature using a Bruker AM 500 (500 MHz) spectrometer, with the residual proton resonance of the deuterated solvent acting as the internal standard. High-resolution solid-state 13C NMR experiments were carried out at room temperature using a Bruker DSX-400 Spectrometer operating at resonance frequencies of 399.53 and 100.47 MHz for 1H and 13C, respectively. Molecular weights and molecular weight distributions were determined through gel permeation chromatography (GPC) using a Waters 510 high-performance liquid chromatograph (HPLC) equipped with a 410 differential refractometer and three Ultrastyragel columns (100, 500, and 103 Å) connected in series, with DMF as the eluent (flow rate: 0.4 mL min−1). Thermal analysis through differential scanning calorimetry (DSC) was performed using a TA-Q20 operated at a scan rate of 10 °C min−1 over a temperature range of −90 to 200 °C under a N2 atmosphere. FTIR spectra of the polymer films were recorded using the conventional KBr disk method. The films used in this study were sufficiently thin to obey the Beer–Lambert law. FTIR spectra were recorded using a Bruker Tensor 27 FTIR spectrophotometer; 32 scans were collected at a spectral resolution of 1 cm−1. Because polymers containing amidegroups are hygroscopic, pure N2 gas was used to purge the spectrometer's optical box to maintain dry sample films. A mass spectrum was obtained using a Bruker Daltonics Autoflex III MALDI-TOF mass spectrometer. The following voltage parameters were employed: ion source 1, 19.06 kV; ion source 2, 16.61 kV; lens, 8.78 kV; reflector 1, 21.08 kV; and reflector 2, 9.73 kV. X-Ray diffraction (XRD) data were collected on the wiggler beamline BL17A1 of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. A triangular bent Si (111) single crystal was employed to obtain a monochromated beam with a wavelength (λ) of 1.3344 Å. The XRD patterns were collected using an imaging plate (IP; Fuji BAS III; area = 20 × 40 cm2) curved with a radius equivalent to the sample-to-detector distance (280 mm). The two-dimensional (2D) XRD patterns observed for the sample (typical diameter: 10 mm; thickness: 1 mm) were circularly averaged to obtain a one-dimensional (1D) diffraction profile I(Q). The value of Q was calibrated using standard samples of Ag-behenate and Si powder (NBS 640b). SAXS experiments were performed using the SWAXS instrument at the BL17B3 beamline of the NSRRC, Taiwan; the X-ray beam had a diameter of 0.5 mm and a wavelength (λ) of 1.24 Å. The blend samples (thickness: 1 mm) were sealed between two thin Kapton windows (thickness: 80 μm) and analyzed at room temperature.
Results and discussion
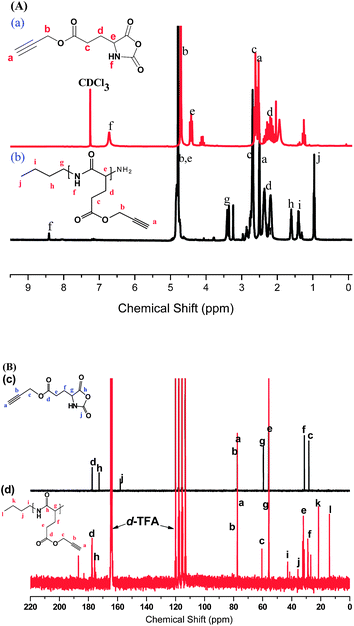
Fig.
1(A)-(a) presents the 1H NMR spectrum of the PLG monomer in CDCl3. The singlet at 2.50 and 4.70 ppm corresponds to HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C (protona) and CC–CH2 (protonb), respectively. We assigned the singlet at 6.72 ppm to the proton on the ring nitrogen atom; the other alkyl CH2protons appeared upfield as multiplets between 2.0 and 2.6 ppm. Fig. 1(A)-(b) displays the 1H NMR spectrum of PPLG5 in d-TFA. The signal for the proton on the nitrogen atom of PPLG appeared as a singlet at 8.41 ppm; the singlet at 2.50 and 4.78 ppm corresponded to the HCC–C and CC–CH2protons, respectively, which is close to the PLG monomer. The signals of the butyl groups of butylamineinitiator were located at 0.95 (CH3C3H6, triplet), 1.40 (CH3CH2C2H4, multiplets), 1.60 (C2H5CH2CH2, multiplets), and 3.38 (C3H7CH2NH, triplet) ppm. We determined the molar masses of PPLG from their 1H NMR spectra using the equation:31,32
C–C (protona) and CC–CH2 (protonb), respectively. We assigned the singlet at 6.72 ppm to the proton on the ring nitrogen atom; the other alkyl CH2protons appeared upfield as multiplets between 2.0 and 2.6 ppm. Fig. 1(A)-(b) displays the 1H NMR spectrum of PPLG5 in d-TFA. The signal for the proton on the nitrogen atom of PPLG appeared as a singlet at 8.41 ppm; the singlet at 2.50 and 4.78 ppm corresponded to the HCC–C and CC–CH2protons, respectively, which is close to the PLG monomer. The signals of the butyl groups of butylamineinitiator were located at 0.95 (CH3C3H6, triplet), 1.40 (CH3CH2C2H4, multiplets), 1.60 (C2H5CH2CH2, multiplets), and 3.38 (C3H7CH2NH, triplet) ppm. We determined the molar masses of PPLG from their 1H NMR spectra using the equation:31,32
where Ig and Ic are intensities of the signals of the methyleneprotonsb on the side chains of PPLG and the methyleneprotonsg of the butylamineinitiator, respectively. Mbutylamine is the molar mass of the butylamineinitiator. Table 1 lists the DPs of PPLG used in this study as determined using 1H NMR spectroscopy.
Table 1 Characterizations of PPLG used in this study
|
Polymer
|
M
n
|
M
n
|
PDIb |
M
n
|
PDIc |
|
Determined from 1H NMR spectra.
Determined from GPC analysis.
Determined from MALDI-TOF.
|
|
PPLG5
|
830 |
3830 |
1.07 |
980 |
1.11 |
|
PPLG15
|
2500 |
7740 |
1.11 |
3600 |
1.04 |
|
PPLG30
|
5080 |
9860 |
1.08 |
4550 |
1.02 |
|
PPLG50
|
8350 |
21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 200 |
1.07 |
9020 |
1.01 |
Fig.
1(B)-(c) shows the 13C NMR spectrum of the PLG monomer in d-TFA because the alkyne carbons overlapped with d-chloroform. The signals of the alkynecarbon atoms of the PLG monomer appear at 77 ppm, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbon atom signals appear at 158, 172, and 177 ppm. All other carbon assignments are summarized in Fig. 1(B)-(c). Fig. 1(B)-(d) shows the 13C NMR spectrum of PPLG5 in d-TFA, revealing that the CO and amidecarbon atom signals appeared in the PPLG5 at 177 and 175 ppm, respectively, while those of the alkynecarbon atoms remain at 77 ppm. The signals for the amino acid α-carbon atoms (NHCOC) appeared at 55.7 ppm. Fig. 1(B)-(d) provides assignments of the remaining signals for the carbon atoms of PPLG.
O carbon atom signals appear at 158, 172, and 177 ppm. All other carbon assignments are summarized in Fig. 1(B)-(c). Fig. 1(B)-(d) shows the 13C NMR spectrum of PPLG5 in d-TFA, revealing that the CO and amidecarbon atom signals appeared in the PPLG5 at 177 and 175 ppm, respectively, while those of the alkynecarbon atoms remain at 77 ppm. The signals for the amino acid α-carbon atoms (NHCOC) appeared at 55.7 ppm. Fig. 1(B)-(d) provides assignments of the remaining signals for the carbon atoms of PPLG.
Fig. 2 displays FTIR spectra of the PLG-NCA monomers and corresponding polymers, PPLG, recorded at room temperature. The acetyleneCC stretching vibration at 2130 cm−1 and two typical CO stretching bands—at 1857 and 1786 cm−1, corresponding to the CO units b and c, respectively—confirm the formation of the NCA ring.29 The CO stretching bands at 1739 cm−1 correspond to free CO units d of the PLG NCA monomers. After ring-opening polymerization of the NCA monomer, the FTIR spectrum revealed the absence of the absorbances from the CO groupsb and c of the NCA rings and the appearance of new absorbance values at 1653, 1626 (e), and 1543 (f) cm−1, representing the amide bonds in the polymer backbone.28 In addition, the acetylene CC stretching vibration at 2130 cm−1 remained in PPLG. Our 1H, 13C NMR and FTIR spectroscopic analyses confirmed the successful synthesis of PPLG.
Fig. 3 shows the MALDI-TOF-MS of PPLG obtained from the different ratios of PLGNCA monomer to butylamineinitiator. It can be observed that the variation of the PPLG molecular weight based on the MALDI-TOF-MS spectrum matches well with that from GPC. The signals of every series in the MALDI-TOF-MS spectrum are separated by 167 Da, the molecular weight of a PLG unit. As observed by both MALDI-TOF-MS and GPC (Fig. S1†), peak maxima of these spectra clearly shift to high molecular weights with increasing ratios of the PLGNCA monomer to butylamine. Table 1 also lists the DPs and PDI of PPLG used in this study that was determined using GPC and MALDI-TOF MS analyses. The molecular weight determined from MALDI-TOF mass is lower than that from GPC but agrees well with the 1H NMR. This is most likely due to the systematic deviation of the overall elution volume of the PPLG from that of the standard PS.33
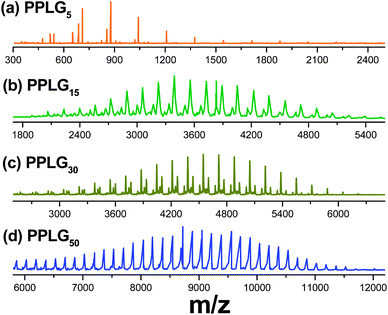 |
| | Fig. 3
MALDI-TOF MS of (a) PPLG5, (b) PPLG15, (c) PPLG30 and (d) PPLG50. | |
Synthesis of PPLG-g-POSS
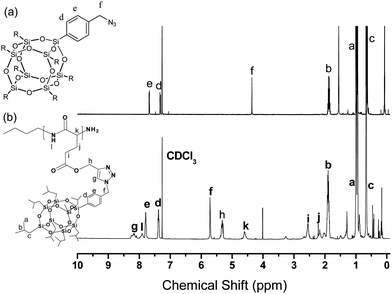
Fig. 4 presents the 1H NMR spectra of N3-POSS and PPLG-g-POSS. The signal of the benzyl CH2group connected to the azide atom at 4.36 ppm of N3-POSS shifted downfield significantly to 5.70 ppm for PPLG-g-POSS. In addition, the signals for the isobutyl groups attached to the silicon atoms of the POSS moiety appeared at approximately 0.63, 0.93, and 1.85 ppm. The proton on the nitrogen atom of PPLG resonated as a singlet at 7.90 ppm. The resonance at 8.20 ppm was due to the protons of the triazole structures resulting from the click reaction, confirming the successful synthesis of PPLG-g-POSS. Fig. 5 presents the 13C NMR spectra of PPLG and PPLG-g-POSS. Fig. 1(B) already provided the peak assignments for the spectra of PPLG. Fig. 5 reveals that the CO and amidecarbon atom signals appeared in the 13C NMR spectrum of PPLG-g-POSS at 172.0 ppm and 175.0 ppm, while those of the phenyl ring from N3-POSS appeared at 128.1, 132.2, 135.4, and 140.0 ppm. The signals for the benzylic carbon atom (C-h), the estercarbon atom (C-k) and the amino acid α-carbon atoms (NHCOC) appear at 50.0, 58.4 and 55.0 ppm (α-helical conformation), respectively. The signals for the alkanecarbon atoms from butylamine are absent, but those of the isobutylcarbon atoms of the POSS moiety were present at 0.2, 25.0, and 29.0 ppm (the latter two peaks overlapped with d-THF). The signals of the alkynecarbon atoms of PPLG that appeared at 73.4 ppm disappeared, but two new peaks at 125.1 ppm and 144.0 ppm were observed. These peaks are the carbons of the triazole structures resulting from the click reaction, confirming the successful synthesis of PPLG-g-POSS. The 29Si NMR spectrum shows two sharp signals at −66.7 and −66.1 ppm corresponding to the silicone atoms of the POSS molecules (Fig. S2†.). This observation supported the conclusion that PPLG-g-POSS was successfully obtained. FTIR spectroscopic analysis (Fig. 6) also confirmed the complete disappearance of the characteristic signals for the azido and acetylenegroups.
 |
| | Fig. 5
13C NMR spectra of (a) PPLG5 (in d-TFA) and (b) PPLG5-g-POSS (in d-THF). | |
 |
| | Fig. 6
FTIR spectra of (a) PPLG5, (b) PPLG5-g-POSS and (c) N3-POSS. | |
The signal at 2105 cm−1, corresponding to the absorbance of the azido group of N3-POSS, and the signal at 2130 cm−1, corresponding to the acetylenegroup of PPLG, were absent in the spectra of the PPLG-g-POSS. The absorption band of the Si–O–Si (siloxane) groups of the POSS moiety at 1100 cm−1 and the amide I group of the PPLG at 1655 cm−1 appeared in the PPLG-g-POSS spectra, indicating that the azido and acetylene functionalities had participated in the click reactions. The well-defined structure was confirmed by the MALDI-TOF MSspectra in Fig. 7, which shows only one apparent distribution of both PPLG and PPLG-g-POSS. It is clear from the spectrum that the mass difference between all adjacent two peaks is m/z 167 for PPLG and m/z 1103 for PPLG-g-POSS, exactly that of the PPLG and PPLG-g-POSS repeating unit, respectively. Taken together, the 1H NMR, 13C NMR, 29Si NMR, FTIR and MALDI-TOF MSspectra all confirmed the successful synthesis of PPLG-g-POSS.
Thermal analyses of PPLG-g-POSS copolymers
Fig. 8 presents the DSC thermograms of the PPLG and PPLG-g-POSS systems. During the second heating run of each of these systems, we observed only one glass transition temperature (Tg, at ca. 20 °C), which slightly increased upon increasing the degree of polymerization for PPLG.2 After grafting the POSS moiety onto PPLG, a dramatic change in the second heating run was observed by DSC analyses. Firstly, the glass transition behavior of PPLG-g-POSS significantly increased to ca. 90 °C. The value of Tg for PPLG-g-POSS was higher (by ca. 70 °C) than that of the PPLG. The value was also slightly increased with the degree of polymerization for PPLG-g-POSS. Secondly, the exothermic peak at ca. 120–140 °C and the endothermic peak at ca. 185 °C corresponded to the crystallization and the melt of the isobutyl-POSS moiety, respectively.33 The significant Tg increase of PPLG-g-POSS is due to the nano-reinforcement effect and rigid cubic silsesquioxane core of POSS on the PPLG segments that effectively restricts molecular chain motion.34–38 In addition, specific interactions may occur between POSS and PPLG in these graft polymers. The amidegroup of PPLG may possess hydrogen bonding interactions with the siloxanegroup of POSS and change the secondary structure of PPLG. This is most likely due to the β-sheet conformation of PPLG transforming into the α-helix rigid rod like structure after incorporation of the POSS unit in the PPLG-g-POSS system. The secondary structure transformation between PPLG and PPLG-g-POSS was characterized by Fourier transform infrared spectroscopy (FTIR), solid state nuclear magnetic resonance (NMR) and wide-angle X-ray diffraction (WAXD) analysis.
 |
| | Fig. 8
DSC traces (second heating runs) of (a) PPLG and (b) PPLG-g-POSS. | |
Conformation study of the peptide segment
FTIR studies at room temperature were performed to obtain information about the conformation of the peptide segment of the PPLG and PPLG-g-POSS as shown in Fig. 9. These spectra were first analyzed by the second derivative technique4 and indicated that the amideI band at 1655 cm−1 was characteristic of the α-helical secondary structure. For polypeptides possessing a β-sheet conformation, the position of the amideI band shifted to 1627 cm−1. Random coil or turn population is located at 1693 cm−1. The free carbonyl of the side chain group of PPLG was at 1740 cm−1 (Fig. 9(A)), and the peak at 1609 cm−1 corresponded to the benzene stretching of the side chain group of POSS (Fig. 9(B)). The second step was to use the deconvolution technique in a series of Gaussian distributions to quantify the fraction of each of the peaks, as shown in the inset of Fig. 9(A). Fig. 9(C) summarizes the curve fitting results of the amide I group for the β-sheet, α-helical and random coil structures. Clearly, the fraction of α-helical secondary structure increased with the increase in degree of polymerization of PPLG. This result is similar to the report by Papadopoulos et al. for PBLG.2 At low degrees of polymerization (DP < 18), both secondary structures are present, but as the degree of polymerization increases, the α-helical secondary structure is favored. In addition, all PPLG-g-POSS molecules after click reaction show α-helical secondary structure, even those with a relatively lower degree of polymerization (PPLG5-g-POSS). The result was different with POSS-b-PBLG; POSS was only located on the chain end of polypeptide, and the amount of POSS in the polypeptide was extremely low. As a result, the POSS-b-PBLG5 and even POSS-b-PBLG10 only had 51.4% and 74.2% α-helical conformation, respectively.26 Jeon et al. reported that the mobility of the side chain groups of polyglutamates affected the α-helical conformation.39 Their experimental data revealed that longer flexible side chains induced weaker hydrogen bonds between the CO groups and the amide linkages of the α-helical conformation. A corollary is that the rigid POSS moiety might enhance hydrogen bonding between the CO groups and the amide linkages of the α-helical conformation. The secondary structures of PPLG also can be identified on the basis of the distinctly different resonances observed in their solid state NMR spectra. The 13C CP/MASspectra of PPLG5 and PPLG5-g-POSS at room temperature are shown in Fig. 10. The peak assignments are similar to Fig. 5, but a broad band in solid state NMR was observed due to the dipole–dipole interaction and chemical shift anisotropy. The signals of the alkynecarbon atoms of PPLG at 75 ppm disappeared, but new peaks at 126 and 133 ppm were observed corresponding to the phenyl group of N3-POSS for PPLG-g-POSS, also indicating the synthesis of PPLG-g-POSS was successful. The different 13C chemical shifts of the Cα and amide CO resonances are attributed to the local conformations of individual amino acid residues characterized by the dihedral angles and the type of intermolecular and intramolecular hydrogen bonding interactions. In the case of PPLG, the chemical shift of the Cα and amide CO resonances corresponding to the α-helical secondary structure are at 57.5 and 176 ppm, respectively. In the β-sheet conformation, these chemical shifts are displayed upfield by ca. 4–5 ppm relative to the α-helical values at 52.7 and 172 ppm.2 Since the amide CO resonance in the β-sheet conformation partially overlaps with the signal from the side-chain ester, the distinction of the peptide secondary structure is best from the distinctly different Cα resonances. Clearly, PPLG5-g-POSS after click reaction has a higher fraction of α-helical secondary structure than the corresponding PPLG5 with the same degree of polymerization of polypeptide. This phenomenon can also be characterized from wide-angle X-ray diffraction at 393 K, identifying the secondary structure change of PPLG and PPLG-g-POSS as shown in Fig. 11.
 |
| | Fig. 9
FTIR spectra of (A) PPLG species incorporating (a) PPLG5, (b) PPLG15, (c) PPLG30 and (d) PPLG50, (B) PPLG-g-POSS species incorporating (e) PPLG5-g-POSS, (f) PPLG15-g-POSS, (g) PPLG30-g-POSS and (h) PPLG50-g-POSS, and (C) secondary structures of PPLG and PPLG-g-POSS systems featuring different degrees of polymerization. | |
 |
| | Fig. 11
WAXD patterns (recorded at 393 K) of (a) PPLG and (b) PPLG-g-POSS species featuring with different degrees of polymerization. | |
The effects of chain length after grafting of POSS onto PPLG and the type of secondary structure are discussed. For PPLG as shown in Fig. 11(a), the diffraction pattern of PPLG at degree of polymerization (DP = 5 and 15) shows the presence of β-sheet secondary structures. The first peak at q = 0.48 reflects the distance (d = 1.30 nm) between backbones in the antiparallel β-pleated sheet structure, and the reflection at q = 1.34 (d = 0.468 nm) represents the intermolecular distance between adjacent peptide chains within one lamella. Increases in the degree of polymerization of PPLG (DP = 50) result in the diffraction peaks at q = 0.48 associated with the disappearance of the β-sheet secondary structure. This suggests the absence of that particular conformation from longer peptides. The three reflections at higher angles, with positions 1:31/2:41/2 relative to the primary peak (q*), are indexed according to the (10), (11), and (20) reflections of a 2D hexagonal packing of cylinders composed of 18/5 α-helices with a cylinder distance of 1.18 nm.2 The structure of PPLG has been described as a nematic-like paracrystal with a periodic packing of α-helices in the direction lateral to the chain axis. The broad amorphous region at q = 1.54 originates mainly from the long amorphous side chain. Additionally, the α-helical conformations are better packed in the longer peptide. Decreasing the length of the PPLG chain (DP = 5 and 15) leads to a destabilization of the α-helical secondary structure. The β-sheet secondary structure was observed at q = 0.48 and 1.34. These results are in agreement with the FTIR analyses. From the FTIR and WAXD observations, we obtained the following information on PPLG: for low degree of polymerization, β-sheet secondary structures are favored, but as the degree of polymerization increases, the α-helical secondary structure is favored.
The WAXD patterns of the PPLG-g-POSS recorded at 393 K are presented in Fig. 11(b). For PPLG-g-POSS, there is a dramatic change in the diffraction pattern by X-ray analyses compared with PPLG. For a considerable fraction of the peptide segments, a sharp peak at q = 1.34 was not observed that corresponded to the β-sheet secondary structure when decreasing the length of the peptide segment (DP < 15) of PPLG-g-POSS. This result is different than that obtained for PPLG. Clearly, after click reaction, the PPLG-g-POSS had a higher fraction of the α-helical secondary structure than PPLG, which is consistent with FTIR analyses. As a result, the incorporation of the POSS moiety on the PPLG side chain end leads to the formation of the PPLG α-helix.
This conformation is stabilized through intramolecular hydrogen bonding interactions. From this information, it can be explained that even if peptides partly adopt the α-helical and β-sheet conformation when anchored to POSS as a side chain polymer, conformational stabilization occurs, and most of the peptide segments are constrained in the α-helical secondary structure. The strong Bragg peak at the diffraction angle of q = 0.58 in Fig. 11(b) is close to the α-helical conformation for PPLG. However, this peak should correspond to the POSS crystal, not the α-helical conformation of PPLG. This peak accompanies two major diffraction angles and d-spacing distance, where (dhkl)-q = 0.58 (10.82 Å), q = 0.77 (8.15 Å), q = 1.34 (4.69 Å), that corresponded to the (hkl) diffraction planes (101), (110), and (113) for a hexagonal POSS crystal with following lattice parameters: γ = 120°, α = β = 90°, a = b = 16.4 Å; c = 17.4 Å.40 This result is consistent with DSC analyses that reveal endothermic peaks at ca. 185 °C corresponding to the melt of the POSS moiety. Structurally speaking, with the incorporation of the rigid and bulky POSS moiety on the PPLG side chain, the distance of the 2D hexagonal packing of cylinders of α-helices should be expanded or swollen for PPLG-g-POSS. Fig. 11(b) reveals that the WAXD pattern of PPLG50-g-POSS features more than one strong additional diffraction angle (at q = 0.2) and a weak diffraction angle (at q = 0.35) relative to that of pure PPLG. These two reflections with relative position 1:31/2 correspond to 2D hexagonal packing of the cylinders for PPLG-g-POSS. To observe the correlation between the self-assembly of the hexagonally packed polypeptides and the crystallization of the POSS units, we conducted SAXS analyses of the PPLG-g-POSS systems (Fig. S3†.) The broad peak, assumed to be a Bragg reflection, at a value of q = 0.2 corresponded to an ordered spacing of ca. 31.4 Å for PPLG-g-POSS. This phenomenon is consistent with the WAXD data. Increasing the length of the PPLG chain did not change the diffraction peak position. Rather, it increased the scattering intensity, indicating a corresponding POSS moiety increase. Structurally speaking, PPLG tethers the POSS units on the side chain from its crystalline domains; meanwhile, the scattered POSS moieties gather and lock into the copolymers to form crystalline entities. The α-helical conformation cylinder organization of PPLG and PPLG-g-POSS is schematically depicted in Scheme 2. A 2D hexagonal packing of cylinders with a cylinder distance of 1.18 nm for PPLG (Scheme 2(a)), as calculated from the WAXD data, and the characteristic spacing (31.4 Å) observed from the SAXS data are consistent with the hexagonal packing cylinder structure of the PPLG-g-POSS copolymer (Scheme 2(b)).
 |
| | Scheme 2
Self-assembly structure of (a) PPLG and (b) PPLG-g-POSS. | |
Temperature-dependent FTIR studies were performed to obtain information about the conformation of the peptide segment. Fig. 12 summarizes the results of the FTIR studies on PPLG15 and PPLG15-g-POSS. The amide I and amide II bands at 1655 and 1545 cm−1, respectively, corresponded to the α-helical secondary structure. The amideI band shifted to 1626 cm−1, and the amideII band shifted to 1520 cm−1, both corresponding to the β-sheet conformation. Here, we chose PPLG15 oligomers because they possess both α-helical and β-sheet conformation. The longer PPLG50 nearly adopt the α-helical secondary structure, which is insensitive to changes in temperature. Clearly, the PPLG15-g-POSS indicated that the α-helical secondary structure is significantly stabilized upon attachment of the rigid POSS structure in comparison with the FTIR spectra of the PPLG homopeptide. The fraction of α-helical secondary structure of PPLG15 significantly decreased with the increase of temperature (Fig. 12(A)). The secondary structure almost did not change with the increase of temperature for PPLG-g-POSS (Fig. 12(B)). This result revealed that the degree of polymerization, the temperature, and the attachment of POSS all strongly influence the secondary structure. Increasing the degree of polymerization for all polypeptides would result in the stabilization of the α-helical secondary structure. Compared with pure PPLG, the attachment of POSS could also stabilize the α-helical secondary structure with the increase in temperature.
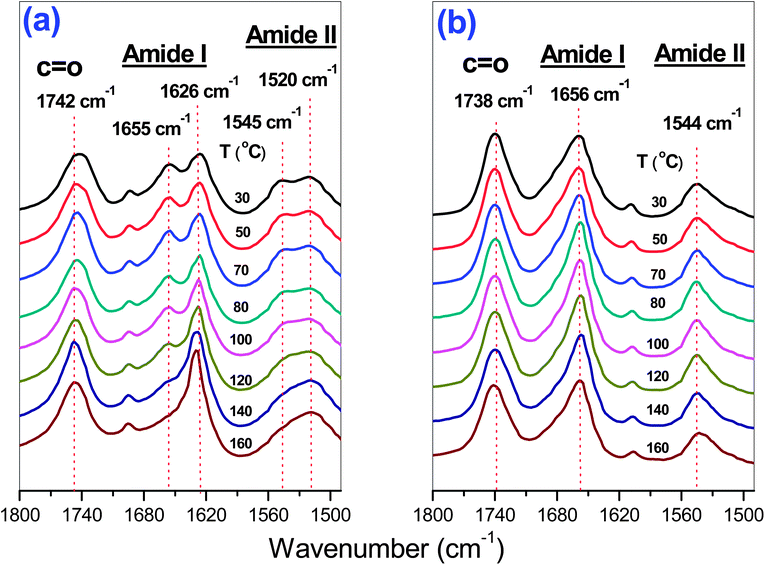 |
| | Fig. 12
FTIR spectra of (a) PPLG15 species and (b) PPLG15-g-POSS species at different temperatures. | |
Conclusions
We have prepared well-defined PPLG-g-POSS copolymers through ROP of Glu-NCA followed by click reactions with a monofunctional azide-POSS. After attaching the PPLG to the POSS nanoparticles on the side chain, the fraction of α-helical secondary structures increased. The PPLG-g-POSS systems exhibited greater conformation stability and superior thermal properties, including Tg behavior, relative to those of pure PPLG. PPLG-g-POSS underwent hierarchical self-assembly, which was characterized using small-angle X-ray scattering, to form a hexagonal packing cylinder nanostructure featuring α-helical conformations and POSS aggregates. Based on temperature-dependent FTIR analyses, the attachment of POSS could stabilize the α-helical secondary structure with the increase of temperature in comparison with pure PPLG.
Acknowledgements
This study was supported financially by the National Science Council, Taiwan, Republic of China, under contracts NSC 97-2221-E-110-013-MY3 and NSC 99-2628-E-110-003.
Notes and references
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- P. Papadopoulos, G. Floudas, H. A. Klok, I. Schnell and T. Pakula, Biomacromolecules, 2004, 5, 81–91 CrossRef CAS.
- A. Gitsas, G. Floudas, M. Mondeshki, H. W. Spiess, T. Aliferis, H. Iatrou and N. Hadjichristidis, Macromolecules, 2008, 41, 8072–8080 CrossRef CAS.
- A. Sanchez-Ferrer and R. Mezzenga, Macromolecules, 2010, 43, 1093–1100 CrossRef CAS.
- Q. H. Zhou, J. K. Zheng, Z. H. Shen, X. H. Fan, X. F. Chen and Q. F. Zhou, Macromolecules, 2010, 43, 5367–5646 Search PubMed.
- H. F. Lee, H. S. Sheu, U. S. Jeng, C. F. Huang and F. C. Chang, Macromolecules, 2005, 38, 6551–6558 CrossRef CAS.
- P. Papadopoulos, G. Floudas, I. Schnell, T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2005, 6, 2352–2361 CrossRef CAS.
- J. Rao, Y. Zhang, J. Zhang and S. Liu, Biomacromolecules, 2008, 9, 2586–2593 CrossRef CAS.
- E. Ibarboure, E. Papon and J. Rodriguez-Hernandez, Polymer, 2007, 48, 3717–3725 CrossRef CAS.
- E. Ibarboure and J. Rodriguez-Hernandez, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4668–4679 CrossRef CAS.
- H. A. Klok, J. F. Langenwalter and S. Lecommandoux, Macromolecules, 2000, 33, 7819–7826 CrossRef CAS.
- S. Lecommandoux, M. F. Achard, J. F. Langenwalter and H. A. Klok, Macromolecules, 2001, 34, 9100–9111 CrossRef CAS.
- J. S. Crespo, S. Lecommandoux, R. Borsali, H. A. Klok and V. Soldi, Macromolecules, 2003, 36, 1253–1256 CrossRef CAS.
- P. Papadopoulos, G. Floudas, I. Schnell, I. Lieberwirth, T. Q. Nguyen and H. A. Klok, Biomacromolecules, 2006, 7, 618–626 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Polym. Sci., 2006, 202, 75–111 CrossRef CAS.
- G. Floudas, P. Papadopoulos, H. A. Klok, G. W. M. Vandermeulen and J. Rodriguez-Hernandez, Macromolecules, 2003, 36, 3673–3683 CrossRef CAS.
- I. K. Kang, Y. Ito, M. Sisido and Y. Imanishi, Biomaterials, 1988, 9, 349–355 CrossRef CAS.
- C. Hua, C. M. Dong and Y. Wei, Biomacromolecules, 2009, 10, 1140–1148 CrossRef CAS.
- C. J. Huang and F. C. Chang, Macromolecules, 2008, 41, 7041–7052 CrossRef CAS.
- B. Perly, A. Douy and B. Gallot, Makromol. Chem., 1976, 177, 2569–2589 CrossRef CAS.
- A. Nakajima, T. Hayashi, K. Kugo and K. Shinoda, Macromolecules, 1979, 12, 840–843 CrossRef CAS.
- S. W. Kuo, H. F. Lee and F. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3108–3119 CrossRef CAS.
- X. Kong and S. A. Jenekhe, Macromolecules, 2004, 37, 8180–8183 CrossRef CAS.
- S. W. Kuo, H. F. Lee, W. J. Huang, K. U. Jeong and F. C. Chang, Macromolecules, 2009, 42, 1619–1626 CrossRef CAS.
- S. W. Kuo and H. T. Tsai, Polymer, 2010, 51, 5605–5704 CrossRef.
- Y. C. Lin and S. W. Kuo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2127–2137 CrossRef CAS.
- A. C. Engler, H. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS.
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS.
- J. G. Ray, J. T. Ly and D. A. Savin, Polym. Chem., 2011, 2, 1536–1541 RSC.
- H. Tang and D. Zhang, Polym. Chem., 2011, 2, 1542–1551 RSC.
- W. Agut, D. Taton and S. Lecommandoux, Macromolecules, 2007, 40, 5653–5661 CrossRef CAS.
- W. Agut, R. Agnaou, S. Lecommandoux and D. Taton, Macromol. Rapid Commun., 2008, 29, 1147–1155 CrossRef CAS.
- W. B. Zhang, Y. Li, X. Li, X. Dong, X. Yu, C. L. Wang, C. Wesdemiotis, R. P. Quirk and S. Z. D. Cheng, Macromolecules, 2011, 44, 2589–2596 CrossRef CAS.
- Y. Ishida, T. Hira, R. Goseki, M. Tokita, M. A. Kakimoto and T. Hayakawa, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2653–2663 CrossRef CAS.
- S. W. Kuo and F. C. Chang, Prog. Polym. Sci., 2011, 36, 1649–1696 CrossRef CAS.
- Y. C. Wu and S. W. Kuo, Polymer, 2010, 51, 3948–3955 CrossRef CAS.
- K. W. Huang and S. W. Kuo, Macromol. Chem. Phys., 2010, 211, 2301–2311 CrossRef CAS.
- H. Xu, S. W. Kuo, J. S. Lee and F. C. Chang, Macromolecules, 2002, 35, 8788–8793 CrossRef CAS.
- S. Jeon, J. Choo, D. Sohn and S. N. Lee, Polymer, 2001, 42, 9915–9920 CrossRef CAS.
- C. H. Lu, S. W. Kuo, C. F. Huang and F. C. Chang, J. Phys. Chem. C, 2009, 113, 3517–3524 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1py00381j |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.