Development of functional amino acid-based star polymers
Adrian
Sulistio
a,
Anton
Blencowe
a,
Adrian
Widjaya
a,
Xiaoqing
Zhang
b and
Greg
Qiao
*a
aDepartment of Chemical and Biomolecular Engineering, University of Melbourne, Parkville, Melbourne, VIC 3010, Australia. E-mail: gregghq@unimelb.edu.au; Fax: +61 3 8344 4153; Tel: +61 3 8344 8665
bCSIRO Material Science and Engineering, Private Bag 33, Clayton South, MDC 3169, VIC, Australia
First published on 15th November 2011
Abstract
Highly functionalized core cross-linked star (CCS) polymers composed entirely of naturally-occurring amino acids were prepared via the sequential ring-opening polymerisation (ROP) of amino acidN-carboxyanhydride (NCA) derivatives in a facile one-pot, arm-first strategy. The formation of the star polymers was investigated using side-chain protected poly(ε-Z-L-lysine) (PZLL) and poly(γ-benzyl-L-glutamic acid) (PBGA) macroinitiators with various molecular weights in combination with a cystine NCA cross-linker to afford poly(ε-Z-L-lysine)armspoly(L-cystine)core (PZLLarmsPLCcore) and poly(γ-benzyl-L-glutamic acid)armspoly(L-cystine)core (PBLGarmsPLCcore) stars, respectively. As the cross-linker to macroinitiator ratio or macroinitiator molecular weights were increased the molecular weights, average number of arms and core size of the resulting stars also increased. Core-isolated NCA moieties remaining after star formation provided a facile approach to core-functionalization with primary amines bearing different functionalities, including aminomethyl pyrene, propargylamine and hexylamine. UV-vis spectroscopic analysis of PZLLarmsPLCcore and PBLGarmsPLCcore stars core-functionalised with aminomethyl pyrene provided high loadings of 240 and 128 mol/mol stars, respectively. Furthermore, stars with alkyne functionalized cores were capable of undergoing further click reactions with azido derivatives, demonstrating the accessibility of the core-isolated moieties. Deprotection of the PZLLarmsPLCcore and PBLGarmsPLCcore stars yielded water soluble CCS polymers with poly(L-lysine) and poly(L-glutamic acid) arms, respectively, and functionalised cores. In addition, direct hydrazinolysis of the PBLGarmsPLCcore star provided hydrazide functionalities along the arms, which allow for conjugation of drug molecules via pH sensitive hydrazone linkers. These results open up exciting opportunities for the development of star polymerdrug delivery systems, whereby a lack of functionality, biocompatibility and biodegradability are often limiting factors.
1. Introduction
Since the discovery of amino acidN-carboxyanhydrides (NCAs) by Leuchs in 19061 and the first account of well-controlled metal-catalysed ring-opening polymerization (ROP) of NCAs by Deming,2 several controlled ROP systems of NCAs have been developed.3–7 Consequently, these have provided synthetic pathways to produce well-defined homo/block polypeptides,4,8 hybrids,3,9 star-shaped polymers10,11 and polypeptides with complex architectures.12 Two excellent reviews by Kricheldorf13 and Hadjichristidis et al.14 provide comprehensive summaries of the different polymeric architectures that have been synthesized from amino acid NCAs.Polymers with star-shaped architectures having several linear arms connected to a central core are of particular interest as a result of their structure related properties, including low solution viscosities,15 encapsulation capabilities16 as well as enhanced and compartmentalized functionalities.17–19 The two most commonly employed approaches to synthesize star-shaped polymers involve the use of multifunctional-initiators (core-first approach) or the linking of preformed linear polymers using cross-linkers or multifunctional-linking agents with complementary functionalities (arm-first approach). In the core-first approach, multifunctional compounds which can concurrently initiate the polymerization of monomers to form several arms are used. However, the resulting star polymers are difficult to characterize since the molecular weight characteristics of the arms cannot be measured directly.14,19 In contrast, the arm-first approach is viewed to be more efficient because it allows for a high degree of control at every stage of the synthetic process and the arms can be characterized easily prior to star formation.14,19 The preparation of so-called core cross-linked star (CCS) polymersvia the arm-first approach, involving the reaction of macroinitiators with suitable cross-linkers, has grown in popularity as it is applicable to a wide range of controlled polymerisation techniques and compositionally diverse precursors. Furthermore, it provides access to star polymers with relatively large cores suitable for site-specific isolation of functionalities and large capacity encapsulation capabilities.19
To date, a limited number of star polymers based on amino acid derivatives have been prepared11,13,14 and until recently no examples were known of star polymers comprised entirely (core and arms) from amino acids.20,21 Several amino acid-based stars have been prepared via the core-first approach. For example, Inoue et al. employed the core-first approach to synthesize star polymers containing six PBLG arms attached to a hexakis(4-aminophenoxy) cyclotriphosphazene core. After deprotection of the benzyl groups and functionalisation with a triethylene glycol derivative the stars were used to prepare enantioselective membranes for separation of tryptophan, tyrosine and phenylalanine.11 Similarly, poly(amido amide) and poly(propylene imine) dendrimers have also been used to prepare star-shaped polymersvia the core-first approach.13,14 These amphiphilic stars have shown great potential as drug delivery vehicles as they have the capacity to encapsulate hydrophobic drug molecules within the core and stabilize them in aqueous media.16
Recently, we introduced the synthesis of CCS polymers prepared entirely from amino acid building blocks in a one-pot, arm-first approach.20 These CCS polymers have selectively degradable cores and possess hierarchical functionalities spanning from the peripheral groups, along the arms, as well as on the core itself and have been shown to encapsulate hydrophobic drugs, such as pirarubicin in the core. These star were prepared via ROP of protected lysine NCA using hexamethyldisilazane (HMDS) as the initiator to yield poly(ε-Z-L-lysine) (PZLL), which served as the macroinitiator for star formation.6 The subsequent addition of cystine NCA cross-linker afforded the poly(ε-Z-L-lysine)armspoly(L-cystine)core (PZLLarmsPLCcore) CCS polymer. Herein we demonstrate the versatility of this approach by preparing amino acid-based CCS polymers composed of different amino acids and possessing various arm and core functionalities. Furthermore, a detailed study investigating the effect of reaction parameters, such as the cross-linker to macroinitiator ratio and macroinitiator molecular weight, on the properties of the resulting star polymers is presented.
2. Results and discussion
2.1 Synthesis of amino acidN-carboxyanhydrides (NCAs)
In the current study glutamic acid and lysine were chosen as precursors for star synthesis since their side group functionality would be translated to the arms of the resulting stars, although in theory any amino acid that can be converted to a NCA derivative could be employed. Functional groups such as amines or carboxylic acids along the stars' arms allows for post-polymerization functionalisation with different moieties suited to various applications. In comparison, the choice of cross-linker is limited to amino acids that can be converted to di-NCA monomers, thus the only naturally occurring amino acid which is suitable is L-cystine; a dimer of L-cysteine covalently linked via a disulfide bond. The amino acids were converted to their NCA derivatives using either phosphorus pentachloride or triphosgene. For carboxybenzyloxy (Cbz or Z) protected amino acids, di-Z-L-lysine and di-Z-L-cystine, phosphorus pentachloride (PCl5) was used (Scheme 1A and B, respectively), whereas triphosgene was used for γ-benzyl L-glutamic acid (Scheme 1C).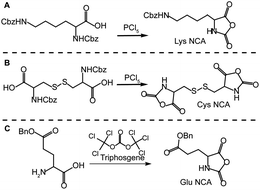 | ||
| Scheme 1 Synthesis of amino acidN-carboxyanhydrides (NCAs): (A) Nε-Z-lysineNCA (Lys NCA) and (B) cystineNCA (Cys NCA) using phosphorus pentachloride and (C) γ-benzyl glutamic acidNCA (Glu NCA) using triphosgene. | ||
2.2 Synthesis of CCS polymers
The formation of the CCS polymers was achieved via a one-pot, arm-first strategy using sequential ROP of NCA derivatives. Initially, Lys NCA or Glu NCA were polymerized using the secondary amineinitiator, hexamethyldisilazane (HMDS), to afford linear polymer, which served as the macroinitiator (MI) for star formation.6 Unlike conventional amineinitiators, HMDS provides excellent control over the ROP process, yielding polypeptides with expected molecular weights and low polydispersities (PDI).6 The subsequent addition of the cross-linker (CL), Cys NCA, resulted in the formation of CCS polymers1, comprised entirely of amino acid building blocks (Scheme 2).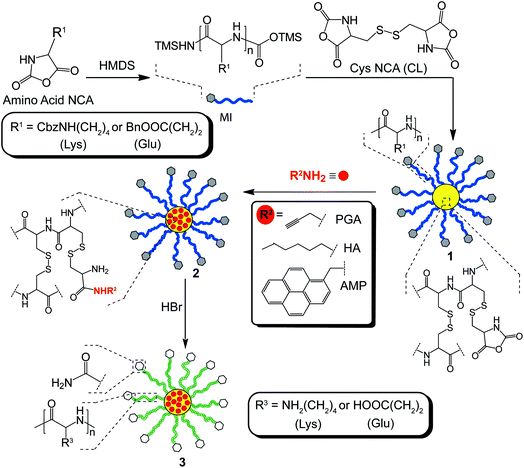 | ||
| Scheme 2 Synthesis of amino acid-based CCS polymers having hierarchical functionalities via a one-pot, arm-first strategy. | ||
For synthesis of poly(ε-Z-L-lysine)armspoly(L-cystine)core (PZLLarmsPLCcore) CCS polymer1a, having PZLL arms, Lys NCA was used to prepare the MI. The formation of linear PZLL using HMDS was followed via1H NMR spectroscopy and gel permeation chromatography (GPC) (Fig. 1). As the methineproton (δH 4.8 ppm) of the strained Lys NCA (circled in Fig. 1) has a distinctly different chemical shift to its open polymeric form (δH 4.2 ppm) it was possible to follow its consumption by monitoring the decrease in the resonance intensity. Thus, the resonance at δH 4.8 ppm gradually decreases over time as the Lys NCA is converted to its polymeric form (Fig. 1A), which results in the appearance of a new resonance at 4.2 ppm corresponding to the methineproton on the peptide backbone.
![(A) 1H NMR spectra (d7-DMF) taken every 15 min over a 6 h period showing decrease in the intensity of the Lys NCA methineproton resonance (δH 4.8 ppm) over the course of the polymerization. (B) Monomer conversion versus time and (C) plot of ln([Mo]/[M]) versus time calculated from NMR signal integration. (D) GPC UV (λ = 260 nm) chromatograms showing the reduction of the Lys NCA absorbance at 27 min and the formation of PZLL at ca. 22 min over time. (E) GPCRIchromatograms showing the formation of PZLL over time.](/image/article/2012/PY/c1py00436k/c1py00436k-f1.gif) | ||
| Fig. 1 (A) 1H NMR spectra (d7-DMF) taken every 15 min over a 6 h period showing decrease in the intensity of the Lys NCA methineproton resonance (δH 4.8 ppm) over the course of the polymerization. (B) Monomer conversion versus time and (C) plot of ln([Mo]/[M]) versus time calculated from NMR signal integration. (D) GPC UV (λ = 260 nm) chromatograms showing the reduction of the Lys NCA absorbance at 27 min and the formation of PZLL at ca. 22 min over time. (E) GPCRIchromatograms showing the formation of PZLL over time. | ||
To ensure accurate integration of the resonances the methyleneprotons of the Cbzprotecting group (δH 5.3 ppm) were used as a reference for calculation of the monomer (M) conversion with time (Fig. 1B), which reached 83% after 7 h. A plot of ln([Mo]/[M]) against time gave initially a linear relationship, which indicated that the polymerization was well controlled, although at higher conversions (t > 200 min) deviation was observed indicating slight loss of control (Fig. 1C). This deviation may result from trace impurities present in the Lys NCA.22 The conversion of Lys NCA was also confirmed by GPCUV-vischromatograms (Fig. 1D) recorded at λ = 260 nm, which revealed a decrease in the peak at high retention time (27 min) and the appearance of a peak at low retention time, corresponding to the Lys NCA and PZLL, respectively. GPCRIchromatograms (Fig. 1E) recorded over the course of reaction revealed a continuous increase in the molecular weight (MW) to afford, after 7.5 h, PZLL with a weight average molecular weight (Mw) of 15.0 kDa (PDI = 1.09). Lower MW PZLL (Mw = 6.6 kDa, PDI = 1.03) was also prepared by variation of the monomer to initiator (M/I) ratio and displayed similar kinetic profiles.
The formation of well-defined PZLL was also indicated by MALDI ToF MS (Fig. 2), which confirmed negligible side reactions. The major series correlates to the expected PZLL product after hydrolysis of the α-trimethylsilyl and ω-trimethylsilyl carbamate end groups resulting from exposure to atmospheric conditions.6 Interestingly, a minor series was also observed that is tentatively assigned to PZLL after fragmentation, for it has been demonstrated that side chain functionalized lysine residues are susceptible to free-radical induced fragmentation during ionization processes.23 Most importantly, no series were observed for side products, such as those encountered for the synthesis of polypeptidehomopolymers using primary24 and secondary alkyl amineinitiators.25
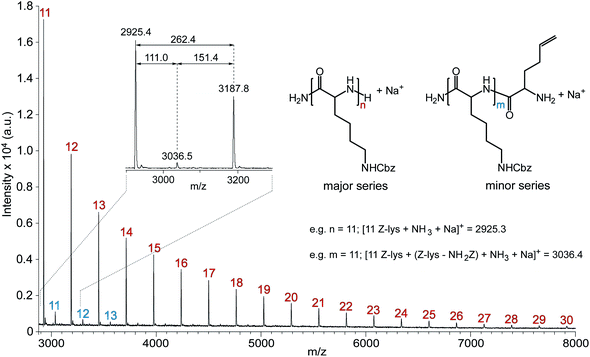 | ||
| Fig. 2 MALDI ToF MSspectrum of PZLL prepared via HMDS-mediated polymerisation recorded in linear/positive mode using α-CHCA/NaI. The numbers on the MSspectrum denote the number of repeating units of PZLL (repeat unit = 262.3 m/z); Inset shows expanded section of MSspectrum complete with peak m/z values and m/z differences. | ||
Subsequently, the PZLLarmsPLCcore CCS polymer1a was synthesized by the addition of the Cys NCACL to the PZLL MI. The optimum CL/MI ratio for star formation was found to be highly dependent on the MI MW, as previously reported for the formation of CCS polymersvia other controlled polymerisation techniques.19 For PZLL with Mws of 6.6 and 15.0 kDa the optimum CL/MI ratios were 15 and 35, and yielded star polymers1aL (Mw = 182 kDa; PDI = 1.77; average number of arms (f) ≈ 17) and 1aH (Mw = 523 kDa; PDI = 1.63; f ≈ 21), respectively (Table 1). Whereas below the optimum CL/MI ratio lower molecular weight stars were formed, higher ratios led to star-star coupling and gelation. The core size of the stars was also found to increase as the CL/MI ratio increased. This demonstrates that the overall size, f and core size of the CCS polymers could be tailored by changing the degree of polymerisation of the arms or the CL/MI ratio (Table 1).
| Polymer | MI | CL/MI | Star | ||||
|---|---|---|---|---|---|---|---|
| M w (kDa)a | PDIa | M w (kDa)a | PDIa | f | Core Size (%)c | ||
| a M w and PDI determined viaGPC multiangle laser light scattering (MALLS); dn/dcPZLL = 0.101 mL g−1 (25 °C), dn/dcPBLG = 0.085 mL g−1 (25 °C). b Average number of arms. c Percentage core size calculated from the CL/MI with respect to the overall molecular weight of the star polymer. All reactions were conducted at room temperature for 7 h. | |||||||
| 1aL | 6.56 | 1.03 | 5 | 29 | 1.28 | 3 | 24 |
| 15 | 182 | 1.77 | 17 | 40 | |||
| 25 | 3820 | 1.20 | 349 | 59 | |||
| 35 | Gel | Gel | Gel | Gel | |||
| 1aH | 15 | 1.09 | 17.5 | 81 | 1.43 | 3 | 25 |
| 20 | 89 | 1.46 | 4 | 32 | |||
| 22.5 | 114 | 1.56 | 5 | 34 | |||
| 25 | 129 | 1.62 | 5 | 32 | |||
| 30 | 183 | 1.83 | 7 | 35 | |||
| 35 | 523 | 1.63 | 21 | 40 | |||
| 40 | Gel | Gel | Gel | Gel | |||
| 1b | 8.7 | 1.03 | 20 | 217 | 1.36 | 17 | 32 |
| 22.5 | 245 | 1.40 | 18 | 35 | |||
| 25 | 297 | 1.49 | 22 | 38 | |||
| 27.5 | 515 | 1.62 | 36 | 40 | |||
| 32.5 | 2330 | 1.40 | 152 | 44 | |||
| 50 | Gel | Gel | Gel | Gel | |||
In all cases 1H NMR spectroscopic analysis of the star formation revealed that the Cys NCACL was completely consumed after 3 h.20 The disappearance of the CL resonances are a good indication of star formation since the reduced segmental mobility of the resulting cross-linked core leads to a reduction and broadening of the cystine signals making them indistinguishable from the baseline;26 however, it is not possible to determine if complete star formation and growth coincides with CL consumption. Therefore, the reaction was monitored over time viaGPC (Fig. 3) using a PZLL MI with a Mw of 12.4 kDa (PDI = 1.06) and a CL/MI ratio of 32.5, which revealed that initially the MW increased rapidly up to 6.5 h and then more gradually up to 23 h, after which no further increase was observed. Thus, even after complete CL consumption the star continues to grow, implying that after 3 h the coupling together of preformed low MW stars becomes the dominant process, although the slight decrease in the peak at a retention time of 21.5 min (Fig. 3), which is attributed to MI chain extended with CL, suggests that these prepolymers are also slowly incorporated into the preformed stars.
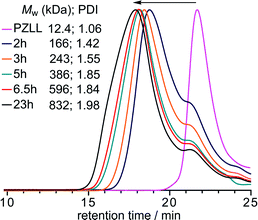 | ||
| Fig. 3 GPC RI chromatograms following star formation over time, using a PZLL MI with a Mw of 12.4 kDa (PDI = 1.06) and a CL/MI ratio of 32.5. | ||
For synthesis of poly(γ-benzyl-L-glutamic acid)armspoly(L-cystine)core (PBLGarmsPLCcore) CCS polymer1b having PBLG arms, Glu NCA was used to prepare the MI followed by the addition of Cys NCA CL. Optimization of the star 1b formation was conducted similarly to that described for 1a, by variation of the CL/MI ratio (Table 1). For a PBLG MI with a Mw of 8.7 kDa (PDI = 1.03) the Mw of the resulting stars was found to increase gradually from 217 to 297 kDa as the CL/MI ratio was increased from 20 to 25. At higher ratios the Mw increased more sharply, with star-star coupling becoming prominent at a ratio of 32.5 and gelation occurring at a ratio of 50. The optimum CL/MI ratio to obtain stars with a large number of arms before significant star-star coupling occurred was found to be 27.5, which resulted in a star with a Mw of 515 kDa (PDI = 1.62) and f of 36.
The GPC RI chromatograms for stars 1aL, 1aH and 1b (Fig. 4A–C, respectively) revealed the presence of unincorporated MI/prepolymers regardless of the CL/MI ratio employed, which can be seen as slight shoulders or small peaks towards higher retention times, similar to those for the MI. This observation is common for CCS polymer preparations, which generally result in incomplete conversion of the MI to star product, regardless of the controlled polymerisation technique employed.18,19,27 For star 1b the improved resolution between the star and prepolymer peaks allowed the MI to star conversion to be determined by deconvolution of the RIchromatograms using Gaussian functions, which revealed that regardless of the CL/MI ratio employed the conversion was ca. 93%.
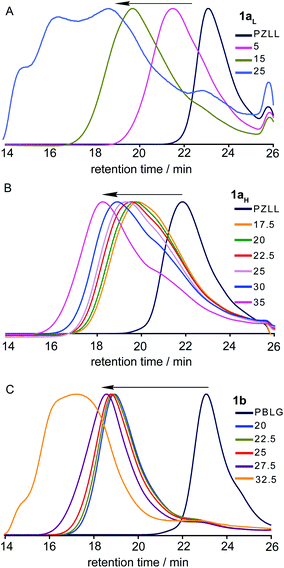 | ||
| Fig. 4 GPC RI traces for the optimization of CCS polymer (A) 1aL, (B) 1aH and (C) 1b prepared at different CL/MI ratios. | ||
At this stage, both CCS polymers1a and 1b could be isolated viaprecipitation or core-functionalized through reaction with primary amines. Isolation of star 1a by precipitation resulted in the formation of a small amount of an insoluble component and led to significant increases in the MW of the soluble component as a result of star-star coupling as described previously.20 Attempts to directly isolate stars 1a and 1bvia removal of the polymerisationsolvent resulted in the formation of insoluble materials.
2.3 Core functionalized CCS polymers
Facile core-functionalization of stars 1a and 1b was achieved by addition of primary amines directly to the polymerisation after star formation, which ring-openings the unreacted pendent NCAs in the core, resulting in the release of CO2 and the formation of cystine-based amide and aminegroups (Scheme 1). Although an excess of primary amine is used it is conceivable that the cystine-based amines generated in the early stage of this reaction reinitiate the ROP of the pendent NCAs leading to further intramolecular cross-linking of the core or even intermolecular cross-linking between the cores of adjacent stars. Core-functionalization was conducted by reacting star 1a (with either a Mw of 596 kDa (PDI = 1.65) or 832 kDa (PDI = 1.98)) and 1b (Mw = 503 kDa; PDI = 1.55) with primary amines bearing different functional groups, including propargylamine (PGA), hexylamine (HA) and aminomethyl pyrene (AMP) to yield core-functionalized stars 2a and 2b, respectively (Scheme 2). In all cases the star MW and PDI increased after core-functionalization (Fig. 5), although the observed increases were greater than could be accounted for solely through functionalisation of the core. Therefore, it appears likely that the increase in MW probably results from a combination of core-functionalization and star-star coupling caused by intermolecular cross-linking.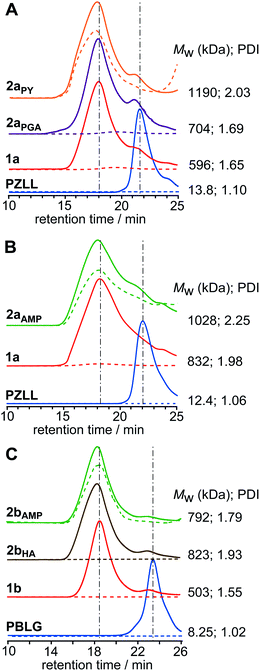 | ||
| Fig. 5 GPC RI (solid) and UV (dashed) chromatograms of (A) PGA and PY core-functionalized CCS polymers2aPGA and 2aPY, and their precursor PZLL MI and star 1a, (B) AMP core-functionalized CCS polymer2aAMP, and its precursor PZLL MI and star 1a, and (C) AMP and HA core-functionalized CCS polymers2bAMP and 2bHA, and their precursor PBLG MI and star 1b. All polymer concentrations were 15 mg mL−1 and UV-Vis spectra were recorded at λ = 340 nm. | ||
Attachment of pyrene to the cores of stars 1a and 1b was achieved through reaction of AMP with the unreacted pendent NCAs to afford stars 2aAMP and 2bAMP, respectively. GPC UV analysis of 2aAMP and 2bAMP revealed an enhanced absorbance at λ = 340 nm relative to the unfunctionalized precursor stars and provided evidence for the successful incorporation of AMP (Fig. 5B and C, respectively). The extent of this functionalization was quantified using UV-Vis spectroscopic analysis and provided AMP loadings of 240 and 128 mol/mol star for 2aAMP and 2bAMP, respectively, which theoretically equates to increases in MW of ca. 45 and 23 kDa; however, the increases in MW observed from GPC analysis implies that star-star coupling occurs to a minor extent. Fluorescently labeled amino acid star polymers such as 2aAMP and 2bAMP have huge potential in imaging and sensor applications, as well as for the study of polymer molecular dynamics; for example, the study of polymer-tissue/cell interactions28 or the detection of metal ions.29
Unfortunately, UV-Vis spectroscopy could not be employed to determine the extent of core-functionalization of stars 2aPGA and 2bHA with PGA and HA, respectively (Fig. 5A and C), as these amines do not possess distinctive UV-Vis absorbances. However, the core isolated alkyne moieties of star 2aPGA provided the opportunity to further functionalize the core via click chemistry. Therefore, to determine the number of accessible alkynegroups within the core of star 2aPGA it was reacted with an azidopyrene derivative (3-azidopropyl 4-oxo-4-(pyren-4-yl) butanoate) under standard copper catalyzed click conditions to yield star 2aPY (Scheme 3).
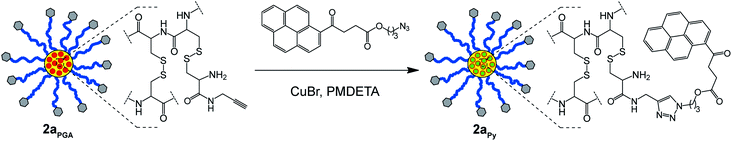 | ||
| Scheme 3 Copper catalyzed click reaction between PGA functionalized CCS polymer2aPGA and 3-azidopropyl 4-oxo-4-(pyren-4-yl) butanoate to yield star 2aPY. | ||
GPC UV analysis confirmed the successful incorporation of pyrene into the star (Fig. 5A) and UV-Vis spectroscopic analysis provided a pyrene loading of 117 mol/mol star, implying that the core of star 2aPGA contains at least 117 propargyl groups. Surprisingly, GPC analysis revealed a further increase in Mw from 704 (PDI = 1.69) for 2aPGA to 1190 kDa (PDI = 2.05) for 2aPY after the click reaction (Fig. 5A), suggesting that star-star coupling continues to occurs to a minor extent during reaction or upon isolation. Nevertheless, this experiment demonstrates that the core was successfully functionalized with PGA and that the resulting core-isolated alkynes are accessible for further reactions. These core-isolated alkyne functionalities provide the ability to selectively functionalize the star with azido derivatives and could potentially be employed to attach drug molecules or ligands to the star for biomedical or catalysis applications.
2.4 Arm-functionalized CCS polymers
Arm-functionalized CCS polymers could be prepared by acid-mediated hydrolysis of the protecting groups along the arms, or in the case of PBLGarmsPLCcore star 2b, via direct conversion of the benzylesters to their hydrazide derivatives. Thus, hydrazinolysis was performed by addition of hydrazine (2 molar equiv. relative to glutamic acidrepeat units) to star 2bAMP, which resulted in quantitative replacement of the benzylprotecting groups to afford the hydrazide functionalized star 2bAMP-HYD, as confirmed by 1H NMR spectroscopic analysis (Fig. 6). The 1H NMR spectrum of 2bAMP-HYD revealed the disappearance of resonances corresponding to the benzylprotecting groupprotons of 2bAMP at δH 5.2 and 7.2 ppm (Fig. 6A) and the formation of new resonances at δH 3.6 and 9.0 ppm corresponding to the hydrazideprotons (Fig. 6B). The resulting hydrazide-functionalised star 2bAMP-HYD was no longer soluble in DMF, but became soluble in mildly acidic or basic aqueous solutions. Hydrazinolysis of benzylesters provides a facile route towards hydrazide functionalized polymers, which can be used to conjugate drugs (e.g., doxorubicin, which is commonly used for the treatment of bladder,33 breast,34 and liver35 cancers) via acid-labile hydrazone linkages. These linkages are readily cleaved at acidic pH values, similar to the endosomal/lysosomal environment of cancer cells, which allows for site-specific release of conjugated drugs.36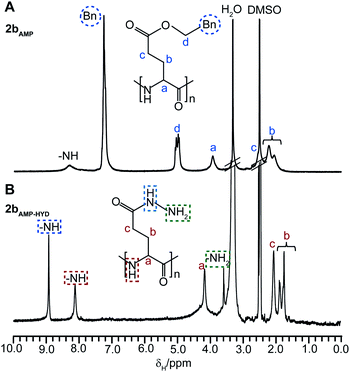 | ||
| Fig. 6 1H NMR spectra (d6-DMSO) and the assigned structures of (A) benzyl protected CCS polymer2bAMP and (B) hydrazide functionalized CCS polymer2bAMP-HYD. | ||
Water soluble amino acid-based stars were prepared from stars 2a and 2bvia deprotection of the side chain groups of the arms (Cbz for 2a and Bn for 2b) with 33 wt% HBr in acetic acid.37 The resulting degradable CCS polymers have amide peripheral groups, poly(L-lysine) (PLL) arms (3a) or poly(L-glutamic acid) (PLGA) (3b) arms, and functionalized PLC cores. Analysis of the stars 3a and 3bvia1H NMR spectroscopy confirmed the removal of the majority of the Cbz and Bnprotecting groups (∼88% removal for both) and displayed similar chemical shifts as their linear PLL and PLGA analogues (Fig. 7A and B, respectively). As before, the core-functionalities were also not visible due to the reduced segmental mobility of the core.26 In addition, the water soluble star 3aAMP has been shown to encapsulate the water insoluble drugpirarubicinvia physical interactions such as π–π stacking.20
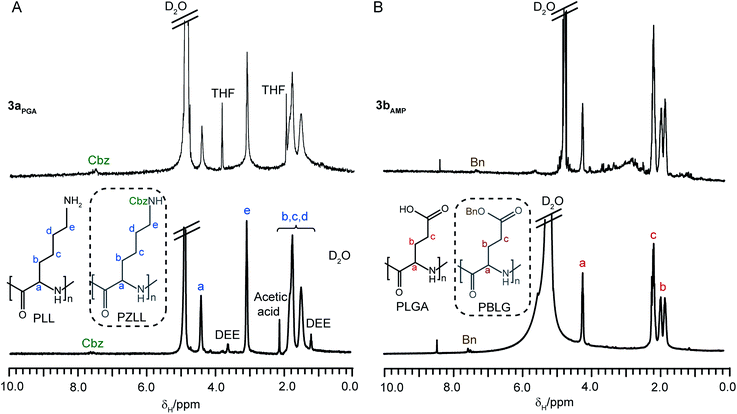 | ||
| Fig. 7 1H NMR spectra (D2O) of (A) water soluble CCS polymer3aPGA and linear PLL and (B) water soluble CCS polymer3bAMP and linear PLGA. The structures in the dotted boxes represent the unprotected PZLL (A) and PBLG (B) respectively. | ||
2.5 CCS polymerdegradation
Previously it was demonstrated20 that the disulfide bonds located centrally within the cross-links responsible for forming the core could be successfully cleaved using excess DTT in DMF under argon to afford poly(Z-L-lysine-b-L-cysteine) (P(ZLL-b-LC) copolymers resulting from the cleavage of the star 1aH (Mw = 1140 kDa; PDI = 1.33; f = 52) into its constituent components.30,31 When the star 1aHdegradation solution was exposed to the atmosphere the resulting P(ZLL-b-LC) copolymer rapidly formed organogels. This behaviour is not observed for the PZLL MI, which is readily soluble in DMF, and therefore, is attributed to intermolecular thiol/disulfide exchange that occurs in the presence of oxygen between the polycysteine blocks to create a gel network (Fig. 8).32 This not only demonstrates that the stars are degradable, but also possess the ability to undergo chemically triggered rearrangement to form insoluble macroscopic structures.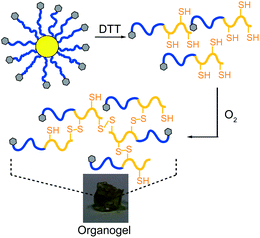 | ||
| Fig. 8 Organogel formed after DTT cleavage of star 1aH to form P(ZLL-b-LC), followed by oxygen-induced gelation. | ||
In comparison, cleavage of the AMP core-functionalized star 2aAMP did not result in gel formation upon exposure to the atmosphere, although the solution became very viscous after 24 h. GPC analysis of this degradation solution after 1 and 24 h exposure to the atmosphere revealed very high MW species (ca. 5 and 12 MDa, respectively) approaching the exclusion limits of the columns. It is proposed that formation of the gel network is disrupted by the AMP-cysteine conjugate products that were also cleaved off from the core when the disulfide bonds were reduced. Comparison of the 1H NMR spectra of the star 2aAMP before and after cleavage (the cleaved products were separated from excess DTTviaprecipitation of the 1 h degradation solution into 2-propanol) confirmed that a mixture of block copolymers and the AMP-cysteine conjugate were formed after degradation of the core with DTT (Fig. 9A and B, respectively).
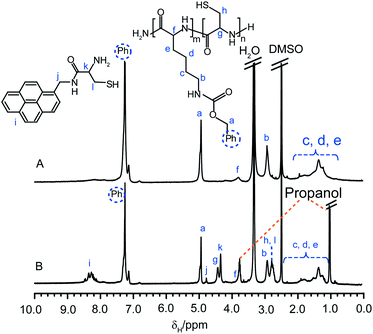 | ||
| Fig. 9 1H NMR spectra (d6-DMSO) of CCS polymer2aAMP (A) before and (B) after cleavage with DTT and their assigned structures. | ||
3. Experimental section
3.1 Materials
Z-L-Lys(Z)-OH (Bachem), H-L-Glu(OBn)-OH (Bachem), L-cystine (Aldrich), hexamethyldisilazane (HMDS) (99.9%, Aldrich), benzyl chloroformate (95%, Aldrich), sodium bicarbonate (NaHCO3) (99%, Ajax Fine Chemical), magnesium sulfate (MgSO4) (>98%, Scharlau)), bis(trichloromethyl)carbonate (triphosgene) (>99%, Aldrich), phosphorus pentachloride (PCl5) (>99%, Merck), dithiothreitol (DTT) (>99%, A.G. Scientific), 3-bromopropanol (97%, Aldirch), sodium azide (>99.5%, BioUltra), tetrabutylammoniumhydrogen sulfate (TBAHS) (>99%, Aldrich), γ-Oxo-1-pyrenebutyric acid (Aldrich), potassium hydroxide (KOH) (> 85%, Biolab), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDCI) (>98%, Fluka), 4-(dimethylamino)pyridine (DMAP) (99%, Aldrich), pirarubicin (>95%, Aldrich), propargylamine (98%, Aldrich), aminomethylpyrene hydrochloride (95%, Aldrich), propylamine (Merck), hexylamine (99%, Aldrich), hydrazine hydrate (99%, BDH), copper(I) bromide (CuBr) (98%, Aldrich), N,N,N′,N′′,N′′-pentamethyldiethylenetriamine (PMDETA) (98%, Fluka), hydrobromic acid (33% in acetic acid) (Aldrich), trifluoroacetic acid (TFA) (98%, Aldrich), hydrochloric acid (37%, Scharlau), lithium bromide (99%, Aldrich), tetrahydrofuran (THF) (99.9%, Scharlau) and N,N-dimethylformamide (DMF) (anhyd., Aldrich) were used as received. n-Hexane, ethyl acetate, dimethylsulfoxide (DMSO), chloroform and methanol were purchased from Chem-Supply and used as received. 1,4-Dioxane (Fluka) and diethyl ether (Chem-Supply) were used to make binary solvent (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) and dried for 48 h prior to use over 3 Å molecular sieves. Acetonitrile (HPLC grade, B&J) was stored over 3 Å molecular sieves. Dichloromethane (99.5%, Chem-Supply) was distilled from CaH2. THF (99%, Lab Scan) was distilled from sodium benzophenone ketal. Milli Q water (18.2 MΩ.cm) was obtained from a Millipore Synergy Water System. Acetone-d6 (99.9%), DMSO-d6 (99.9%), DMF-d7 (99.5%) and D2O (99.9%) were purchased from Cambridge Isotope and used as received. MALDI ToF MS matrix α-cyano-4-hydroxycinnamic acid (α-CHCA) (≥99.5%) and cationisation agent NaI (99.999%) were purchased from Fluka and Aldrich, respectively and were used as received. Z-L-Lysine-NCA (Lys NCA), di-Z-L-cystine ((Z-Cys-OH)2), L-cystine NCA (Cys NCA), aminomethylpyrene (AMP), 3-azidopropanol, 3-azidopropyl 4-oxo-4-(pyren-4-yl) butanoate and click functionalized star 2aPY were prepared according to previously published procedures.20
:3 v/v) and dried for 48 h prior to use over 3 Å molecular sieves. Acetonitrile (HPLC grade, B&J) was stored over 3 Å molecular sieves. Dichloromethane (99.5%, Chem-Supply) was distilled from CaH2. THF (99%, Lab Scan) was distilled from sodium benzophenone ketal. Milli Q water (18.2 MΩ.cm) was obtained from a Millipore Synergy Water System. Acetone-d6 (99.9%), DMSO-d6 (99.9%), DMF-d7 (99.5%) and D2O (99.9%) were purchased from Cambridge Isotope and used as received. MALDI ToF MS matrix α-cyano-4-hydroxycinnamic acid (α-CHCA) (≥99.5%) and cationisation agent NaI (99.999%) were purchased from Fluka and Aldrich, respectively and were used as received. Z-L-Lysine-NCA (Lys NCA), di-Z-L-cystine ((Z-Cys-OH)2), L-cystine NCA (Cys NCA), aminomethylpyrene (AMP), 3-azidopropanol, 3-azidopropyl 4-oxo-4-(pyren-4-yl) butanoate and click functionalized star 2aPY were prepared according to previously published procedures.20
3.2 Characterization
GPC analysis was performed on a Shimadzu size exclusion chromatogram fitted with Wyatt DAWN HELEOS LSdetector (λ = 658 nm), Shimadzu RID-10 refractometer (λ = 633 nm), and Shimadzu SPD-20AUV-Visdetector using three identical PLgel columns (5 μm, MIXED-C) in series and HPLC grade DMF with 0.05 M LiBr (70 °C, 1 mL min−1) as mobile phase. Astra software (Wyatt Technology Corp.) was used to determine the molecular weight characteristics using known dn/dc values for PZLL (dn/dcPZLL = 0.101 mL g−1 (25 °C)) and PBLG (dn/dcPBLG = 0.085 mL g−1 (25 °C)) in DMF. The dn/dc values of densely branched CCS polymers and linear polymers of the same monomeric constitution have been reported to be comparable;31 thus, the dn/dc values of linear PZLL or PBLG were used to determine the molecular weights of the CCS polymers. 1H NMR spectroscopy was performed using Varian Unity400 (400 MHz) spectrometer using the deuterated solvent as reference and a sample concentration of ca. 20 mg mL−1. DLS measurements were performed on a Malvern high performance particle sizer (HPPS) with a He-Ne laser (633 nm) at an angle of 173° and a temperature of 25 ± 0.1 °C, initial sample concentrations of 10 mg mL−1 in DMF were used then serial dilutions were performed until stable spectra were obtained. All sample solutions were filtered using 0.45 μm filters. MALDI ToF MS was performed on a Bruker Autoflex III Mass Spectrometer operating in positive/linear mode. The analyte, matrix (α-CHCA) and caternisation agent (NaI) were dissolved in THF at concentration of 10 mg mL−1, 10 mg mL−1 and 1 mg mL−1, respectively, and then mixed in a ratio of 10:1:1. 0.3 μL of this solution was then spotted onto a ground steel target plate and the solvent allowed to evaporate prior to analysis. FlexAnalysis (Bruker) was used to analyses the data. UV-Visspectrophotometry was performed on a Shimadzu UV-2101PCspectrometer using quartz cuvettes with a 1 cm path length.
3.3 Methods
:1 ethyl acetate:hexane (20 mL) 2 times to afford Glu NCA (1.73 g, 78%). 1H NMR (400 MHz, d6-DMSO) δH 1.87–2.10 (m, 2H, CH2), 2.52 (t, 2H, J = 7.6 Hz, CH2), 4.45 (dd, 1H, J = 5.6 & 8.0 Hz, CHN), 5.10 (s, 2H, CH2O), 7.31–7.40 (m, 5H, ArH), 9.09 (s, 1H, NH) ppm; 13C NMR (100 MHz, d6-DMSO) δC 26.3 (CH2), 29.0 (CH2), 56.1 (CHN), 65.6 (CH2), 127.9 (3 ArCH), 128.4 (2 ArCH), 135.9 (ArCC), 151.8 (NHCO2), 171.2 (CHCO2), 171.6 (CH2CO2) ppm.
:1 v/v, 10 times the volume of DMF) or core-functionalised via the addition of primary amines.
Synthesis of CCS 1aL. Starting from Lys NCA (200 mg, 0.654 mmol) and HMDS (M/I = 20, 6.81 μL, 32.7 μmol) in anhydrous DMF (2 mL), PZLL (Mw = 6.56 kDa, PDI = 1.03) was obtained after 6 h. Addition of Cys NCA (CL/MI = 15, 143 mg, 0.490 mmol) afforded after 7 h PZLLarmsPLCcore CCS polymer1aL (Mw = 182 kDa, PDI = 1.77, f ≈ 19). Precipitation of the polymer solution into water:methanol (1
:1, 20 mL) followed by isolation viacentrifugation and drying (0.1 mbar) afford 1aL (Mw = 500 kDa, PDI = 2.0, f ≈ 52) as a pale yellow solid, 173 mg (63%).
Synthesis of CCS 1aH. Starting from Lys NCA (200 mg, 0.654 mmol) and HMDS (M/I = 40, 3.41 μL, 16.4 μmol) in anhydrous DMF (2 mL), PZLL (Mw = 15.0 kDa, PDI = 1.09, dh = 10.8 nm) was obtained after 7 h. Addition of Cys NCA (CL/MI = 35, 170 mg, 0.582 mmol) afforded after 7 h PZLLarmsPLCcore CCS polymer1aH (Mw = 523 kDa, PDI = 1.63, f ≈ 24, dh = 74 nm). Precipitation of the polymer solution into water:methanol (1
:1, 20 mL) followed by isolation viacentrifugation and drying (0.1 mbar) afford 1aH (Mw = 1440 kDa, PDI = 1.33, f ≈ 66, dh = 180 nm) as a pale yellow solid, 257 mg (89%).
Synthesis of CCS 1b. Starting from Glu NCA (200 mg, 0.760 mmol) and HMDS (M/I = 40, 3.96 μL, 18.9 μmol) in anhydrous DMF (2 mL), PBLG (Mw = 8.70 kDa, PDI = 1.03) was obtained after 7 h. Addition of Cys NCA (CL/MI = 27.5, 153 mg, 0.522 mmol) afforded after 7 h PBLGarmsPLCcore CCS polymer1b (Mw = 515 kDa, PDI = 1.65, f ≈ 36). Precipitation of the polymer solution into water:methanol (1
:1, 20 mL) followed by isolation viacentrifugation and drying (0.1 mbar) afford 1b as a pale yellow solid, 207 mg (70%). GPC analysis was not possible because the isolated product was not soluble in DMF.
Core-functionalisation with PGA to yield 2aPGA. PGA (20 equiv. relative to Cys NCA used, 0.340 mL, 5.31 mmol) was dissolved in DMF (10 mL) and added to a solution of CCS polymer1a (Mw = 596 kDa, PDI = 1.65, 2 mL) and stirred for 3 h to afford, after isolation, CCS polymer2aPGA (Mw = 704 kDa, PDI = 1.69).
Core-functionalisation with AMP to yield 2aAMP. AMP (1.3 equiv. relative to Cys NCA used, 1.42 g, 6.15 mmol) was dissolved in DMF (10 mL) and added to a solution of CCS polymer1a (Mw = 832 kDa, PDI = 1.98, 2 mL) and stirred for 3 h to afford, after isolation, CCS polymer2aAMP (Mw = 1028 kDa, PDI = 2.25).
Core-functionalisation with HEX to yield 2bHEX. HEX (10 equiv. relative to Cys NCA used, 0.312 mL, 2.37 mmol) was dissolved in DMF (10 mL) and added to a solution of CCS polymer1b (Mw = 503 kDa, PDI = 1.55, 1 mL) and stirred for 1 h to afford, after isolation, CCS polymer2bHEX (Mw = 823 kDa, PDI = 1.93).
Core-functionalisation with AMP to yield 2bAMP. AMP (2.2 equiv. relative to Cys NCA used, 1.20 g, 6.15 mmol) was dissolved in DMF (10 mL) and added to a solution of CCS polymer1b (Mw = 503 kDa, PDI = 1.55, 1 mL) and stirred for 3 h to afford, after isolation, CCS polymer2bAMP (Mw = 792 kDa, PDI = 1.79).
Conclusions
Core-functionalized CCS polymers composed entirely from naturally occurring amino acids can be readily prepared in one-pot using sequential reagent addition and the arm-first approach. Two different CCS polymers were synthesized, having either poly(Z-L-lysine) or poly(Bn-L-glutamic acid) arms. Facile core-functionalization followed by deprotection of the star's arms ultimately yields water soluble, biocompatible and biodegradable CCS polymers with a hierarchy of functionalities spanning from the core, along the arms, to the periphery. The core-isolated moieties are accessible for further reaction as demonstrated by the click reaction of alkyne core-functionalized stars with an azidopyrene derivative. Variation of the reaction parameters allows the stars molecular weights, average number of arms and core sizes to be tailored, providing access to stars with selective loading capacities. The degradability of the amino acid-based CCS polymers coupled with the ease of site-specific functionalization and their encapsulation abilities makes this type of star a very promising candidate for the development of biocompatible and biodegradable polymer therapeutics.Notes and references
- H. Leuchs, Ber. Dtsch. Chem. Ges., 1906, 39, 857 CrossRef; H. Leuchs and W. Manasse, Ber. Dtsch. Chem. Ges., 1907, 40, 3235 CAS; H. Leuchs and W. Geiger, Ber. Dtsch. Chem. Ges., 1908, 41, 1721 CrossRef CAS.
- T. J. Deming, Nature, 1997, 390, 386 CrossRef CAS.
- I. Dimitrov and H. Schlaad, Chem. Commun., 2003, 2944 RSC.
- T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2004, 5, 1653–1656 CrossRef CAS.
- W. Vayaboury, O. Giani, H. Cottet, A. Deratani and F. Schué, Macromol. Rapid Commun., 2004, 25, 1221 CrossRef CAS.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114 CrossRef CAS; H. Lu and J. Cheng, J. Am. Chem. Soc., 2008, 130, 12562 CrossRef CAS.
- Y. Peng, S. Lai and C. Lin, Macromolecules, 2008, 41, 3455 Search PubMed.
- N. Higashi, T. Koga and M. Niwa, Langmuir, 2000, 16, 3482 CrossRef CAS; P. Papadopoulos, G. Floudas, T. Aliferis, H. Iatrou and N. Hadjichristidis, Biomacromolecules, 2005, 6, 2352 CrossRef CAS; J. R. Hernandez and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1167 CrossRef; J. R. Cha, G. D. Stucky, D. E. Morse and T. J. Deming, Nature, 2000, 403, 289 CrossRef CAS; H. R. Kricheldorf, C. Lossow and G. Schwarz, Macromol. Chem. Phys., 2004, 205, 918 CrossRef CAS.
- J. F. Lutz, D. Schütt and S. Kubowicz, Macromol. Rapid Commun., 2005, 26, 23 CrossRef CAS; T. J. Deming, Adv. Drug Delivery Rev., 2002, 54, 1145 CrossRef CAS; L. Wang, S. Wang and J. Z. Bei, Polym. Adv. Technol., 2004, 15, 617 CrossRef CAS; H. R. Kricheldorf and K. Hauser, Biomacromolecules, 2001, 2, 1110 Search PubMed; H. Schlaad and M. Antonietti, Eur. Phys. J. E, 2003, 10, 17 CrossRef CAS.
- H.-A. Klok, J. R. Hernandez, S. Becker and K. Müllen, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1572 Search PubMed.
- K. Inoue, A. Miyahara and T. Itaya, J. Am. Chem. Soc., 1997, 119, 6191 CrossRef CAS; K. Inoue, S. Horibe, M. Fukae, T. Muraki, E. Ihara and H. Kayama, Macromol. Biosci., 2003, 3, 26 CrossRef CAS.
- J. Li, W. He, N. He, S. Han, X. Sun, L. Li and B. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1450 CrossRef CAS; J. Rao, Y. Zhang, J. Zhang and S. Liu, Biomacromolecules, 2008, 9, 2586 CrossRef CAS; H. Lu, J. Wang, Y. Lin and J. Cheng, J. Am. Chem. Soc., 2009, 131, 13582 CrossRef CAS.
- H. R. Kricheldorf, Angew. Chem., Int. Ed., 2006, 45, 5752 CrossRef CAS.
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and G. Sakellariou, Chem. Rev., 2009, 109, 5528 CrossRef CAS.
- A. K. Ho, P. A. Gurr, M. F. Mills and G. G. Qiao, Polymer, 2005, 46, 6727 CrossRef CAS; T. K. Goh, K. D. Coventry, A. Blencowe and G. G. Qiao, Polymer, 2008, 49, 5095 CrossRef CAS.
- J. T. Wiltshire and G. G. Qiao, Aust. J. Chem., 2007, 60, 699 CrossRef CAS; C. Kojima, K. Kono, K. Maruyama and T. Takagishi, Bioconjugate Chem., 2000, 11, 910 CrossRef CAS.
- A. Blencowe, T. K. Goh, S. P. Best and G. G. Qiao, Polymer, 2008, 49, 825 CrossRef CAS; J. T. Wiltshire and G. G. Qiao, Macromolecules, 2008, 41, 623 CrossRef CAS; C. T. Adkins and E. Harth, Macromolecules, 2008, 41, 3472 CrossRef CAS; H. Gao and K. Matyjaszewski, Macromolecules, 2007, 40, 399 Search PubMed; B. Helms, S. J. Guillaudeu, Y. Xie, M. McMurdo, C. J. Hawker and J. M. Fréchet, Angew. Chem., Int. Ed., 2005, 44, 6384 CrossRef CAS.
- A. Spiniello, A. Blencowe and G. G. Qiao, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2422 CrossRef CAS.
- A. Blencowe, J. F. Tan, T. K. Goh and G. G. Qiao, Polymer, 2009, 50, 5 CrossRef CAS.
- A. Sulistio, A. Widjaya, A. Blencowe, X. Zhang and G. G. Qiao, Chem. Commun., 2011, 47, 1151 RSC.
- A. Sulistio, J. Lowenthal, A. Blencowe, M. N. Bongiovanni, L. Ong, S. L. Gras, X. Zhang and G. G. Qiao, Biomacromolecules, 2011 DOI:10.1021/bm200604h , Article ASAP.
- D. Ballard and C. Bamford, J. Am. Chem. Soc., 1957, 79, 2336 Search PubMed; P. Doty and R. Lundberg, J. Am. Chem. Soc., 1957, 79, 2338 Search PubMed.
- H. Yin, A. Chacon, N. A. Porter, H. Yin and D. S. Masterson, J. Am. Soc. Mass Spectrom., 2007, 18, 807 CrossRef CAS.
- D. L. Pickel, N. Politakos, A. Avgeropoulos and J. M. Messman, Macromolecules, 2009, 42, 7781 CrossRef CAS.
- W. N. E. van Dijk-Wolthuis, L. van de Water, P. van de Wetering, M. J. van Steenbergen, J. J. Kettenes-van den Bosch, W. J. W. Schuyl and W. E. Hennink, Macromol. Chem. Phys., 1997, 198, 3893 CrossRef.
- M. Sawamoto, K. Y. Baek and M. Kamigaito, Macromolecules, 2001, 34, 215 CrossRef CAS; J. T. Wiltshire and G. G. Qiao, Macromolecules, 2006, 39, 4828.
- N. Hadjichristidis, S. Pispas, M. Pitsikalis, H. Iatrou and C. Vlahos, Adv. Polym. Chem., 1999, 142, 71 Search PubMed.
- A. J. Limer, A. K. Rullay, V. San Miguel, C. Peinado, S. Keely, E. Fitzpatrick, S. D. Carrington, D. Brayden and D. M. Haddleton, React. Funct. Polym., 2006, 66, 51 CrossRef CAS; T. K. Georgiou, M. Vamvakaki, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2005, 6, 2990 CrossRef CAS.
- M. A. R. Meier and U. S. Schubert, Chem. Commun., 2005, 36, 610 Search PubMed.
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995 CrossRef CAS.
- D. L. Rabenstein and P. L. Yeo, J. Org. Chem., 1994, 59, 4223 CrossRef CAS; D. L. Rabenstein and K. H. Weaver, J. Org. Chem., 1996, 61, 7391 CrossRef CAS.
- H. Kawano, S. Komaba, T. Yamasaki, M. Maeda, Y. Kimura, A. Maeda and Y. Kaneda, Cancer Chemother. Pharmacol., 2008, 616, 973 Search PubMed.
- L. Livi, I. Meattini, C. De Luca Cardillo, V. Scotti, B. Agresti, C. Franzese, L. Sanchez, J. Nori, S. Bertocci, S. Cassani, S. Bianchi, L. Cataliotti and G. Biti, J. Chemotherapy, 2011, 23, 36 Search PubMed.
- J. A. Munoz-Gamez, R. Quiles-Perez, A. Ruiz-Extremera, A. B. Martin-Alvarez, L. Sanjuan-Nunez, A. Carazo, J. Leon, F. J. Oliver and J. Salmeron, Cancer Lett., 2011, 301, 47 Search PubMed.
- K. Kono, C. Kojima, N. Hayashi, E. Nishisaka, K. Kiura, S. Watarai and A. Harada, Biomaterials, 2008, 29, 1664 CrossRef CAS.
- J. Lee, S. J. Lee, J. Y. Choi, J. Y. Yoo and C. H. Ahn, Eur. J. Pharm. Sci., 2005, 24, 441 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |