End-linked, amphiphilic, degradable polymer conetworks: synthesis by sequential atom transfer radical polymerization using a bifunctional, cleavable initiator
Maria
Rikkou-Kalourkoti
a,
Elena
Loizou
a,
Lionel
Porcar
b,
Krzysztof
Matyjaszewski
c and
Costas S.
Patrickios
*a
aDepartment of Chemistry, University of Cyprus, P. O. Box 20537, 1678, Nicosia, Cyprus. E-mail: chp5mr1@ucy.ac.cy; elenaloizou@gmail.com; costasp@ucy.ac.cy
bInstitut Laue-Langevin, B. P. 156, F-38042, Grenoble Cedex 9, France. E-mail: porcar@ill.fr
cDepartment of Chemistry, Carnegie Mellon University, 4400 Fifth Avenue, Pittsburgh, PA 15213, USA. E-mail: km3b@andrew.cmu.edu
First published on 19th October 2011
Abstract
End-linked, degradable, amphiphilic polymer conetworks (APCNs) of various compositions and architectures were synthesized by sequential atom transfer radical terpolymerization of monomers and cross-linker using a cleavable, bifunctional initiator, bearing two acid-labile hemiacetal ester groups. The incorporation of the initiator residue into the conetworks rendered them cleavable in the middle of their elastic polymer chains. The temporal evolution of the swollen mass of the conetworks in acidified THF–water mixtures was studied and it was determined that the APCNhydrolysis rates were dependent on both conetwork composition and architecture. Regarding the former, hydrophobic APCNs degraded slowly or they even did not dissolve at all. Regarding the latter, conetworks with hydrophobic monomer and hydrophobic cross-linker units distributed around the initiator fragment also dissolved more slowly or they did not degrade at all. Finally, characterization of the star copolymers, produced from conetwork hydrolysis, allowed the determination of the APCN core functionality to be around 30.
Introduction
Degradable polymers represent a field of growing importance in polymer science, with applications in medicine and biotechnology for the fabrication of erodible matrices for drug delivery1–3 and tissue engineering,4–9 in the microelectronics industry for the preparation of positive or negative resists for the manufacture of integrated circuits,10–13 and in environmental protection via the manufacture of degradable packaging material.14–16 The degradable components in these polymers are usually the monomer repeating units,17 and less frequently the cross-linker units.18–40 Even less frequently is the degradable polymer component the initiator fragment,41–62 in cases when polymer synthesis was performed using a cleavable initiator under controlled polymerization conditions.63In this last case, the labile initiator fragment confers to the polymer chain degradability at a precise location, allowing the controlled reduction in molecular weight and the degree of branching, but also the formation of certain functional end-groups. Regarding reduction in size and degree of branching upon degradation, polymers made using degradable initiators have been transformed to smaller entities of simpler architecture, with most notable examples the scission of linear homopolymers into two equal pieces,42,45,47,52,53,60 the conversion of ABA triblocks into diblocks,43,44,51 the decomposition of star polymers into the constituting arms,48–50 the separation of dumbbell polymers to two star polymers,46,54 and the conversion of homopolymer model networks to star homopolymers.57,59,61,62 Regarding the formation of functional end-groups, these are the residues of the labile initiator fragment after its degradation, remaining in the polymer. Thus, thiol end-functional groups resulted from the reductive degradation of polymers made using initiators bearing disulfidegroups41–53 or the aminolysis of polymers prepared using initiators carrying symmetrical trithiocarbonates,54–56aldehyde end-functional groups were produced upon the ozonolysis of olefin-bearing initiators,59 diene and dienophile end-groups were released upon the thermolysis of initiators containing Diels–Alder adducts,60 and, finally, nitroso-aldehyde and carboxylic acid end-groups resulted upon the photocleavage of initiators with ortho-nitrobenzyl estergroups.61,62
Owing to their remarkable tolerance to the various degradable groups, controlled radical polymerization methods were employed in most of the above polymer syntheses. These mainly included atom transfer radical polymerization (ATRP),64–67 but also reversible addition–fragmentation chain transfer (RAFT) polymerization68–71 and, to a smaller extent, nitroxide-mediated polymerization (NMP).72 For ATRP, the most frequently used degradable group was the disulfide,41–46 whose cleavage can be achieved using reducing reagents.73 Although these reagents may be rather common compounds, such as phosphines, glutathione, or dithiothreitol, groups cleavable by even more common compounds might be advantageous. Such an example is the hemiacetal ester group, resulting from the addition of a carboxylic acid onto a vinyl ether, and which can be readily cleaved using dilute mineral acid or alcohols,74 or even, sometimes, pure water.75 The use of the hemiacetal ester group in polymer science has mainly been restricted to the protection of carboxylic acid-bearing monomers and their facile post-polymerization deprotection.76–89 However, there are some rare examples of the use of the labile hemi(acetal ester)group in the polymer backbone,90 in the cross-linker18 and in the initiator.57,58 The aim in the present investigation is to prepare, for the first time, a hemiacetal ester-bearing ATRP bifunctional initiator and use it to synthesize degradable, end-linked amphiphilic polymer conetworks (APCN). After their characterization, these APCNs were hydrolyzed and the hydrolysis products, novel amphiphilic star copolymers, were also characterized.
Experimental
Materials and methods
The monomers 2-(dimethylamino)ethyl methacrylate (DMAEMA, 99%) and methyl methacrylate (MMA, 99%), and the cross-linker ethylene glycol dimethacrylate (EGDMA, 98%) were purchased from Aldrich, Germany. Copper(I) bromide (99%), copper(II) bromide (99%), 2,2′-bipyridine (bpy, 99%), 2-bromoisobutyric acid (98%), ethylene glycol divinyl ether (97%), calcium hydride (CaH2, 90–95%), 2,2-diphenyl-1-picrylhydrazyl hydrate (DPPH, 95%), and basic alumina (98%) were also purchased from Aldrich and were used as received. N,N-Dimethylformamide (DMF, Merck 99%) was dried over CaH2 and was freshly distilled under reduced pressure. The DMAEMA and MMA monomers and the EGDMA cross-linker were passed through basic alumina columns to remove the polymerizationinhibitor and any other acidic impurities, and they were stirred overnight over CaH2 to remove the last traces of moisture and protonic impurities. This was done in the presence of added DPPH, a free radical inhibitor, to avoid undesired thermal polymerization. The monomers and the cross-linker were freshly distilled prior to the polymerization.Preparation and characterization of the degradable, bifunctional ATRPinitiator
For the synthesis of the degradable bifunctional initiator1,2-bis[1-(2-bromo-2-methyl-1-oxo-propanyloxy)ethoxy]ethane (bBMOPE), 1.25 equivalents of 2-bromoisobutyric acid (16.72 g, 0.10 mol) were reacted with 5 mL of ethylene glycol divinyl ether (4.57 g, 0.04 mol) at 65 °C for 24 h until all divinyl ether was consumed, as confirmed by 1H-NMR spectroscopy. Afterwards, the mixture was dissolved in 60 mL of THF and the solution was passed through a column of basic alumina to remove the excess of 2-bromoisobutyric acid. Finally, the THF was evaporated off using a rotary evaporator and the desired bifunctional initiator was isolated at 65% yield as a yellowish liquid. The structure of the initiator was confirmed by 1H and 13C-NMR spectroscopy.1H NMR (300 MHz, CDCl3, δ): 5.96 (t, 2 × 1H, −OCH(CH3)O−), 3.86 (dd, 2 × 2H, −OCH2CH2O–), 1.92 (s, 4 × 3H, –C(CH3)2Br), 1.46 (d, 2 × 3H, –OCH(CH3)O–). 13C NMR (300 MHz, CDCl3, δ): 171.4 (2C, –COOCH(CH3)O–), 97.8 (2C, –OCH(CH3)O–), 65.1 (2C, –OCH2CH2O–), 54.3 (2C, –C(CH3)2Br), 32.7 (4C, –C(CH3)2Br), 18.9 (2C, –OCH(CH3)O–).
Homopolymer network preparation
All end-linked homopolymer networks of this study were prepared by sequential ATRP. The polymerization procedure for the synthesis of the conetwork EGDMA0.5-grad-MMA25-bBMOPE-MMA25-grad-EGDMA0.5 is described below. To a 25 mL Schlenk flask kept under a dry nitrogen atmosphere were added 44.8 mg CuBr (3.1 × 10−4 mol), 2.0 mg CuBr2 (9.0 × 10−6 mol), 0.10 g bpy (6.4 × 10−4 mol), 1.20 mL MMA (1.13 g, 11.3 × 10−3 mol) and 2.0 mL DMF. The mixture was degassed by two vacuum-nitrogen cycles. The reaction flask was heated to 50 °C and a deoxygenated solution of the bBMOPE bifunctional initiator (0.1 g, 2.23 × 10−4 mol) in 0.5 mL DMF was added (monomer concentration 3.0 M). After 50 min of reaction, at a monomer conversion of 84%, samples were extracted for size exclusion chromatography (SEC) and 1H NMR spectroscopy analyses (polymer number-average molecular weight = Mn = 6760 g mol−1 compared with 4450 g mol−1 which was the theoretically expected molecular weight; polydispersity index = PDI = Mw/Mn = 1.43; Mw is the polymer weight-average molecular weight). Then, the cross-linker EGDMA (0.04 mL, 0.04 g, 2.23 × 10−4 mol, at a cross-linker![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :initiator molar ratio of 1:1) was added, leading to network formation within 20 min. The polymerization reaction was kept at 50 °C for another 2 h after network formation was observed. Homopolymer networks with different degrees of polymerization (DPs) of MMA were prepared by varying the monomer loading (but at a constant MMA monomer concentration of 3.0 M in all cases) at constant loadings of the degradable bifunctional initiator, the copper salts and the bpy ligand.
:initiator molar ratio of 1:1) was added, leading to network formation within 20 min. The polymerization reaction was kept at 50 °C for another 2 h after network formation was observed. Homopolymer networks with different degrees of polymerization (DPs) of MMA were prepared by varying the monomer loading (but at a constant MMA monomer concentration of 3.0 M in all cases) at constant loadings of the degradable bifunctional initiator, the copper salts and the bpy ligand.
Conetwork preparation
All end-linked amphiphilic copolymer conetworks of this study were also prepared by sequential ATRP. The polymerization procedure for the synthesis of the conetwork EGDMA0.5-grad-MMA12.5-grad-DMAEMA12.5-bBMOPE-DMAEMA12.5-grad-MMA12.5-grad-EGDMA0.5 is described below. To a 25 mL Schlenk flask kept under a dry nitrogen atmosphere were added 96.0 mg CuBr (6.7 × 10−4 mol), 5.0 mg CuBr2 (2.2 × 10−5 mol), 0.21 g bpy (1.3 × 10−3 mol), 1.88 mL DMAEMA (1.75 g, 0.011 mol) and 0.5 mL DMF. The mixture was degassed by two vacuum-nitrogen cycles. The reaction flask was heated to 50 °C and a deoxygenated solution of the bBMOPE bifunctional initiator (0.2 g, 4.5 × 10−4 mol) in 0.35 mL DMF was added. After 141 min of reaction, at a monomer conversion of 97%, samples were extracted for SEC and 1H NMR spectroscopy analyses (polymerMn = 6040 g mol−1 compared with 4260 g mol−1 which was the theoretically expected; PDI = 1.56). Then, the second monomer, MMA (1.19 mL, 1.12 g, 0.011 mol) was added and was allowed to react for another 214 min until the MMA conversion reached 78%. After sampling for SEC and 1H NMR spectroscopy analyses (polymerMn = 9600 g mol−1 compared with the theoretically expected value of 6330 g mol−1; PDI = 1.63; MMA content determined by 1H NMR = 46% mol, compared with the theoretically expected value of 50%), EGDMA (0.08 mL, 0.09 g, 4.46 × 10−4 mol, cross-linker:initiator molar ratio equal to 1:1) was added, leading to conetwork formation within 116 min. The polymerization reaction was kept at 50 °C for another 18 h after conetwork formation was observed. Conetworks of different compositions were prepared by varying the relative amounts of the two comonomers, whereas conetworks of different architectures were obtained by varying the addition sequences. In all cases, at each step of the sequential procedure, the monomers were allowed to react until conversions between about 80 and 95% were reached before proceeding to the next step.
Size exclusion chromatography (SEC)
Samples of the linear homopolymer and the linear copolymer precursors to the (co)networks obtained during the polymerizations, samples of the sol fraction from the conetworks, and samples from the hydrolysis products of the end-linked homopolymer networks were characterized by SEC to obtain their molecular weight distributions (MWD) and calculate from those the average molecular weights and PDIs. SEC was performed on a Polymer Laboratories chromatograph equipped with an ERC-7515A refractive index (RI) detector and a PL Mixed “D” column (SEC-RI system). The mobile phase was THF delivered at a flow rate of 1 mL min−1 using a Waters 515 isocratic pump. The molecular weight calibration curve was based on eight narrow MWD linear polyMMA standards of molecular weights of 850, 2810, 4900, 11550, 30530, 60150, 138500 and 342900 g mol−1 also from Polymer Laboratories.
Nuclear magnetic resonance (NMR) spectroscopy
The compositions of the conetwork linear precursors and of the sol fraction from the conetworks were determined by 1H-NMR spectroscopy using a 300 MHz Avance Bruker NMR spectrometer equipped with an Ultrashield magnet. The solvent was CDCl3 containing traces of tetramethylsilane (TMS) which was used as an internal reference. 1H NMR spectroscopy was also used to confirm the structure and the purity of the degradable bBMOPE bifunctional initiator, the monomers and the cross-linker, and also to determine monomer and cross-linker conversion. The structure of bBMOPE was also characterized using 13C NMR spectroscopy.Determination and characterization of the sol fraction in the (co)networks
Before extracting the sol fraction from the entire (co)network, a small piece from each (co)network was extracted by being placed in CDCl3 and allowed to equilibrate for one day. The thus-obtained CDCl3 solution of “early extractables” was characterized using 1H-NMR spectroscopy to swiftly determine the final monomer and cross-linker conversions. Subsequently, the entire conetwork was extracted with 100 mL THF for 2 weeks to remove the sol fraction. Next, the resulting THF solution of the extractables was separated from small conetwork pieces by filtration, and the THF was subsequently evaporated off using a rotary evaporator. The recovered extracted polymer was further dried for 48 h in a vacuum oven at room temperature. The sol fraction was calculated as the ratio of the dried mass of the extracted polymer divided by the theoretical mass of the polymer in the (co)network, taken as the sum of the masses of the initiator and the polymerized monomers and cross-linker. The sol fraction was finally characterized in terms of its molecular weight and composition using SEC and 1H NMR spectroscopy, respectively.Measurement of the degree of swelling (DS)
The degrees of swelling (DSs) of all the (co)networks were measured first in THF. To this end, small (∼1 cm3) pieces were cut out from each THF-equilibrated (co)network, weighed separately, and dried in a vacuum oven at room temperature for 72 h. Finally, each dried piece was weighed separately and the DS was calculated as the ratio of the swollen divided by the dry (co)network mass. For the measurement of the DSs in pure water, samples from each conetwork were dried from THF in a vacuum oven at room temperature for 72 h to determine their dry mass, and the samples were subsequently equilibrated in pure water for 2 weeks. The mass of the water-swollen conetwork was determined gravimetrically and the DS was calculated again as the ratio of the swollen divided by the dry conetwork mass.Measurement of the evolution of the conetwork apparent DSs in pure water and in acidified water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :THF mixtures during hydrolysis
:THF mixtures during hydrolysis
For the measurements of the apparent DSs as a function of time, a dry sample from each (co)network (∼55 mg) was placed in a glass vial with ∼5 mL of the appropriate solvent (either 5 mL of pure water, or a mixture composed of 2.5 mL of THF and 2.5 mL of pure water plus ∼100 mg of neat HCl added as a 10 M HCl aqueous solution). The measurements were performed at room temperature (∼23 °C), and, in the case of the experiments in pure water, the pH was around 7.3, a result of the presence of the weakly basic DMAEMA units in the (co)networks. At regular time intervals, the solvent was removed via a disposable syringe and the mass of the (co)network was determined gravimetrically. The apparent DSs at different times were calculated again as the ratio of the swollen divided by the dry (co)network mass. Finally, the moment of complete (co)network dissolution was noted.
Characterization of the (co)network hydrolysis products. Static light scattering
The absolute Mw of the star polymers that resulted from the hydrolysis of the homopolymer end-linked networks of MMA was measured using static light scattering (SLS) in an SEC configuration (SEC-SLS system). To this end, a Brookhaven Molecular Weight Analyzer, BI-MwA, equipped with a 30 mW red diode laser emitting at 673 nm and a multi-angle detector, was used to determine the intensity of scattered light at 7 different angles—35°, 50°, 75°, 90°, 105°, 130° and 145°—whereas a PL-RI 800 RI detector was used to simultaneously measure the RI signal. A Polymer Laboratories PL-LC1120 isocratic pump was used to deliver the THFmobile phase at a flow rate of 1 mL min−1 through a PL-Mixed “D” column, also supplied by Polymer Laboratories. The analysis for the calculation of the absolute Mw was conducted using the PSS-WinGPC 7 SLS-flow software. The star polymers were dissolved in HPLC-grade THF at a 2% w/v polymer concentration and the resulting solutions were filtered through a 0.45 μm pore size syringe filter. The RI increment (dn/dc) of the star polymer solutions in THF was determined using an ABBE refractometer and was found to be 0.087 mL g−1.It was not possible to characterize the molecular weight of the hydrolysis products of the DMAEMA-containing conetworks using SEC for various reasons. First of all, these copolymers were insoluble in THF (a result of the ionization of the DMAEMA units by the added HCl), which precluded characterization using the SEC-RI or the SEC-SLS systems in THF. Although these copolymers were soluble in DMF and in acidified water (pH ≈ 4), the copolymers did not elute from the respective columns (organic and aqueous), even when the mobile phases were modified via the addition of salt (0.5 M potassium hexafluorophosphate or 0.5 M Na2NO3, respectively). Nonetheless, an estimation of the molecular weights of these star copolymers was obtained through their characterization using small angle-neutron scattering (SANS) in deuterated dimethyl sulfoxide (d6-DMSO) as detailed in the following paragraph.
Small-angle neutron scattering (SANS)
All star copolymers formed from the acid hydrolysis of the APCNs were characterized using SANS in d6-DMSO. The SANS measurements were performed on the 30 m NG7 instrument at the Center for Neutron Research of the National Institute of Standards and Technology (NIST) in Gaithersburg, Maryland, USA. The incident wavelength was λ = 6 Å. Three sample to detector distances of 1, 4.5 and 13.5 m were employed, covering a q-range [q = 4π/λ sin (θ/2)] from 0.0335 nm to 5.375 nm−1. The samples were loaded in 1 mm gap thickness quartzcells. The scattering patterns were isotropic, and, therefore, the measured counts were circularly averaged. The averaged data were corrected for empty cell and background. The SANS data were extrapolated to q = 0 to determine the scattered intensity at zero angle, which was used to calculate the absolute Mw of the star copolymers using eqn (1) given below:91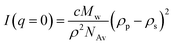 | (1) |
Results and discussion
Novelty of the synthetic strategy
The copolymer conetworks produced in this study represent the first degradable, amphiphilic, end-linked conetworks prepared by ATRP. End-linked networks based on degradable initiators have already been synthesized by ATRP, but these networks were composed of only hydrophobic homopolymers.59,61,62 Degradable APCNs comprising degradable monomer repeating units either in the macro-cross-linker92,93 or in the main chain94 have also been prepared by ATRP, but those were not end-linked. Our group has recently reported the preparation of degradable, amphiphilic end-linked conetworks, but that synthesis was performed by group transfer polymerization (GTP).58 In the present investigation, we elected to pursue the synthesis of such APCNs viaATRP, a modern, robust controlled radical polymerization method.64–67 The first step in this study was the synthesis of an appropriate initiator. Thus, a bis(hemiacetal ester) bifunctional degradable ATRPinitiator, 1,2-bis[1-(2-bromo-2-methyl-1-oxo-propanyloxy)ethoxy]ethane (bBMOPE), was synthesized by an addition reaction shown in Fig. 1. This compound possesses two ATRP initiating groups and two degradable hemiacetal ester groups that can easily degrade under mildly acidic conditions.74–89 Previously prepared degradable, bifunctional ATRPinitiators bore reducible disulfide,41–46ozonolyzable olefinic,59 thermolyzable Diels–Alder adduct,60 and photocleavable ortho-nitrobenzyl ester61,62groups.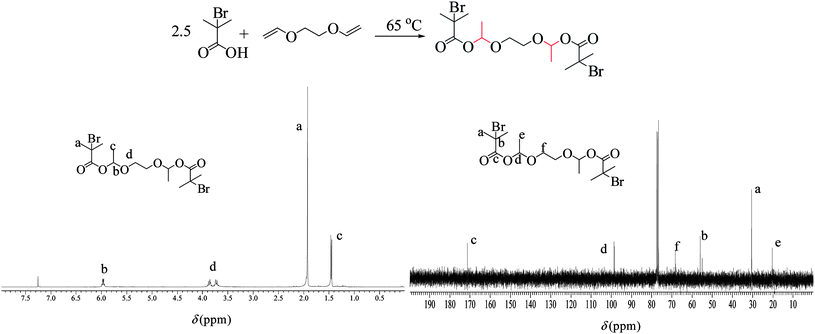 | ||
| Fig. 1 Reaction scheme for the synthesis of the degradable bifunctional ATRPinitiator and its structure confirmation by 1H and 13C NMR spectroscopy. | ||
For the synthesis of the cleavable APCNs, sequential terpolymerization of the two comonomers and the cross-linker was employed, as illustrated in Scheme 1. MMA and DMAEMA were employed as the hydrophobic and the hydrophilic comonomers, respectively, while EGDMA served as the hydrophobic cross-linker.
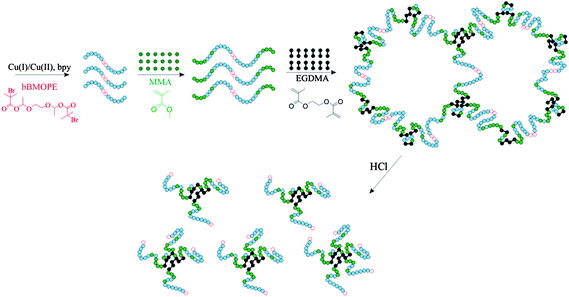 | ||
| Scheme 1 ATRP synthesis and hydrolysis of an end-linked APCN prepared using a degradable bifunctional initiator. The green and blue filled circles represent the MMA and the DMAEMA monomer repeating units, respectively, whereas the black dumbbells indicate the EGDMA cross-linker units. The degradable bifunctional initiator residue is represented by the two adjacent hollow red circles. | ||
(Co)network design
The present conetwork syntheses produced APCNs of different compositions and different architectures. The former were attained by varying the relative loadings of the two comonomers, whereas the latter were afforded by changing the order of comonomer, cross-linker or initiator additions. The conetwork composition was varied in three APCNs (entries 4, 6 and 7 in Table 1) based on MMA-b-DMAEMA-b-MMAtriblock copolymers (“ABA structure”) in which the overall targeted DP of the chain was kept at 50. The conetwork architecture was varied in four equimolar-in-comonomer-composition APCNs with an overall targeted chain DP again of 50 (entries 4, 5, 8 and 9 in Table 1). One of these four APCNs was based on a DMAEMA-b-MMA-b-DMAEMAtriblock copolymer (“BAB structure”), one was based on a DMAEMA-co-MMA statistical copolymer, and the third was the randomly cross-linked conetwork of the statistical copolymer, prepared by the simultaneous terpolymerization of the two comonomers and the cross-linker. The fourth isomeric APCN was the equimolar conetwork with the ABA structure mentioned above. Thus, there were in total four isomeric conetworks with the same composition and different architectures. The three homopolymer networks (entries 1, 2 and 3 in Table 1) were based on MMA with linear precursors of different molecular weights.| No. | Polymer structurea | Conversionb (mol%) | Timec/min | Gelation time/min | Theor. molec. weightd | SEC results | MMA (mol%) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MMA | DMAEMA | EGDMA | M n | M w/Mn | Theor. | 1H NMR | |||||
| a D, M and E are further abbreviations for DMAEMA, MMA and EGDMA, respectively. b Determined using 1H NMR spectroscopy in CDCl3. c Separate reaction time at each step of the sequential addition. d Theor. molec. weight = [monomer]/[initiator] × (monomer conversion) × (monomer molecular weight) + (initiator molecular weight). | |||||||||||
| 1 | M10-I-M10 | 86 | — | — | 50 | — | 2170 | 7260 | 1.43 | 100 | 100 |
| E0.5-grad-M10-I-M10-grad-E0.5 | 100 | — | 100 | 133 | 34 | ||||||
| 2 | M25-I-M25 | 80 | — | 90 | — | 4450 | 6760 | 1.43 | 100 | 100 | |
| E0.5-grad-M25-I-M25-grad-E0.5 | 84 | — | 100 | 140 | 19 | ||||||
| 3 | M35.5-I-M35.5 | 83 | — | — | 150 | — | 6680 | 10000 | 1.41 | 100 | 100 |
| E0.5-grad-M35.5-I-M35.5-grad-E0.5 | 89 | — | 63 | 150 | 13 | ||||||
| 4 | D12.5-I-D12.5 | — | 97 | — | 141 | — | 4260 | 6040 | 1.56 | — | — |
| M12.5-grad-D12.5-I-D12.5-grad-M12.5 | 78 | 100 | — | 214 | — | 6330 | 9600 | 1.63 | 50 | 46 | |
| E0.5-grad-M12.5-grad-D12.5-I-D12.5-grad-M12.5-grad-E0.5 | 85 | 100 | 89 | 1154 | 114 | — | — | — | — | — | |
| 5 | M12.5-I-M12.5 | 93 | — | — | 161 | — | 2780 | 4860 | 1.40 | — | — |
| D12.5-grad-M12.5-I-M12.5-grad-D12.5 | 100 | 55 | — | 125 | — | 4940 | 6990 | 1.47 | 50 | 65 | |
| E0.5-grad-D12.5-grad-M12.5-I-M12.5-grad-D12.5-grad-E0.5 | 100 | 73 | 57 | 1507 | 260 | — | — | — | — | — | |
| 6 | D20-I-D20 | — | 89 | — | 165 | — | 6050 | 8000 | 1.41 | — | — |
| M5-grad-D20-I-D20-grad-M5 | 70 | 94 | — | 172 | — | 6750 | 9250 | 1.45 | 20 | 25 | |
| E0.5-grad-M5-grad-D20-I-D20-grad-M5-grad-E0.5 | 85 | 100 | 72 | 1260 | 54 | — | — | — | — | — | |
| 7 | D5-I-D5 | — | 91 | — | 111 | — | 1880 | 3740 | 1.38 | — | — |
| M20-grad-D5-I-D5-grad-M20 | 86 | 100 | — | 250 | — | 5324 | 29200 | 1.31 | 80 | 85 | |
| E0.5-grad-M20-grad-D5-I-D5-grad-M20-grad-E0.5 | 91 | 100 | 85 | 1160 | 230 | — | — | — | — | — | |
| 8 | (D12.5-co-M12.5)-I-(D12.5-co-M12.5) | 86 | 89 | — | 115 | — | 6100 | 10400 | 1.45 | 50 | 44 |
| E0.5-grad-(D12.5-co-M12.5)-I-(D12.5-co-M12.5)-grad-E0.5 | 94 | 100 | 100 | 1350 | 70 | — | — | — | — | — | |
| 9 | (D12.5-co-M12.5-co-E0.5)-I-(D12.5-co-M12.5-co-E0.5) | 83 | 100 | 95 | 1405 | 33 | — | — | — | — | — |
Characteristics of the linear precursors
Table 1 lists all the data concerning the polymerizations, both for linear polymer and network formation. In particular, next to each polymer structure the table provides the monomer and cross-linker conversion (determined using 1H NMR spectroscopy in CDCl3) at the time the polymerization was stopped, the gelation time for the networks and the molecular weights, PDIs and composition for the linear polymers. As shown in the table, the time required for the monomers to reach 80–95% conversion increased with the targeted molecular weight. For example, focusing on the linear polyMMAs, to afford DPs of 20, 50 and 75, at constant final monomer conversion, the polymerization time had to be increased from 50 to 90 and to 150 min, respectively. Characterization using SEC indicated that the Mn values of the linear precursors of the (co)networks were, in most cases, close to, but systematically higher than the theoretically expected molecular weights due to partial deactivation of the active sites. The PDIs of these precursors were relatively high, but still lower than 1.5. High PDIs were probably the result of partial deactivation of the active sites as well as the relatively low DPs aimed. It is noteworthy that the present PDIs were higher than those of similar (co)polymers synthesized by ATRP but using a non-degradable initiator.95 We also note that MMA was preferred over DMAEMA for performing the study with the homopolymers of different molecular weights, as the hydrolysis products of polyMMA could readily be characterized by using SEC, whereas those of polyDMAEMA would be insoluble in the SECsolvent, THF, as a result of their partial ionization due to uptake of HCl.The compositions of the copolymers were determined from their 1H NMR spectra by dividing the normalized area under the signal from the three methoxyprotons in MMA at 3.6 ppm by that due to the six protons in the two azamethylgroups in DMAEMA at 2.3 ppm and were found to be very close to the compositions calculated on the basis of the comonomer feed ratios and monomer conversions.
Conetwork sol fraction
The sol fractions of all the (co)networks are listed in Table 2. These cover a range of values from 8 to 60%. In the case of the homopolymer networks, as the elastic chain length increased, the sol fraction also increased, indicating less efficient end-linking for longer chains, which can be attributed to increased steric hindrances and to the lower concentration of the active polymerization sites, a result of the lower initiator concentration (note that the initial MMA monomer concentration was kept constant at 3.0 M in these three syntheses). In the APCNs, when the conetwork composition was varied at constant architecture (ABA structures), the sol fraction was almost the same (∼40%). When the conetwork architecture was varied at constant composition, the sol fraction was found to decrease in the more random architectures. In particular, the sol fraction was lowest for the randomly cross-linked conetwork (8%), higher for the end-linked statistical conetwork (26%), even higher for the end-linked MMA-DMAEMA-MMA gradient copolymer-based conetwork (42%), and highest for the end-linked DMAEMA-MMA-DMAEMA gradient copolymer-based conetwork (60%). The very high sol fraction in this last conetwork was probably due to the longer polymerization time necessary for the formation of this conetwork compared to the other amphiphilic conetworks, leading to deactivation of more chains. It is noteworthy that this last APCN, with the BAB structure, was very loose and had a mucous texture rather than a network texture, which made the study of its acid hydrolysis difficult. In addition to architecture and elastic chain length, the sol fraction was also highly influenced by the conversion of the EGDMA cross-linker. For example, in the case of the EGDMA0.5-grad-DMAEMA12.5-grad-MMA25-grad-DMAEMA12.5-grad-EGDMA0.5 conetwork which displayed the highest sol fraction of 60%, this conetwork had the lowest EGDMA conversion (57%) and exhibited very poor mechanical properties, as already mentioned. On the other hand, the randomly cross-linked conetwork, which displayed a high conversion of EGDMA, presented the lowest sol fraction value and was a very compact conetwork. The sol fraction determined for these conetworks was similar to that determined for the degradable amphiphilic conetworks synthesized by GTP reported previously.58| Polymer (co)network structurea | Sol fraction (%) | MMA (mol%) | GPC | |||
|---|---|---|---|---|---|---|
| Theory | 1H-NMR | M p | M n | M w/Mn | ||
| a M: methyl methacrylate; D: 2-(dimethylamino)ethyl methacrylate; E: ethylene glycol dimethacrylate. | ||||||
| E0.5-grad-M10-I-M10-grad-E0.5 | 19 | 100 | 100 | 16300 |
14100 |
1.4 |
| E0.5-grad-M25-I-M25-grad-E0.5 | 46 | 100 | 100 | 57500 |
64800 |
1.3 |
| E0.5-grad-M37.5-I-M37.5-grad-E0.5 | 54 | 100 | 100 | 12700 |
14100 |
4.1 |
| E0.5-grad-M12.5-grad-D12.5-I-D12.5-grad-M12.5-grad-E0.5 | 42 | 50 | 42.0 | 7620 | 4880 | 1.8 |
| E0.5-grad-D12.5-grad-M12.5-I-M12.5-grad-D12.5-grad-E0.5 | 60 | 50 | 40.1 | 3050 | 4310 | 1.4 |
| E0.5-grad-M5-grad-D20-I-D20-grad-M5-grad-E0.5 | 40 | 20 | 18.0 | 8320 | 6210 | 1.5 |
| E0.5-grad-M20-grad-D5-I-D5-grad-M20-grad-E0.5 | 45 | 80 | 84.2 | 32400 |
11300 |
2.3 |
| E0.5-grad-(D12.5-co-M12.5)-I-(D12.5-co-M12.5)-grad-E0.5 | 26 | 50 | 46.6 | 5130 | 4900 | 1.8 |
| (D12.5-co-M12.5-co-E0.5)-I-(D12.5-co-M12.5-co-E0.5) | 8 | 50 | 44.7 | 2330 | 2070 | 1.3 |
SEC measurements indicated that the sol fraction mainly consisted of linear chains with molecular weights lower than those of the corresponding linear precursors (in some cases even lower than those of the linear homopolymer precursors), suggesting that these resulted from termination at the early stages of the polymerization procedure or transfer reaction to monomer in the medium. The same conclusion was also reached by examining the compositions of the extractables from the conetworks, which were found to be richer in the monomer that was added first during the sequential polymerization. It is noteworthy that the APCN with the BAB structure, presenting the highest sol fraction, 60%, displayed the lowest molecular weight, contrary to our expectation for a high molecular weight. It is possible that the sol fraction of this APCN also contained microgels (of much higher molecular weight) which were retained on the paper filter during the separation of the solution of the extractables from the mucous conetwork.
(Co)network DSs in THF and in pure water
The DSs of the (co)networks in THF and in pure water were measured gravimetrically and are listed in Table 3. The DSs in THF of the MMA homopolymer end-linked networks increased with chain molecular weight from 3 to 10. In contrast, the DSs in THF of the ABA triblock copolymer-based end-linked conetworks exhibited values within a narrower range, from 14 to 18, a result of the property of THF to be a non-selective solvent for DMAEMA and MMA and the identical nominal DPs of 50. In fact, the DSs in THF of the three ABA triblock copolymer-based APCNs were inversely proportional to the EGDMA conversion in them (see Table 1), with the DMAEMA-richest homologue displaying the highest DS in THF due to its lowest EGDMA conversion (72%). The isomeric APCNs exhibited a broader range of DSs in THF, from 4 to 20, owing to the wider range of EGDMA conversions, from 57 to 100%, with the APCN with the lowest EGDMA conversion presenting the highest DS in THF. The randomly cross-linked conetwork and the statistical copolymer-based end-linked APCN displayed the highest EGDMA conversions and the lowest DSs in THF.| Polymer (Co)network structurea | DS in THF | DS in water |
|---|---|---|
| a M: methyl methacrylate; D: 2-(dimethylamino)ethyl methacrylate; E: ethylene glycol dimethacrylate. | ||
| E0.5-grad-M10-I-M10-grad-E0.5 | 3.4 ± 0.5 | — |
| E0.5-grad-M25-I-M25-grad-E0.5 | 10 ± 4 | — |
| E0.5-grad-M12.5-grad-D12.5-I-D12.5-grad-M12.5-grad-E0.5 | 13.6 ± 1.5 | 5.4 ± 0.8 |
| E0.5-grad-D12.5-grad-M12.5-I-M12.5-grad-D12.5-grad-E0.5 | 20.1 ± 0.8 | 10 ± 2 |
| E0.5-grad-M5-grad-D20-I-D20-grad-M5-grad-E0.5 | 17.7 ± 0.2 | 5.5 ± 0.7 |
| E0.5-grad-M20-grad-D5-I-D5-grad-M20-grad-E0.5 | 15 ± 2 | 2.1 ± 0.7 |
| E0.5-grad-(D12.5-co-M12.5)-I-(D12.5-co-M12.5)-grad-E0.5 | 4.93 ± 0.17 | 4 ± 2 |
| (D12.5-co-M12.5-co-E0.5)-I-(D12.5-co-M12.5-co-E0.5) | 4.32 ± 0.13 | 2.1 ± 0.7 |
The DSs of the APCNs in water were always lower than those determined in THF because water is a selective solvent for the DMAEMA units. The DSs in water of the isomeric APCNs of different architectures (and all with the same hydrophobic content) reflected the different EGDMA conversions. However, the DSs in water of the ABA triblock copolymer-based end-linked APCNs reflected both the EGDMA conversion and the MMA content. For example, the MMA-richest ABA triblock copolymer-based end-linked APCN displayed the lowest DS in water even though it did not display the lowest DS in THF, a result of its high hydrophobic content of 80 mol%.
The DSs in THF and in water from Table 3 for the APCNs are plotted in Fig. 2. Fig. 2a shows the effect of copolymer composition, while Fig. 2b presents the effect of conetwork architecture.
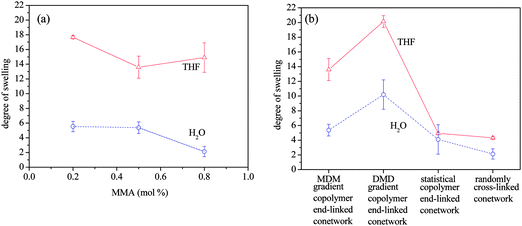 | ||
| Fig. 2 Degree of swelling of the conetworks in THF and in water. (a) Effect of copolymer composition. (b) Effect of copolymer architecture. | ||
(Co)network degradation
After (co)network characterization in terms of their DSs and their sol fraction, we proceeded with their hydrolysis using mineral acid. In the cases of the three MMA homopolymer networks, the hydrolysis was performed in THF in the presence of aqueous HCl. When pieces of the three networks were placed in this solution, in two of the three cases the networks dissolved due to the hydrolysis of the initiator residue. The homopolymer network with the lowest DP (E0.5-grad-MMA20-grad-E0.5) did not dissolve because the short length of the elastic chains allowed the formation of a second network entirely based on cross-linker, which could not be degraded,57 even though the contained initiator residues probably degraded.For the APCNs, hydrolysis was first attempted in pure water, because this solvent could readily hydrolyze most of the APCNs synthesized by GTP based on a degradable initiator also bearing two hemiacetal ester groups.58 Thus, the time-dependence of the DSs in pure water of all APCNs was followed and the results are plotted in Fig. 3. Fig. 3a displays the effect of copolymer composition by plotting the swelling profiles of all the APCNs based on MMA-grad-DMAEMA-grad-MMA triblock copolymers with different compositions, whereas Fig. 3b shows the effect of conetwork architecture by plotting the swelling profiles of the four isomeric conetworks with the same composition but different distribution of the comonomer and cross-linker units. As can be observed from Fig. 3, in no case did conetwork hydrolysis occur in pure water. This is because after a brief period, the apparent DSs of all conetworks did not fall down to zero but were stabilized to certain values. These values were relatively low and increased with the DMAEMA content in the case of the conetworks with different compositions, a result of the water-solubility of the DMAEMA units. In the case of the conetworks with different architectures, higher DSs were observed when the cross-linker conversion was lower.
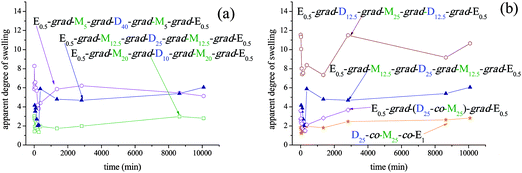 | ||
| Fig. 3 Temporal evolution of the apparent degrees of swelling of all the conetworks in pure water. (a) Effect of copolymer composition. (b) Effect of copolymer architecture. | ||
The failure of the present conetworks to hydrolyze in pure water might be due to their increased overall hydrophobicity and/or micelle-like arrangement of the constituting (gradient) block copolymer chains, hindering some part of the APCN and preventing sufficient access of water to the labile initiator residues. Thus, conetwork hydrolysis was performed using a 50:50 v/v THF:H2O mixture with added HCl (final HCl concentration in the solvent mixture = 0.16 M). The hydrolysis of the APCNs could not be performed in acidified (with neat HCl) THF (without added water) due to the insolubility in THF of the charged DMAEMA units, leading to the collapse of the conetworks in this solvent which would inhibit their hydrolysis. The temporal evolution of the swollen mass of all the conetworks in the acidified THF/H2O mixture was followed, from which the apparent DSs were calculated and are plotted as a function of time in Fig. 4.
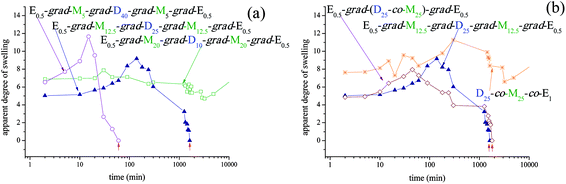 | ||
| Fig. 4 Temporal evolution of the apparent degrees of swelling of all the conetworks in 50:50 v/v water–THF mixtures in the presence of HCl. (a) Effect of copolymer composition. (b) Effect of copolymer architecture. The vertical red arrows indicate the time at which complete conetwork dissolution was observed. | ||
Fig. 4a displays the time-dependence of the apparent DSs of the three APCNs based on end-linked MMA-grad-DMAEMA-grad-MMA triblock copolymers of different compositions. Whereas the apparent DSs of the MMA-rich APCN displayed low and time-invariant DSs, a result of the protection of the labile initiator residue from HCl by the higher mol fraction of the hydrophobic MMA monomer repeating units, the two other end-linked APCNs exhibited a swelling maximum: increasing apparent DSs at early times arising from the absorption of large amounts of solvent and some conetwork hydrolysis, followed by a large reduction in the apparent DS because of extensive conetwork hydrolysis and washing away of both polymer and solvents. In the case of the DMAEMA-rich APCN, complete conetwork dissolution was observed within 1 h, while in the case of the equimolar APCN, hydrolysis was much delayed (27 h) compared to the former conetwork. The presence of more hydrophobic MMA units in the equimolar APCN provided more protection to the labile initiator residue, thus further delaying the hydrolysis of this conetwork.
Fig. 4b shows the apparent DSs in the acidified THF:water mixture of the three isomeric APCNs of different polymer architectures as a function of time. These include the APCN based on the end-linked MMA-grad-DMAEMA-grad-MMA triblock copolymer (taken from Fig. 4a), the one based on the end-linked DMAEMA-co-MMA statistical copolymer, and the randomly cross-linked APCN of the statistical copolymer. The poor mechanical properties of the APCN based on the end-linked DMAEMA-grad-MMA-grad-DMAEMA triblock copolymer made very difficult the monitoring of the apparent DSs of this conetwork during the course of its acid hydrolysis, and, therefore, such data were not collected. The apparent swelling profile of the end-linked conetwork based on the statistical copolymer was similar to that based on the triblock copolymer with a polyDMAEMA midblock, taking only slightly longer for its full dissolution, 30 h compared to 27 h in the case of the latter APCN. In the statistical conetwork, the distribution of the two monomers was random, and, unlike the APCN based on the triblock copolymer with a polyDMAEMA midblock, the initiator residue was surrounded also by MMA units, thus providing a slightly better protection from hydrolysis. Finally, the randomly cross-linked conetwork of the statistical copolymer gradually swelled without completely dissolving, as the initiator residues in this architecture were also protected by the hydrophobic EGDMA cross-linker units which were also randomly distributed.
Star polymer molecular weights and numbers of arms
The hydrolysis products of the two degradable end-linked MMA homopolymer networks were characterized in terms of their molecular weights using SEC-RI and SEC-SLS, and the results are summarized in Table 4. The SEC-RI traces of the hydrolysis products bore two peaks: one at earlier retention times due to the expected star polymers, and one at later times due to the (cleavable) dangling chains. The possibility that this latter peak was due to the extractables was excluded since those were thoroughly washed out before network hydrolysis. The fraction of dangling chains was estimated from the relative peak areas in the chromatograms and was about 27%. The star polymers were also characterized in terms of their absolute Mws using SEC-SLS. The SLSMws were higher than the SEC-RI molecular weights due to the compact nature of the star polymers compared to the linear polyMMA SEC-RI calibration standards. From the SLSMws, the absolute numbers of arms were calculated to be 106 and 33 for the two star polymer products and decreasing with the parent network elastic chain length due to increasing steric hindrances opposing core–core coupling. This number of arms is equal to the core functionality of the star and also equal to that of the network, an important structural feature of these polymer systems, not known a priori. It is noteworthy that the MMA homopolymer end-linked network with the shortest chains, E0.5-grad-M10-I-M10-grad-E0.5, did not dissolve upon its reaction with the HCl solution, manifesting exactly the presence of extensive inter-core connections (arising from the very weak steric hindrance), holding the network together even after the cleavage of the hemi(acetal ester) linkages.57 Some of these core-core interconnections were also present in the MMA homopolymer end-linked network with polymer chains of intermediate length, E0.5-grad-M25-I-M25-grad-E0.5, as evidenced by a high molecular weight shoulder/tail in the SEC trace of its hydrolysis (star polymer) product; no such shoulder was observed in the SEC trace of the hydrolysis product of the MMA homopolymer end-linked network with the longest chains, E0.5-grad-M37.5-I-M37.5-grad-E0.5, as stronger steric hindrances in this case averted core–core coupling.| Network structurea | Linear precursor | Network hydrolysis product | ||||
|---|---|---|---|---|---|---|
| SEC-RI | SEC-RI | SEC-SLS | No. of arms | |||
| M n/g mol−1 | M w/Mn | M n/g mol−1 | M w/Mn | M w/g mol−1 | ||
| a M: methyl methacrylate; E: ethylene glycol dimethacrylate. | ||||||
| E0.5-grad-M50-grad-E0.5 | 6760 | 1.43 | 139000 |
2.16 | 274000 |
106 |
| 7290 | 1.22 | |||||
| E0.5-grad-M75-grad-E0.5 | 10020 |
1.41 | 69044 |
1.34 | 165000 |
33 |
| 7791 | 1.15 | |||||
As already mentioned, the SEC characterization in THF, DMF or acidic water of the hydrolysis products of the DMAEMA-containing APCNs was not possible due to sample insolubility or sample retention on the SEC columns. Thus, these star copolymers were characterized in terms of their molecular weights using SANS in d6-DMSO. The absolute Mw values of the copolymers were calculated from the (extrapolated) values of the scattered intensity to zero angle, as initial efforts to fit the whole SANSspectrum to the Guinier model were not successful. These values of zero-angle scattered intensity were subsequently used for the calculation of the absolute Mws from eqn (1) (ref. 91) given in the Experimental section. The Mws determined for the star copolymers obtained from this equation are listed in Table 5. Table 5 also presents the numbers of arms of the star copolymers calculated as the ratio of the Mw of the star copolymers as determined from SANS divided by one-half the Mn of the linear precursors to the conetworks measured using SEC-RI. No results are presented for the two star copolymers obtained from the hydrolysis of conetworks E0.5-grad-M20-grad-D5-I-D5-grad-M20-grad-E0.5 and E0.5-grad-D12.5-grad-M12.5-I-M12.5-grad-D12.5-grad-E0.5, as the d6-DMSO solutions of these samples did not sufficiently scatter.
| Star copolymer structurea | SEC-RI results | SANS results | Number of arms |
|---|---|---|---|
| M n, linear precursors/g mol−1 | M w, star polymer /g mol−1 | ||
| a M: methyl methacrylate; D: 2-(dimethylamino)ethyl methacrylate; E: ethylene glycol dimethacrylate. | |||
| D20-grad-M5-grad-E0.5 | 9250 | 156000 |
34 |
| (D12.5-co-M12.5)-grad-E0.5 | 10400 |
166000 |
32 |
| D12.5-grad-M12.5-grad-E0.5 | 9600 | 132000 |
28 |
The number of arms of the star copolymers resulting from the hydrolysis of the three end-linked conetworks listed in Table 5 was approximately 30. This value agrees well with the number of arms determined by SEC-SLS for the MMA homopolymer network with linear precursor chain DP equal to 75 and linear precursor chain molecular weight similar to those of the three APCNs. As already mentioned, these numbers of arms are equal to the core functionality of the conetworks. The core functionality of the conetworks synthesized in the present study was higher than the core functionality (∼20) of the conetworks synthesized in our previous study viaGTP.57 This can be attributed to the synthetic procedure in the present study in which the cross-linker was added in the presence of unreacted MMA monomer, leading to the formation of larger cores composed of both cross-linker and monomer, and, therefore, capable of incorporating more arms.
Characterization using 1H NMR spectroscopy in D2O of the star copolymers obtained from the hydrolysis of the conetworks allowed the determination of their compositions which are listed in Table 6. The results show that the calculated compositions were sufficiently close to those expected theoretically based on the comonomer feed ratios and monomer conversions.
| No. | Star copolymer structurea | M (mol%) | |
|---|---|---|---|
| 1H-NMR | Theory | ||
| a M: methyl methacrylate; D: 2-(dimethylamino)ethyl methacrylate; E: ethylene glycol dimethacrylate. | |||
| 1 | D20-grad-M5-grad-E0.5 | 18 | 20 |
| 2 | D12.5-grad-M12.5-grad-E0.5 | 39 | 50 |
| 3 | (D12.5-co-M12.5)-grad-E0.5 | 48 | 50 |
Conclusions
The first ATRP synthesis of degradable, amphiphilic end-linked copolymer conetworks of various compositions and architectures was presented. A newly designed bifunctional ATRPinitiator, bearing two labile hemiacetal ester groups, was synthesized and used in the polymerizations to ensure conetwork cleavage at precise locations within the conetwork chains under acidic conditions. Conetwork composition greatly influenced the hydrolysis rates. In particular, the more hydrophobic APCNs degraded more slowly or they even did not dissolve at all. Conetwork architecture also affected the hydrolysis rates. For example, when the initiator residue was surrounded by large hydrophilic blocks, hydrolysis of the conetworks occurred faster than in the cases where the surrounding blocks also contained some hydrophobic monomers and cross-linker units that provided some protection to the initiator fragment from mineral acid attack. Upon acid hydrolysis of the conetworks, star polymers were produced and their characterization allowed the determination of the core functionality of the (co)networks, an originally unknown property of the system.Acknowledgements
We thank the Cyprus Research Promotion Foundation and the EU Structural and Cohesion Funds for Cyprus for supporting this work in the form of a PENEK2008 Doctoral Research Fellowship (project code: ENISX/03/08/045) to M. R.-K. We are also grateful to the A. G. Leventis Foundation for a generous donation that enabled the purchase of the NMR spectrometer of the University of Cyprus. Furthermore, we are indebted to our colleague Dr Paul D. Butler of NIST for his help with the interpretation of the SANS data. Finally, NIST and the U.S. Department of Commerce are thanked for providing the neutron research facilities used in this work.Notes and references
- K. E. Uhrich, S. M. Cannizzaro, R. S. Langer and K. M. Shakesheff, Chem. Rev., 1999, 99, 3181 CrossRef CAS.
- K. Ulbrich, V. Šubr, L. W. Seymour and R. Duncan, J. Controlled Release, 1993, 24, 181 CrossRef CAS.
- M. Vert, J. Mater. Sci.: Mater. Med., 2009, 20, 437 CrossRef CAS.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 43, 3 CrossRef.
- J. Kim, K.-W. Lee, T. E. Hefferan, B. L. Currier, M. J. Yaszemski and L. Lu, Biomacromolecules, 2008, 9, 149 CrossRef CAS.
- P. J. Martens, S. J. Bryant and K. S. Anseth, Biomacromolecules, 2003, 4, 283 CrossRef CAS.
- E. Farng and O. Sherman, J. Arthrosc. Relat. Surg., 2004, 20, 273 CrossRef.
- B. Saad, T. D. Hirt, M. Welti, G. K. Uhlschmid, P. Neuenschwander and U. W. Suter, J. Biomed. Mater. Res., 1997, 36, 65 CrossRef CAS.
- Z. Zhang, R. Kuijer, S. K. Bulstra, D. W. Grijpma and J. Feijen, Biomaterials, 2006, 27, 1741 CrossRef CAS.
- E. Reichmanis and L. F. Thompson, Chem. Rev., 1989, 89, 1273 CrossRef CAS.
- T. Li, M. Mitsuishi and T. Miyashita, Thin Solid Films, 2001, 389, 267 CrossRef CAS.
- M. F. Montague and C. J. Hawker, Chem. Mater., 2007, 19, 526 CrossRef CAS.
- B. K. Long, B. K. Keitz and C. G. Willson, J. Mater. Chem., 2007, 17, 3575 RSC.
- Y. Poirier, C. Nawrath and C. Somerville, Bio/Technology, 1995, 13, 142 CrossRef CAS.
- G. E. Sheldrick and O. Vogl, Polym. Eng. Sci., 1976, 16, 65 CAS.
- Y. Merle, L. Merle-Aubry and J. E. Guillet, Macromolecules, 1984, 17, 288 CrossRef CAS.
- D. Eglin and M. Alini, Eur. Cells Mater., 2008, 16, 80 CAS.
- E. Ruckenstein and H. Zhang, Macromolecules, 1999, 32, 3979 CrossRef CAS.
- S. Kaihara, S. Matsumura and J. P. Fisher, Macromolecules, 2007, 40, 7625 CrossRef CAS.
- D. S. Muggli, A. K. Burkoth, S. A. Keyser, H. R. Lee and K. S. Anseth, Macromolecules, 1998, 31, 4120 CrossRef CAS.
- A. K. Burkoth and K. S. Anseth, Macromolecules, 1999, 32, 1438 CrossRef CAS.
- D. Mawad, L. A. Poole-Warren, P. Martens, L. H. Koole, T. L. B. Slots and C. S. J. van Hooy-Corstjens, Biomacromolecules, 2008, 9, 263 CrossRef CAS.
- W. Yin, E. O. Akala and R. E. Taylor, Int. J. Pharm., 2002, 244, 9 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155 CrossRef CAS.
- H. Gao, N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 5995 CrossRef CAS.
- J. K. Oh, C. Tang, H. Gao, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 5578 CrossRef CAS.
- Y. Li, B. S. Lokitz, S. P. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726 CrossRef CAS.
- B. D. Mather, S. R. Williams and T. E. Long, Macromol. Chem. Phys., 2007, 208, 1949 CrossRef CAS.
- M. J. Bruining, H. G. T. Blaauwgeers, R. Kuijer, E. Pels, R. M. M. A. Nuijts and L. H. Koole, Biomaterials, 2000, 21, 595 CrossRef CAS.
- S. Yang, J. S. Chen, H. Korner, T. Breiner, C. K. Ober and M. D. Poliks, Chem. Mater., 1998, 10, 1475 CrossRef CAS.
- L. Kilian, Z. H. Wang and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3083 CrossRef CAS.
- H. Okamura, T. Terakawa and M. Shirai, Res. Chem. Intermed., 2009, 35, 865 CrossRef CAS.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS.
- W. H. Heath, F. Palmieri, J. R. Adams, B. K. Long, J. Chute, T. W. Holcombe, S. Zieren, M. J. Truitt, J. L. White and C. G. Willson, Macromolecules, 2008, 41, 719 CrossRef CAS.
- J. A. Moss, S. Stokols, M. S. Hixon, F. T. Ashley, J. Y. Chang and K. D. Janda, Biomacromolecules, 2006, 7, 1011 CrossRef CAS.
- S. Kim and K. E. Healy, Biomacromolecules, 2003, 4, 1214 CrossRef CAS.
- M. Kurisawa, Y. Matsuo and N. Yui, Macromol. Chem. Phys., 1998, 199, 705 CrossRef CAS.
- H. Brøndsted and J. Kopeček, Biomaterials, 1991, 12, 584 CrossRef.
- L. Bromberg, M. Temchenko, V. Alakhov and T. A. Hatton, Langmuir, 2005, 21, 1590 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2005, 38, 3087 CrossRef CAS.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009 CrossRef CAS.
- C. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510 CrossRef CAS.
- J. Madsen, S. P. Armes, K. Bertal, H. Lomas, S. MacNeil and A. L. Lewis, Biomacromolecules, 2008, 9, 2265 CrossRef CAS.
- K. Yoshimoto, T. Hirase, J. Madsen, S. P. Armes and Y. Nagasaki, Macromol. Rapid Commun., 2009, 30, 2136 CrossRef CAS.
- T. He, D. J. Adams, M. F. Butler, A. I. Cooper and S. P. Rannard, J. Am. Chem. Soc., 2009, 131, 1495 CrossRef CAS.
- H. Kitano, H. Nakada and K. Mizukami, Colloids Surf., B, 2008, 61, 17 CrossRef CAS.
- J. Liu, H. Liu, Z. Jia, V. Bulmus and T. P. Davis, Chem. Commun., 2008, 6582 RSC.
- E. Setijadi, L. Tao, J. Liu, Z. Jia, C. Boyer and T. P. Davis, Biomacromolecules, 2009, 10, 2699 CrossRef CAS.
- J. Liu, L. Tao, J. Xu, Z. Jia, C. Boyer and T. P. Davis, Polymer, 2009, 50, 4455 CrossRef CAS.
- N. C. Kalarickal, S. Rimmer, P. Sarker and J. C. Leroux, Macromolecules, 2007, 40, 1874 CrossRef CAS.
- X. Hou, Q. Li, L. Jia, Y. Li, Y. Zhu and A. Cao, Macromol. Biosci., 2009, 9, 551 CrossRef CAS.
- N. L. Hill, J. L. Jarvis, F. Pettersson and R. Braslau, React. Funct. Polym., 2008, 68, 361 CrossRef CAS.
- Z. Ge, D. Chen, J. Zhang, J. Rao, J. Yin, D. Wang, X. Wan, W. Shi and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1432 CrossRef CAS.
- Y. Liu and K. A. Cavicchi, Macromol. Chem. Phys., 2009, 210, 1647 CrossRef CAS.
- Y. You, C. Hong, W. Wang, P. Wang, W. Lu and C. Pan, Macromolecules, 2004, 37, 7140 CrossRef CAS.
- M. D. Rikkou and C. S. Patrickios, Macromolecules, 2008, 41, 5957 CrossRef CAS.
- M. D. Rikkou, E. Loizou, L. Porcar, P. Butler and C. S. Patrickios, Macromolecules, 2009, 42, 9412 CrossRef CAS.
- A. Johnson, D. R. Lewis, D. D. Diaz, M. G. Finn, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 6564 CrossRef.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978 RSC.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromolecules, 2007, 40, 3589 CrossRef CAS.
- J. A. Johnson, J. M. Baskin, C. R. Bertozzi, J. T. Koberstein and N. J. Turro, Chem. Commun., 2008, 3064 RSC.
- M. D. Rikkou and C. S. Patrickios, Prog. Polym. Sci., 2011, 36, 1079 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721 CrossRef CAS.
- M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- G. Moad, Aust. J. Chem., 2006, 59, 661 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 6692 Search PubMed.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079 CrossRef CAS.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- S. F. Betz, Protein Sci., 1993, 32, 3178 Search PubMed.
- R. R. Gallucci and R. C. Going, J. Org. Chem., 1982, 47, 3517 CrossRef CAS.
- S. Vanwetswinkel, V. Carlier, J. Marchand-Brynaert and J. Fastrez, Tetrahedron Lett., 1996, 37, 2761 CrossRef CAS.
- C. S. Patrickios, W. R. Hertler, N. L. Abbott and T. A. Hatton, Macromolecules, 1994, 27, 930 CrossRef CAS.
- C. S. Patrickios, A. B. Lowe, S. P. Armes and N. C. Billingham, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 617 CrossRef CAS.
- A. B. Lowe, N. C. Billingham and S. P. Armes, Macromolecules, 1998, 31, 5991 CrossRef CAS.
- E. Demosthenous, S. C. Hadjiyannakou, M. Vamvakaki and C. S. Patrickios, Macromolecules, 2002, 35, 2252 CrossRef CAS.
- S. N. Georgiades, M. Vamvakaki and C. S. Patrickios, Macromolecules, 2002, 35, 4903 CrossRef CAS.
- G. Hadjikallis, S. C. Hadjiyannakou, M. Vamvakaki and C. S. Patrickios, Polymer, 2002, 43, 7269 CrossRef CAS.
- R. Hoogenboom, U. S. Schubert, W. Van Camp and F. E. Du Prez, Macromolecules, 2005, 38, 7653 CrossRef CAS.
- K. V. Bernaerts, N. Willet, W. Van Camp, R. Jérôme and F. E. Du Prez, Macromolecules, 2006, 39, 3760 CrossRef CAS.
- T. K. Georgiou, L. A. Phylactou and C. S. Patrickios, Biomacromolecules, 2006, 7, 3505 CrossRef CAS.
- G. Kali, T. K. Georgiou, B. Iván, C. S. Patrickios, E. Loizou, Y. Thomann and J. C. Tiller, Macromolecules, 2007, 40, 2192 CrossRef CAS.
- G. Kali, T. K. Georgiou, B. Iván, C. S. Patrickios, E. Loizou, Y. Thomann and J. C. Tiller, Langmuir, 2007, 23, 10746 CrossRef CAS.
- T. K. Georgiou and C. S. Patrickios, Biomacromolecules, 2008, 9, 574 CrossRef CAS.
- G. Kali, T. K. Georgiou, B. Iván and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4289 CrossRef CAS.
- K. S. Pafiti, Z. Philippou, E. Loizou, L. Porcar and C. S. Patrickios, Macromolecules, 2011, 44, 5352 CrossRef CAS.
- H. Otsuka and T. Endo, Macromolecules, 1999, 32, 9059 CrossRef CAS.
- J. S. Higgins and H. C. Benoit, Polymers and Neutron Scattering, Clarendon Press, Oxford, 1996, p. 398 Search PubMed.
- L. Mespouille, O. Coulembier, D. Paneva, P. Degée, I. Rashkov and Ph. Dubois, Chem.–Eur. J., 2008, 14, 6369 CrossRef CAS.
- L. Mespouille, O. Coulembier, D. Paneva, P. Degée, I. Rashkov and Ph. Dubois, J. Polym. Sci., Part A: Polym. Chem., 2008, 14, 6369 CAS.
- J. Zednik, R. Riva, P. Lussis, C. Jérôme, R. Jérôme and P. Lecomte, Polymer, 2008, 49, 697 CrossRef CAS.
- M. D. Rikkou, M. Kolokasi, K. Matyjaszewski and C. S. Patrickios, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1878 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |