DOI:
10.1039/C1PY00376C
(Paper)
Polym. Chem., 2012,
3, 99-104
Synthesis and photovoltaic properties of D–A copolymers of benzodithiophene and naphtho[2,3-c]thiophene-4,9-dione
Received
24th August 2011
, Accepted 2nd October 2011
First published on 19th October 2011
Abstract
Two donor–acceptor (D–A) alternative copolymers of benzodithiophene (BDT) donor unit and an acceptor unit of naphtho[2,3-c]thiophene-4,9-dione (NTDO) with different alkyl side chains, PBDTNTDO-1 and PBDTNTDO-2, were synthesized for application as donor materials in polymer solar cells (PSCs). The copolymers show good solubility in common organic solvents, broad visible absorption from 350 nm to 670 nm, and relatively lower HOMO energy levels at −5.14 eV for PBDTNTDO-1 and −5.19 eV for PBDTNTDO-2. The PSCs based on PBDTNTDO-2 as donor and PC70BM as acceptor demonstrated power conversion efficiency of 1.52% with an open circuit voltage of 0.88 V and a short circuit current of 5.67 mA cm−2, under the illumination of AM1.5, 100 mW cm−2.
Introduction
Polymer solar cells (PSCs) based on bulk heterojunction (BHJ) structure of polymer donors and fullerene derivative acceptors have been attracting considerable attention due to their unique advantages of low cost, light weight, and mechanical flexibility.1–5 The central concern of the studies of PSCs is to increase the power conversion efficiency (PCE) of the devices. As we know, the PCE is proportional to the open circuit voltage (Voc), short circuit current (Jsc) and fill factor (FF) of the PSCs. Therefore, for the conjugated polymer donor materials in the active layer of PSCs, the key issues we should consider in their molecular design are broad absorption spectra to enhance sunlight harvest for higher Jsc, appropriately lower HOMO (the highest occupied molecular orbital) energy levels to maximize the Voc, and higher hole mobility for higher Jsc and higher FF of the PSCs.
Donor–acceptor (D–A) copolymers have drawn great attentions for the conjugated polymer donor materials in recent years, because of easy tuning of their bandgap, absorption spectra and HOMO energy level by using different donor and acceptor units in the copolymers.6–15Benzo[1,2-b:3,4-b′] dithiophene (BDT) is the most successful donor unit in constructing high efficiency photovoltaic donor copolymers, not only in the narrow bandgap copolymers with thieno[3,4-b]thiophene unit16–19 but also in the D–A copolymers with different acceptor units.20–23 The advantages of the BDT units include its planar conjugated structure and easy purification of BDT monomers, and the BDT-based conjugated polymers show high hole mobility, for example, the hole mobility of a copolymer of BDT and thiophene reached 0.25 cm2 V−1 s−1.24
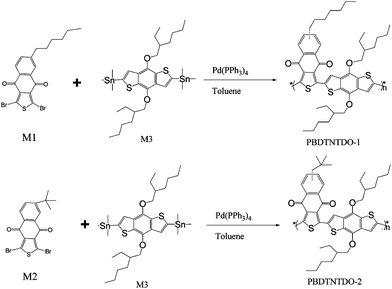
In order to enrich the family of the BDT-based D–A copolymers and look for new conjugated polymers for the application as donor materials in PSCs, here we designed and synthesized two new BDT-based D–A copolymers with an acceptor unit of naphtho[2,3-c]thiophene-4,9-dione (NTDO), PBDTNTDO-1, PBDTNTDO-2 (see Scheme 1). NTDO is easy to synthesize, and possesses a relatively simple and planar structure which could be beneficial to the electron delocalization when it is incorporated into the D–A copolymers. Besides, its relatively strong electron-withdrawing ability would lead to lower HOMO and LUMO energy levels of the D–A copolymers, which is desirable for obtaining higher open circuit voltage (Voc) of the PSCs. The difference of the two copolymers is the alkyl chain substituent on the NTDO unit. The two copolymers show good solubility in common organic solvents, broad visible absorption from 350 nm to 670 nm and relatively lower HOMO energy levels. The PSCs based on PBDTNTDO-2 as donor and PC70BM as acceptor demonstrated PCE of 1.52% with a Voc of 0.88 V and a Jsc of 5.67 mA cm−2, under the illumination of AM1.5, 100 mW cm−2.
 |
| | Scheme 1 Synthetic route for the PBDTNTDOcopolymers. | |
Results and discussion
Synthesis and thermal stability
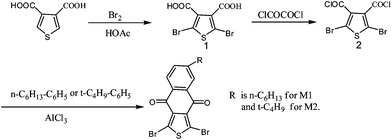
The general synthetic routes for naphtho[2,3-c]thiophene-4,9-dione (NTDO) monomer 1 (M1) and monomer 2 (M2) are shown in Scheme 2. Benzo[1,2-b:3,4-b′] dithiophene (BDT) was synthesized according to the procedure reported in literature.16,25 The copolymers of PBDTNTDO-1 and PBDTNTDO-2 were synthesized by Stille coupling reaction, as shown in Scheme 1. The two copolymers possess good solubility in common organic solvents such as chloroform, toluene, and chlorobenzene. Molecular weights of the polymers were measured by gel permeation chromatography (GPC) analysis with a polystyrene standard calibration. The number-average molecular weight (Mn) is 24 K for PBDTNTDO-1 and 13 K for PBDTNTDO-2. The polydispersity indices (PDIs) of PBDTNTDO-1 and PBDTNTDO-2 are 2.48 and 3.07 respectively. The molecular weights and PDI values of the copolymers were listed in Table 1.
 |
| | Scheme 2 Synthetic route for the NTDO monomer. | |
Table 1 Molecular weights and thermal properties of the polymers
|
Polymers
|
M
w
a
|
M
n
a
|
PDIa |
T
d
b (°C) |
|
M
n, Mw, and PDI of the polymers were determined by GPC using polystyrene standards in THF.
The 5% weight-loss temperatures under inert atmosphere.
|
|
PBDTNTDO-1
|
59862 |
24130 |
2.48 |
308 |
|
PBDTNTDO-2
|
40132 |
13035 |
3.07 |
334 |
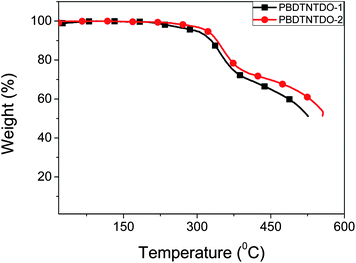
Thermal stability of the copolymers was investigated by thermogravimetric analysis (TGA), as shown in Fig. 1. The TGA analysis reveals that the onset temperatures with 5% weight-loss (Td) of PBDTNTDO-1 and PBDTNTDO-2 are 308 and 334 °C respectively. The thermal stability of the two copolymers is similar, and it is adequate for the applications in PSCs and other optoelectronic devices.
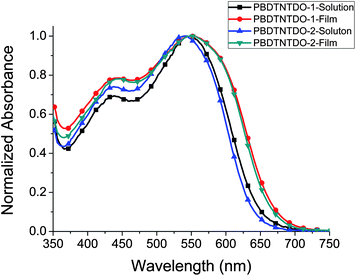
Fig. 2 shows the ultraviolet-visible (UV-vis) absorption spectra of the polymer dilute solutions in chloroform and films spin-coated on quartz substrates. PBDTNTDO-1 and PBDTNTDO-2 solutions display broad absorption spectra in the wavelength range of 350–650 nm with the absorption maxima at 549 and 541 nm respectively. The absorption peak of PBDTNTDO-1 solution is 8 nm red-shifted compared to that of PBDTNTDO-2 solution. PBDTNTDO-1 and PBDTNTDO-2 films show almost same absorption peak wavelength at ca. 554 nm. In comparison with the absorption spectra in chloroform solutions, absorption spectra of the polymer thin films shift to a longer wavelength region, which results from the interchain interaction in the polymer films. The absorption edges of PBDTNTDO-1 and PBDTNTDO-2 films are 681 and 677 nm, corresponding to the bandgaps (Eg) of 1.82 and 1.83 eV respectively. The optical data and bandgap of the copolymers were listed in Table 2 for clear comparison.
Table 2 Optical and electrochemical data of PBDTNTDO-1 and PBDTNTDO-2
|
Polymer
|
UV-vis absorption |
Energy levels |
|
CHCl3 solution |
Film |
|
λ
max(nm) |
λ
max (nm) |
λ
onset (nm) |
E
g (eV) |
HOMO (eV) |
LUMO (eV) |
|
PBDTNTDO-1
|
549 |
554 |
681 |
1.82 |
−5.14 |
−3.28 |
|
PBDTNTDO-2
|
541 |
554 |
677 |
1.83 |
−5.19 |
−3.30 |
Electrochemical properties
Electrochemical cyclic voltammetry was performed to determine the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of conjugated polymers.26Fig. 3 shows the cyclic voltammograms of PBDTNTDO-1 and PBDTNTDO-2 films on glassy carbon electrode in 0.1 mol L−1Bu4NPF6 acetonitrile solution. The onset reduction potentials (Ered) of PBDTNTDO-1 and PBDTNTDO-2 are −1.43 and −1.41 V vs. Ag/Ag+, respectively, while the onset oxidation potentials (Eox) are 0.43 and 0.48 V vs. Ag/Ag+, respectively. From the Eox and Ered of the polymers, HOMO and LUMO energy levels of the polymers were calculated according to the equations.27
| HOMO = −e(Eox + 4.71) (eV) |
| LUMO = −e(Ered + 4.71) (eV) |
Where the units of Eox and Ered are V vs. Ag/Ag+.
The HOMO energy levels of PBDTNTDO-1 and PBDTNTDO-2 are −5.14 and −5.19 eV respectively, and their LUMO energy levels are −3.28 and −3.30 eV respectively. The electronic energy level data of the polymers were also listed in Table 2.
Photovoltaic properties
To investigate and compare the photovoltaic properties of the two copolymers, bulk heterojunction PSC devices with configuration of ITO/PEDOT:PSS/copolymer:PC70BM(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5)/Ca/Al were fabricated. Fig. 4 shows the J–V curves of the PSC under the illumination of AM 1.5, 100 mW cm−2. The corresponding open-circuit voltage (Voc), short-circuit current (Jsc), fill factor (FF), and power conversion efficiency (PCE) of the devices at different device fabrication conditions are summarized in Table 3. It can be seen that the PCE of the PSC based on PBDTNTDO-1:PC70BM (1:1.5, w/w) is 1.09% with Voc = 0.81 V, Jsc = 4.71 mA cm−2, and FF = 28.7%, while the PCE of the PSC based on PBDTNTDO-2:PC70BM (1:1.5, w/w) reaches 1.52% with Voc = 0.88 V, Jsc = 5.67 mA cm−2, and FF = 30.5%, under the illumination of AM 1.5, 100 mW cm−2. The Voc (0.88 V) of the device based on PBDTNTDO-2 is higher than that (0.81 V) based on PBDTNTDO-1 which agrees with the deeper HOMO energy level of PBDTNTDO-2, because the Voc is proportional to the difference between the LUMO level of the acceptor and the HOMO level of the donor.28,29 The Jsc of the PSC based on PBDTNTDO-2 is also higher than that based on PBDTNTDO-1, so that the PSC based on PBDTNTDO-2 displayed a higher PCE of 1.52%. The AFM images of PBDTNTDO-1/PC70BM and PBDTNTDO-2/PC70BM films are very similar, as shown in Fig. 5. And surface roughness of the films for PBDTNTDO-1 and PBDTNTDO-2 are 0.47 and 0.48 nm respectively, which is almost the same. Obviously, the side chains of the NTDO unit influence the morphology of the blend films only a little. The hole mobility of PBDTNTDO-2 with the shorter side chain is probably higher than that of PBDTNTDO-1 with the longer side chain, which results in the higher Jsc for the PSC based on PBDTNTDO-2. The results indicate that NTDO is a promising new acceptor unit for constructing D–A copolymers in the application as photovoltaic donor materials in PSCs, and the short and branched side chain on NTDO in PBDTNTDO-2 is superior than the longer side chain in PBDTNTDO-1 for the photovoltaic application.
:1.5)/Ca/Al were fabricated. Fig. 4 shows the J–V curves of the PSC under the illumination of AM 1.5, 100 mW cm−2. The corresponding open-circuit voltage (Voc), short-circuit current (Jsc), fill factor (FF), and power conversion efficiency (PCE) of the devices at different device fabrication conditions are summarized in Table 3. It can be seen that the PCE of the PSC based on PBDTNTDO-1:PC70BM (1:1.5, w/w) is 1.09% with Voc = 0.81 V, Jsc = 4.71 mA cm−2, and FF = 28.7%, while the PCE of the PSC based on PBDTNTDO-2:PC70BM (1:1.5, w/w) reaches 1.52% with Voc = 0.88 V, Jsc = 5.67 mA cm−2, and FF = 30.5%, under the illumination of AM 1.5, 100 mW cm−2. The Voc (0.88 V) of the device based on PBDTNTDO-2 is higher than that (0.81 V) based on PBDTNTDO-1 which agrees with the deeper HOMO energy level of PBDTNTDO-2, because the Voc is proportional to the difference between the LUMO level of the acceptor and the HOMO level of the donor.28,29 The Jsc of the PSC based on PBDTNTDO-2 is also higher than that based on PBDTNTDO-1, so that the PSC based on PBDTNTDO-2 displayed a higher PCE of 1.52%. The AFM images of PBDTNTDO-1/PC70BM and PBDTNTDO-2/PC70BM films are very similar, as shown in Fig. 5. And surface roughness of the films for PBDTNTDO-1 and PBDTNTDO-2 are 0.47 and 0.48 nm respectively, which is almost the same. Obviously, the side chains of the NTDO unit influence the morphology of the blend films only a little. The hole mobility of PBDTNTDO-2 with the shorter side chain is probably higher than that of PBDTNTDO-1 with the longer side chain, which results in the higher Jsc for the PSC based on PBDTNTDO-2. The results indicate that NTDO is a promising new acceptor unit for constructing D–A copolymers in the application as photovoltaic donor materials in PSCs, and the short and branched side chain on NTDO in PBDTNTDO-2 is superior than the longer side chain in PBDTNTDO-1 for the photovoltaic application.
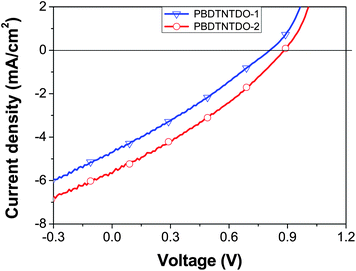 |
| | Fig. 4
J–V curves of the PSCs base on PBDTNTDO:PC70BM (1:1.5, w/w) under the illumination of AM 1.5, 100 mW cm−2. | |
Table 3 Photovoltaic results for the PSCs based on PBDTNTDO:PC70BM (1:1.5, w/w)a
| Active layer |
Layer thickness (nm) |
V
oc (V) |
J
sc (mA cm−2) |
FF (%) |
PCE (%) |
|
Spin-coating at 2000 r min−1, 30 s.
|
|
PBDTNTDO-1:PC70BM = 1:1.5 |
63 |
0.81 |
4.71 |
28.7 |
1.09 |
|
PBDTNTDO-2:PC70BM = 1:1.5 |
56 |
0.88 |
5.67 |
30.5 |
1.52 |
 |
| | Fig. 5
AFM topography (left) and phase (right) images of the blend films of polymer:PC70BM (1:1.5), (a) (b) for PBDTNTDO-1, (c) (d) for PBDTNTDO-2. | |
The external quantum efficiencies (EQEs) of the PSCs based on PBDTNTDO:PC70BM (1:1.5 w/w) are shown in Fig. 6. It can be seen that the response wavelength range of PBDTNTDO-1-based devices is the same as that of PBDTNTDO-2-based devices, which agrees with the absorption spectra of PBDTNTDO-1 and PBDTNTDO-2. The maximum EQE value of the PSC based on PBDTNTDO-2 reached 46% at 480 nm. The higher EQE of the device based on PBDTNTDO-2 than that based on PBDTNTDO-1 is consistent with the higher Jsc of the former.
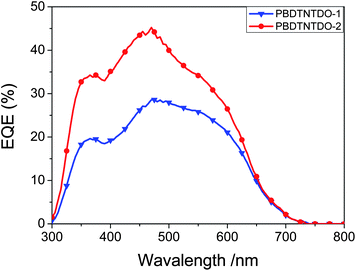 |
| | Fig. 6
EQE plots of the PSCs based on PBDTNTDO: PC70BM (1:1.5, w/w). | |
Conclusion
An acceptor unit of naphtho[2,3-c]thiophene-4,9-dione (NTDO) was used to synthesize D–A copolymers with BDT donor unit, for the application as donor materials in PSCs. The two D–A copolymersPBDTNTDO-1 and PBDTNTDO-2 exhibit good solubility in common organic solvents, broad visible absorption from 350 nm to 670 nm and relatively deeper HOMO energy levels (−5.14 eV for PBDTNTDO-1 and −5.19 eV for PBDTNTDO-2). The PSCs based on PBDTNTDO-2/PC70BM (1:1.5, w/w) showed PCE of 1.52% with Voc of 0.88 V and Jsc of 5.67 mA cm−2, under the illumination of AM1.5, 100 mW cm−2. While the PCE of the device based on PBDTNTDO-1 under the same conditions is 1.09% with Voc of 0.81 V and Jsc of 4.71 mA cm−2. The results indicate that the short and branched alkyl chain on NTDO in PBDTNTDO-2 is beneficial to the photovoltaic application.
Experimental section
Measurements and characterization
All new compounds were characterized by 1H NMR spectroscopy performed on a Bruker DMX-400 spectrometer. For the 1H NMR measurements, CDCl3 was used as the solvent. Chemical shifts in the NMR spectra were reported in ppm relative to the singlet at 7.26 ppm for CDCl3. The molecular weight of the polymers was measured by gel permeation chromatography (GPC), and polystyrene was used as a standard. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TGA-7. Mass spectra were obtained with a Shimadzu QP2010 spectrometer. UV-vis absorption spectra were obtained on a Hitachi U-3010 spectrometer. Cyclic voltammetry was performed on a Zahner IM6e electrochemical workstation with a three-electrode system in a solution of 0.1 M Bu4NPF6 in acetonitrile at a scan rate of 50 mV s−1. The polymer films were coated on a glassy carbon electrode (1.0 cm2) by dipping the electrode into the corresponding solutions and then drying. A Pt wire was used as the counter electrode, and Ag/Ag+ was used as the reference electrode.
Device fabrication and characterization of polymer solar cells
Polymer solar cells (PSCs) were fabricated with ITO glass as a positive electrode, Ca/Al as a negative electrode and the blend film of the polymer/PCBM between them as a photosensitive layer. The ITO glass was pre-cleaned and modified by a thin layer of PEDOT:PSS which was spin-cast from a PEDOT:PSS aqueous solution (Clevious P VP AI 4083 H. C. Stark, Germany) on the ITO substrate, and the thickness of the PEDOT:PSS layer is about 60 nm. The photosensitive layer was prepared by spin-coating a blend solution of polymers and PC70BM in o-dichlorobenzene on the ITO/PEDOT:PSS electrode. Then the Ca/Al cathode was deposited on the active layer by vacuum evaporation under 3 × 10−4 Pa. The effective area of one cell is 4 mm2. The current–voltage (I–V) measurement of the devices was conducted on a computer-controlled Keithley 236 Source Measure Unit. A xenon lamp with AM 1.5 filter was used as the white light source, and the optical power at the sample was 100 mW cm−2.
Synthesis of monomers
The benzo[1,2-b:3,4-b′]dithiophene monomer M3 was synthesized according to the procedure in the literature.16
2,5-Dibromothiophene-3,4-dicarboxylic acid (1).
Thiophene-3,4-dicarboxylic (5 g, 29.1 mmol) and glacial acetic acid (50 mL) were added dropwise. The mixture was stirred overnight. Aqueous sodium bisulfate solution was added until the reddish colour disappeared. The mixture was filtered and washed with 50 mL water, and 9.6 g (yield: 80%) of gray solid compound 1 was obtained. MS: m/z = 330.
1,3-Dibromo-6-hexylnaphtho[2,3-c]thiophene-4,9-dione (M1).
A solution of 2,5-dibromothiophene-3,4-dicarboxylic acid chloride (2) (3.3 g, 9 mmol) in dichloromethane was slowly added to a suspension of aluminum chloride (5.3 g, 39.6 mmol) in dichloromethane (20 mL) maintained at 0 °C. The mixture was stirred at 0 °C for 10 min. n-Hexylbenzene (2 mL, 10.8 mmol) was slowly added. The mixture was stirred for 30 min and then poured into ice. Dichloromethane (100 mL) was added, and the mixture was shaken vigorously. The layers were separated, and the aqueous layer was extracted with dichloromethane. The combined organic portions were washed with a saturated sodium bicarbonate solution and water and then dried over magnesium sulfate. The residue, which remained after the solvent was evaporated, was purified by flash chromatography on silica gel with ethyl acetate/hexane (1:10) to give 2.4 g (58%) of the title compound. 1H NMR (400 MHz, CDCl3), δ (ppm): 8.22∼8.11 (d, 2H), 7.61 (s,1H), 2.79 (s, 2H), 1.69∼1.23 (m, 9H), 0.89 (s, 2H). MS: m/z = 456.
1,3-Dibromo-6-tert-butylnaphtho[2,3-c]thiophene-4,9-dione (M2).
M2 was prepared by the same method as the synthesis of M1, except tert-butylbenzene was used instead of n-hexylbenzene. 1H NMR (400 MHz, CDCl3), δ (ppm): 8.30 (d, 1H), 8.20 (d,1H), 7.80 (d, 1H), 1.38 (s, 9H). MS: m/z = 428.
Synthesis of PBDTNTDO-1 and PBDTNTDO-2
Polymerization of PBDTNTDO-1 and PBDTNTDO-2 was performed by Stille coupling reaction.
PBDTNTDO-1
.
In a 50 mL flask, M1 (0.228 g, 0.5 mmol), and M3 (0.382 g, 0.5 mmol) were dissolved in toluene (16 mL), and flushed with argon for 10 min. Then, Pd(PPh3)4 (0.023 g, 0.020 mmol) was added into the flask, which was then flashed by argon for another 20 min. The solution was heated to reflux and stirred for 24 h under inert atmosphere. Then, the reactant was cooled to room temperature, and the polymer was precipitated by addition of 50 mL of methanol and filtered through a Soxhlet thimble, which was then subjected to Soxhlet extraction with methanol and hexane. The solid was dissolved in chloroform (150 mL) and passed through a column packed with alumina, Celite, and silica gel. The column was eluted with chloroform. The combined polymer solution was concentrated and was poured into methanol. After this, the precipitates were collected and dried under vacuum overnight to get PBDTNTDO-1. Yield: 210 mg (42.9%). 1H NMR (400 MHz, CDCl3), δ (ppm): 8.49∼7.51 (broad, 5H), 4.24 (broad, 4H), 2.77∼0.67 (broad, 43H).
PBDTNTDO-2
.
In a 50 mL flask, M2 (0.214 g, 0.5 mmol) and M3 (0.382 g, 0.5 mmol) were dissolved in toluene (16 mL), and flushed with argon for 10 min. Then, Pd(PPh3)4 (0.023 g, 0.020 mmol) was added into the flask, which was then flashed by argon for another 20 min. The solution was heated to reflux and stirred for 24 h under inert atmosphere. Then, the reactant was cooled to room temperature, and the polymer was precipitated by addition of 50 mL of methanol and filtered through a Soxhlet thimble, which was then subjected to Soxhlet extraction with methanol and hexane. The solid was dissolved in chloroform (150 mL) and passed through a column packed with alumina, Celite, and silica gel. The column was eluted with chloroform. The combined polymer solution was concentrated and was poured into methanol. After this, the precipitates were collected and dried under vacuum overnight to get PBDTNTDO-2. Yield: 220 mg (46.3%). 1H NMR (400 MHz, CDCl3), δ (ppm): 8.74∼7.26 (broad, 5H), 4.38 (broad, 4H), 2.35∼0.88 (broad, 39H).
Acknowledgements
This work was supported by NSFC (Nos. 20874106, 50933003 and 21021091), Ministry of Science and Technology of China, and Chinese Academy of Sciences.
References
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
-
(a) J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS;
(b) A. Facchetti, Chem. Mater., 2011, 23, 733–758 CrossRef CAS;
(c) X. Zhan and D. Zhu, Polym. Chem., 2010, 1, 409–419 RSC.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323 CrossRef CAS.
-
(a) Y. F. Li and Y. P. Zou, Adv. Mater., 2008, 20, 2952–2958 CrossRef CAS;
(b) Y. J. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- E. J. Zhou, S. Yamakawa, K. Tajima, C. H. Yang and K. Hashimoto, Chem. Mater., 2009, 21, 4055–4061 CrossRef CAS.
-
(a) Y.-T. Chang, S.-L. Hsu, M.-H. Su and K.-H. Wei, Adv. Mater., 2009, 21, 2093 CrossRef CAS;
(b) M.-C. Yuan, M.-Y. Chiu, C.-M. Chiang and K.-H. Wei, Macromolecules, 2010, 43, 6270 CrossRef CAS.
-
(a) M. Wang, X. Hu, P. Liu, W. Li, X. Gong, F. Huang and Y. Cao, J. Am. Chem. Soc., 2011, 133, 9638–9641 CrossRef CAS;
(b) Z. He, C. Zhang, X. Xu, L. Zhang, L. Huang, J. W. Chen, H. B. Wu and Y. Cao, Adv. Mater., 2011, 23, 3086–3089 CrossRef CAS.
- A. Gadisa, W. Mammo, L. M. Andersson, S. Admassie, F. L. Zhang, M. R. Andersson and O. Inganas, Adv. Funct. Mater., 2007, 17, 3836 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-plesu, M. Belletete, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732 CrossRef CAS.
- M. J. Zhang, H. J. Fan, X. Guo, Y. J. He, Z.-G. Zhang, J. Min, J. Zhang, G. J. Zhao, X. W. Zhan and Y. F. Li, Macromolecules, 2010, 43, 5706 CrossRef CAS.
- E. J. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. S. Wei, K. Tajima, C. H. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302 CrossRef CAS.
- E. J. Zhou, S. Yamakawa, Y. Zhang, K. Tajima, C. H. Yang and K. Hashimoto, J. Mater. Chem., 2009, 19, 7730 RSC.
- E. J. Zhou, J. Z. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, S. Yamakawa, Q. S. Wei, M. Nakamura, K. Tajima, C. H. Yang and Kazuhito Hashimoto, Macromolecules, 2010, 43, 2873 CrossRef CAS.
- J. H. Hou, M.-H. Park, S. Q. Zhang, Y. Yao, L.-M. Chen, J.-H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS.
-
(a) H.-Y. Chen, J. H. Hou, S. Q. Zhang, Y. Y. Liang, G. Yang, Y. Yang, L. P. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS;
(b) J. H. Hou, H.-Y. Chen, S. Q. Zhang, R. L. Chen, Y. Yang, Y. Wu and G. Li, J. Am. Chem. Soc., 2009, 131, 15586 CrossRef CAS.
- L. J. Huo, S. Q. Zhang, X. Guo, F. Xu, Y. F. Li and J. H. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS.
- Y. Huang, L. J. Huo, S. Q. Zhang, X. Guo, C. C. Han, Y. F. Li and J. H. Hou, Chem. Commun., 2011, 47, 8904–8906 RSC.
-
(a) L. J. Huo, J. H. Hou, S. Q. Zhang, H.-Y. Chen and Y. Yang, Angew. Chem., Int. Ed., 2010, 49, 1500–1503 CrossRef CAS;
(b) L. J. Huo, X. Guo, S. Q. Zhang, Y. F. Li and J. H. Hou, Macromolecules, 2011, 44, 4035–4037 CrossRef CAS.
- Y. P. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS.
-
(a) M. J. Zhang, H. J. Fan, X. Guo, Y. J. He, Z.-G. Zhang, J. Min, J. Zhang, G. J. Zhao, X. W. Zhan and Y. F. Li, Macromolecules, 2010, 43, 8714–8717 CrossRef CAS;
(b) Q. Q. Shi, H. J. Fan, Y. Liu, W. P. Hu, Y. F. Li and X. W. Zhan, J. Phys. Chem. C, 2010, 114, 16843–16848 CrossRef CAS;
(c) M. Yang, B. Peng, B. Liu, Y. P. Zou, K. C. Zhou, Y. H. He, C. Y. Pan and Y. F. Li, J. Phys. Chem. C, 2010, 114, 17989–17994 CrossRef CAS;
(d) Q. Q. Shi, H. J. Fan, Y. Liu, J. M. Chen, L. C. Ma, W. P. Hu, Z. G. Shuai, Y. F. Li and X. W. Zhan, Macromolecules, 2011, 44, 4230–4240 CrossRef CAS.
- H. Pan, Y. Li, Y. Wu, P. Liu, B. S. Ong, S. Zhu and G. Xu, J. Am. Chem. Soc., 2007, 129, 4112 CrossRef CAS.
- J. H. Hou, L.-M. Chen, S. Q. Zhang and Y. Yang, J. Phys. Chem. C, 2009, 113, 21202–21207 CAS.
- Y. F. Li, Y. Cao, J. Gao, D. L. Wang, G. Yu and A. J. Heeger, Synth. Met., 1999, 99, 243–248 CrossRef CAS.
- Q. J. Sun, H. Q. Wang, C. H. Yang and Y. F. Li, J. Mater. Chem., 2003, 13, 800–806 RSC.
- C. J. Brabec, A. Cravino, D. Meissner, N. S. Sariciftci, T. Fromherz, M. T. Rispens, L. Sanchez and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 374 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.