UVA-induced epigenetic regulation of P16INK4a in human epidermal keratinocytes and skin tumor derived cells†
I-Peng
Chen
a,
Stefan
Henning
a,
Alexandra
Faust
a,
Petra
Boukamp
b,
Beate
Volkmer
a and
Rüdiger
Greinert
*a
aDept. Mol. Cell Biology, Center of Dermatology, Elbekliniken, Stade/Buxtehude, Germany. E-mail: ruediger.greinert@elbekliniken.de
bDivision of Genetics of Skin Carcinogenesis, German Cancer Research Center (DKFZ), Heidelberg, Germany
First published on 10th October 2011
Abstract
UVA-radiation (315–400 nm) has been demonstrated to be capable of inducing DNA damage and is regarded as a carcinogen. While chromosomal aberrations found in UVA-irradiated cells and skin tumors provided evidence of the genetic involvement in UVA-carcinogenesis, its epigenetic participation is still illusive. We thus analysed the epigenetic patterns of 5 specific genes that are involved in stem cell fate (KLF4, NANOG), telomere maintenance (hTERT) and tumor suppression in cell cycle control (P16INK4a, P21WAFI/CIPI) in chronically UVA-irradiated HaCaT human keratinocytes. A striking reduction of the permissive histone mark H3K4me3 has been detected in the promoter of P16INK4a (4-fold and 9-fold reduction for 10 and 15 weeks UVA-irradiated cells, respectively), which has often been found deregulated in skin cancers. This alteration in histone modification together with a severe promoter hypermethylation strongly impaired the transcription of P16INK4a (20-fold and 40-fold for 10 weeks and 15 weeks UVA-irradiation, respectively). Analysis of the skin tumor-derived cells revealed the same severe impairment of the P16INK4a transcription attributed to promoter hypermethylation and enrichment of the heterochromatin histone mark H3K9me3 and the repressive mark H3K27me3. Less pronounced UVA-induced epigenetic alterations were also detected for the other genes, demonstrating for the first time that UVA is able to modify transcription of skin cancer associated genes by means of epigenetic DNA and histone alterations.
Introduction
Solar ultraviolet (UV-) radiation, as well as artificial UV (e.g. from sunbeds), represent the most prominent risk factor for the induction of skin cancer.1–6 Due to the ubiquitous nature of solar UV, recreational and occupational behavior of the public and the increasing use of artificial UV in sunbeds (especially by adolescents), skin cancer incidence is steadily increasing, making skin cancer (squamous cell carcinoma, SCC, basal cell carcinoma, BCC, and malignant melanoma, MM) the most frequent cancer in the white population worldwide.6UV-radiation (UVB = 280–315 nm and UVA = 315–400 nm) induces signature mutations in the genome of human skin cells.7 These C-T transitions and CC-TT tandem mutations are characteristic for a misrepair of UV-induced pre-mutagenic DNA lesions.8–11 Interestingly, the recently completed whole genome sequencing of a human melanoma metastasis reveals that a huge majority of detected mutations are of UV-signature type, clearly proving the role of UV-radiation in melanomagenesis.12
The main pre-mutagenic DNA-lesion leading to UV-signature mutations is the cyclobutane–pyrimidine dimer (CPD), which has been shown to be the most prominent DNA-lesion produced by UVB-radiation in human skin cells.10 However, recent results show that UVA is also able to induce CPDs. Although the photochemical mechanisms have still to be elucidated, it turned out that UVA-induced CPDs comprise the main pre-mutagenic lesion in human skin cells.13,14 These results clearly underline an important role of UVA (which comprises 95% of solar UV) in basic mechanisms involved in skin carcinogenesis. The question, however, of which part of the UV-spectrum (UVB, UVA), and which molecular events, pathways and specific genes are involved in photocarcinogenesis has further to be studied in more detail.
It has already been shown that the tumor-suppressor P16INK4a (also known as P16CDKN2a) plays a prominent role in photocarcinogenesis.15 The INK4a/ARF/INK4b locus (chromosomal band 9p21) encodes 3 tumor suppressor genes, P16INK4a, P14ARF and P15INK4b, involved in cell cycle control (proliferation). P16INK4a and P14ARF share common exons 2 and 3, but have alternatively first spliced exons.16P16INK4a functions as a repressor of CDK4 and CDK6 activity and as a positive regulator of RB tumor suppressor activity.17INK4a/ARF/INK4b is regarded as the major gene locus involved in MM pathogenesis and predisposition,18 and has been found to be inactivated in the majority of sporadic MM and represents the most frequently mutated gene locus in familial MM.19
It has been reported that deletions of the INK4a/ARF/INK4b locus are prevalent in familial melanoma.20 Interestingly, later it was found that P16INK4a can also be repressed in non-familial melanoma via epigenetic silencing of its promoter.21P16INK4a levels are low or even undetectable in proliferating cells, but are increased dramatically in senescent cells.22,23 Different expression profiles have been reported for P16INK4a, P14ARF (and also for P15INK4b and P53) in BCCs.24 Several studies have shown that human SCCs harbor unique mutations in the P53 gene and/or inactivated forms of the P16INK4a gene. While mutations in the P53 gene are induced by UV-radiation (UV-signature mutations) and represent tumor initiating events, the majority of the alterations detected in the P16INK4a gene does not appear to be UV-dependent,25 although older work seems to suggest that UV-signature mutations in the P16INK4a gene influence the etiology of aggressive forms of SCC.26 Interestingly, other modifications, including LOH and epigenetic alterations, of the INK4a/ARF/INK4b locus have been demonstrated in non-melanocytic skin cancer (NMSC)15,27 and other types of cancer.28
Recent reports have demonstrated that aberrant CpG island promoter hypermethylation at the INK4a/ARF/INK4b locus independently affects P16INK4a and P14ARF, which are methylated in 27% and 57% of metastatic MM samples, respectively.29 Using genome-scale methylation analysis, Grönniger et al.30 have been able to show that aging and chronic sun exposure cause distinct epigenetic changes in human skin with a significant trend to hypomethylation. Sun exposure related methylation of specific genes could be detected in BCCs and SCCs using methylation specific PCR. It could be shown that methylation commences in UV-exposed human skin at relatively early age and occurs in skin prior to the onset of recognizable preneoplastic changes. Significant differences in methylation were shown especially for the RASSF1A, CDH1 and CAD genes.31 All together, these findings point in a direction of a still underestimated, important role of epigenetic regulation of skin cancer induction and tumor development.
Epigenetic changes are commonly attributed to changes in gene expression without altering the base sequence. These modifications include DNA-methylation, histone modifications, chromatin remodeling32,33 and miRNA-induced regulation of gene expression.34 Epigenetic regulation is mediated e.g. by CpG-island promoter hypermethylation, global DNA-hypomethylation,32 as well as by specific acetylation- and methylation-patterns of certain amino acids of histone proteins.35–38 The pattern of epigenetic DNA and histone modifications dictates an “epigenetic code”, which is “read” by the gene regulatory machinery and allows cells to facilitate or inhibit transcription of target genes.39,40 Stochastic and environment-induced epigenetic modifications are known to be important steps during embryogenesis, the control of stem cell fate, disease development, cancer and aging.35 Promoter CpG island hypermethylation in combination with global DNA hypomethylation has already been termed “hallmark of cancer”.41 Whether epigenetic changes might be induced by UV-radiation and whether these changes might influence photocarcinogenesis has still to be investigated in detail.
However, it is already known that DNA methylation might enhance the mutagenic efficiency of UV-radiation. It has been shown, that DNA methylation actually promotes radiation-induced DNA-damage. The presence of methyl groups in CpG dinucleotides increases the rate at which mutations are induced by UV-radiation, shifting the absorption band of methylated cytosines more to the UVB (280–315 nm) range.42 It has been demonstrated that methylation of CpG sequences creates preferential targets for pre-mutatgenic DNA-lesions (CPDs), leading to UV-specific signature mutations e.g. in the P53 gene during development of skin cancer.43–46
Very recently, Nadakumar et al. clearly demonstrated that UVB-induced aberrant DNA hypermethylation leads to transcriptional silencing of tumor suppressor genes in UVB-exposed mouse skin and UVB-induced skin tumors of mice.47 Whether UVA (315–400 nm) is also able to induce epigenetic changes directly has not been clarified until now.
We have recently reported48 that defined treatment regimes (1 × 20 or 4 × 5 J cm−2 = 1 × 200 or 4 × 50 kJ m−2 per week) of environmentally relevant UVA doses cause tumorigenic conversions of HaCaT skin keratinocytes, allowing them to form well differentiated SCCs in nude mice. When cells of these tumors were re-cultivated, their tumorigenicity was characterized by distinct chromosomal aberrations most probably introduced by UVA-induced DNA single strand and double strand breaks, which have been detected by γ-H2Ax formation, micronuclei induction and alkaline comet assay in UVA-irradiated HaCaT cells and primary human epidermal keratinocytes.48
To further clarify whether UVA-induced epigenetic alterations might contribute to tumorigenicity we investigated UVA-induced DNA-methylation and histone alterations in certain specific genes of UVA-irradiated HaCaT skin keratinocytes and cells re-cultivated from skin tumors, which have been induced by transplantation of UVA-irradiated HaCaT cells into mice.48 Using methylation specific PCR and ChIP-technologies, we were able to show that UVA-irradiation induces a number of epigenetic changes on the histone level (H3K4me3, H3K9me3 and H3K27me3) as well as CpG-island hypermethylation in the promoter region of selected genes in human HaCaT cells. Epigenetic changes were also detected in skin tumor (SCC) derived cells when tumors had been developed in mouse skin after transplantation of UVA-irradiated human keratinocytes. In particular, expression of the tumor suppressor P16INK4a is repressed in UVA-irradiated HaCaT cells, as well as in tumor derived cells.
This investigation focuses on UVA-induced epigenetic modifications of P16INK4a in human keratinocytes and skin cancer derived cells. Other genes, which have also been investigated in this study, only for comparison, have been chosen out of a pool of several genes which our group is studying in the context of UV-induced epigenetic changes in epidermal stem cell fate and their role in photocarcinogenesis. A detailed analysis of these changes, which determine expression and interactions of P21WAF1/CIP1 [known tumor suppressor involved e.g. in cell cycle control49,50], KLF4 and NANOG [regulating stem cell fate51,52], LIN28 [RNA binding protein, association with pluripotency “core” factors, e.g. SOX253,54], DNMT1 [maintenance methyltransferase 155,56], HDAC1 [histone deacetylase 1, crosstalk with epigenetic methylation57,58], HIC 1 [hypermethylated in cancer59,60] and hTERT [human telomerase reverse transcriptase61,62] will be given elsewhere (R. Greinert, in preparation).
Materials and methods
UVA-irradiated HaCaT skin keratinocytes
Cells of sub-confluent state (70–90%) were irradiated with UVA in ice-cold phosphate-buffered saline (PBS) without a lid at a distance of 14 cm with 200 kJ m−2 per week (with 6 days for the recovery) for 10 or 15 weeks. A Phillips HB404 tanning lamp (irradiance 182 W m−2) equipped with an infrared filter (Hoenle) and a UVB-blocking filter UV34 (Hoya) served as the UVA source (for spectral details see Fig. 1). Spectral resolved dosimetry was performed with a Bentham double monochromator (DM 150). In a linear dose effect response 200 kJ m−2 UVA-irradiation resulted in CPD lesions (corresponding to the same amount of lesions caused by ∼100 J m−2 UVB, analyzed by immunofluorescence microscopy), which were repaired within 6 days (data not shown). UVA-irradiation did not significantly impair cell survival. Cell passage was performed weekly. | ||
| Fig. 1 Spectrum of UVA . Infrared- and UVB-free UVA with an irradiance of 182 W m−2 was used as an irradiation source. | ||
Ten and fifteen weeks UVA-irradiated (200 kJ m−2 week−1) HaCaT human skin keratinocytes48 were cultivated in DMEM medium to confluent state for ChIP-qPCR, DNA-methylation and transcriptional analysis. Cells derived from 4 tumors of different mice injected with 15 weeks UVA-irradiated HaCaT cells (200 kJ m−2 week−148) were also grown in DMEM medium to confluent state.
Cell cycle analysis
Control HaCaT and chronically UVA-irradiated cells in a sub-confluent state (70–90%) were subjected to a single UVA-irradiation of 400 kJ m−2. Twenty two hours after irradiation, cells were trypsinized and fixed in 70% EtOH. For flow cytometry analysis (Coulter, EPICS XL) cells were treated with RNase and afterwards DNA was stained with propidium iodide. Additionally, the Sub-G1 fraction of the cell cycle distribution, indicative of apoptotic cells, was determined. The software package MCyclePlus was used to access the fractions of the cell cycle.ChIP-qPCR
ChIP-qPCR (Chromatin ImmunoPrecipitation-quantitative PCR) was applied to access the changes of histone modification gene-specifically upon UVA-irradiation. The levels of H3K4me3 (a permissive histone mark), H3K9me3 (a heterochromatin mark) and H3K27me3 (a repressive mark)37,38 were determined by ChIP-qPCR using the antibodies: Histone H3 trimethyl Lys4 Rabbit pAb (Cat No: 39159, Active Motif), Histone H3 tri methyl K9 Rabbit pAb (Cat No: ab8898, Abcam), Anti-trimethyl-Histone H3 Lys27 (Cat No: 07-449), respectively. ChIP-qPCR using Dynabeads (Invitrogen) was performed according to the protocol by Dahl and Collas.63 Briefly, first DNA was crosslinked with histone by 1% formaldehyde and then the chromatin was fragmented (ranging from 200 bp to 1000 bp) by sonication. Antibody specific chromatin fragments were obtained after incubation of chromatin mixture with Dynabeads protein A–antibody complex on ice and DNA fragments bound to the chromatin were subsequently eluted from Dynabeads protein A–antibody complex. The DNA fragments were purified with peqGold cycle-pure kit (peqLab) and quantified by qPCR Realplex-Mastercycler (Eppendorf). For PCR reactions a SYBR Green containing Mix (SensiMix, Bioline) was applied. Primers used for ChIP-qPCR are given in Table 1.| a ref. 85 b ref. 86 c ref. 87 | |
|---|---|
| ChIP-qPCR primers | |
| KLF4-UP-FOR | G C G C C G A G T T T G T T G A T T T A G |
| KLF4-UP-REV | C T T C C T T C G C T A C A G C C T T T T |
| P14-UP-FOR | G A T G A C C T C G C T T T C C T T T C T |
| P14-UP-REV | C C T A G C T A C A T C C G T C A C C T G |
| P16-UP-FOR | C T C C T G A A A A T C A A G G G T T G A G |
| P16-UP-REV | A C C T T C C T A A C T G C C A A A T T G A |
| P21-CIP1-UP-FOR | C C T C T T C T C T G G G G T C T C A C T |
| P21-CIP1-UP-REV | A A G T G C T G G G A A C A A T G T C A C |
| TERT-FOR | T C A G C G T G C T C A A C T A C G A G |
| TERT-REV | C C A C C T T G A C A A A G T A C A G C T C |
| qMSP primers | |
| MSP Preamplification | |
| P16_F | T T T A G A G G A T T T G A G G G A T A G G |
| P16_R | C T T C T A A A A A C T C C C C A A A A A A |
| MSP qPCR | |
| P16_MSP_M_F a | T T A T T A G A G G G T G G G G C G G A T C G C |
| P16_MSP_M_R a | G A C C C C G A A C C G C G A C C G T A A |
| P16_MSP_U_F a | T T A T T A G A G G G T G G G G T G G A T T G T |
| P16_MSP_U_R a | C A A C C C C A A A C C A C A A C C A T A A |
| Transcriptional analysis primers | |
| DNMT1-FOR | C T T C T T C A G C A C A A C C G T C A |
| DNMT1-REV | G A A G A G C C G G T A G G T G T C A G |
| HDAC1-FOR | T A A A T T C T T G C G C T C C A T C C G |
| HDAC1-REV | A A C A G G C C A T C G A A T A C T G G A |
| HIC1-FOR b | C G A C G A C T A C A A G A G C A G C A |
| HIC1-REV b | T G C A C A C G T A C A G G T T G T C A |
| KLF4-FOR c | A C C A G G C A C T A C C G T A A A C A C A |
| KLF4-REV c | G G T C C G A C C T G G A A A A T G C T |
| LIN28-FOR | C A C T C C A G C C T G G T T A C A G A G |
| LIN28-REV | C C T C C T G A C C C C A C T T T C T A C |
| P14-3519-FOR | C C C T C G T G C T G A T G C T A C T G |
| P14-3519-REV | C A T C A T G A C C T G G T C T T C T A G G A A |
| P16-FOR | G G A G C A G C A T G G A G C C T T C |
| P16-REV | C A T C A T C A T G A C C T G G A T C G |
| P21-CIP1-FOR | T G T G G A C C T G T C A C T G T C T T G |
| P21-CIP1-REV | A A A T C T G T C A T G C T G G T C T G C |
Quantitative methylation specific PCR (qMSP)
Following purification of DNA using the Promega Wizard SV kit 1.5 μg of DNA was treated with bisulfite using the EZ DNA Methylation-Gold™ Kit (Zymo Rearearch). In a nested PCR approach the promoter region of interest was preamplified in the first PCR round (18 cycles) using primer pairs not discriminating between methylated and unmethylated DNA. The first round reaction products were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 and then used as template for the second round of PCR using sequence specific primers for unmethylated (U) and methylated (M) DNA. CT-values of M- and U-primers were used to measure promoter methylation [%] of the analyzed promoter regions. For both PCR reactions a SYBR Green containing Mix (SensiMix, Bioline) was applied. PCR primers used are given in Table 1.
:10 and then used as template for the second round of PCR using sequence specific primers for unmethylated (U) and methylated (M) DNA. CT-values of M- and U-primers were used to measure promoter methylation [%] of the analyzed promoter regions. For both PCR reactions a SYBR Green containing Mix (SensiMix, Bioline) was applied. PCR primers used are given in Table 1.
Transcriptional analysis
Isolation of total RNA (using the “Total RNA Kit” from peqLab) and cDNA synthesis (using the “Enhanced Avian First Strand Synthesis Kit” from Sigma-Aldrich) were performed according to the protocols of the kits. cDNA was quantified with qPCR using Realplex-Mastercycler (Eppendorf). For PCR reactions a SYBR Green containing Mix (SensiMix, Bioline) was applied. HPRT1 (hypoxanthine phosphoribosyltransferase 1) and TBP (TATA box binding protein) were used as house keeping genes for normalisation.64 Fold-change of the transcription upon UVA was obtained by setting the control as one-fold. Two-fold threshold was applied as criterion of altered transcriptional response. Primers used are given in Table 1.Statistical analysis
ChIP-data were analysed with Student's t-test and a two-sided p-value of <0.05 is considered as significant.Results
Sustained epigenetic changes at both the histone and DNA levels in P16INK4a promoter in chronically UVA-irradiated HaCaT skin keratinocytes
Previously, we have shown that chronic UVA-irradiation caused genetic defects and tumorigenic transformation in HaCaT skin keratinocytes.48 Using ChIP-qPCR with the antibodies against H3K4me3 (as a permissive mark), H3K9me3 (as a heterochromatin mark) and H3K27me3 (as a repressive mark), epigenetic alterations in chronically UVA-irradiated HaCaT cells (200 kJ m−2 week−1, 10 and 15 weeks) have now been determined at least over 4 passages. We first analysed the abundance of the three histone marks at promoter region of 5 specific genes that are involved in epidermal stem cell fate (KFL4, NANOG), telomere maintenance (hTERT) or which function as tumour suppressors in cell cycle control (P16INK4a, P21WAF1/CIP1). Our special focus was on P16INK4a, which has been shown to be involved in skin cancer development. ChIP experiments for the P16INK4a promoter revealed no significant deviation for the histone marks H3K9me3 and H3K27me3 between irradiated and un-irradiated cells, whereas a pronounced decline of the permissive mark H3K4me3 at P16INK4a was detected in chronically UVA-treated HaCaT cells (3.8% in control cellsversus 0.7% in 10 weeks and 0.4% in 15 weeks UVA-treated cells, corresponding to a reduction of 80% (10 weeks) and even 90% (15 weeks) in UVA-treated cells) (Fig. 2). | ||
| Fig. 2 Sustained changes in the histone methylation patterns at the KLF4 , P16 INK4a , P21WAF1/CIP1and hTERT promoter regions after chronic UVA-irradiation (200 kJ m−2week−1) of HaCaT human skin keratinocytes. (a) Ten and fifteen weeks chronic UVA-irradiation caused pronounced reduction (decreased ∼80% (10 weeks UVA) and ∼90% (15 weeks UVA)) in the occupancy of the H3K4me3 mark at the promoter region of P16INK4a, as determined by ChIP-qPCR. An increase of H3K4me3 (∼100%) was also obtained for KLF4 with 10 weeks and 15 weeks UVA-treatment. (b) Fifteen weeks UVA-treatment caused reduction (∼60%–∼70%) of the H3K9me3 mark at the KLF4 and P21WAF1/CIP1 promoter regions. (c) A slight reduction (∼40%) of the H3K27me3 mark for KLF4 and hTERT was seen for the HaCaT cells 15 weeks treated with UVA. Error bars indicate standard deviations. N ≥ 3. *: p < 0.05; **: p < 0.01. | ||
Slight changes on the level of the histone marks were obtained for P21WAF1/CIP1, KLF4, and hTERT. Enrichment of the H3K4me3 mark was found at the promoter of KLF4 for the UVA-irradiated cells (∼2% in the control and ∼4% both in the 10 weeks UVA and 15 weeks UVA cells). In addition, reduction of H3K9me3 and H3K27me3 was detected for the 15 weeks UVA cells at KLF4 promoter. However, it is worth mentioning that the occupancies of both marks at this promoter were already rather low in the control (∼0.5%; Fig. 2). A decreased H3K9me3 mark (70% reduction) was observed for P21WAF1/CIP1 after 15 weeks UVA-treatment. Cells treated for 15 weeks with UVA also revealed 40% reduction of the H3K27me3 mark at the hTERT promoter. Levels of the histone marks at the promoter of NANOG (transcription factor involved in stemness) were not altered significantly after UVA-irradiation.
Because epigenetic modifications at the histone level often cooperate with promoter methylation at the DNA level, the promoter methylation at P16INK4a was determined. In accordance with the reduced H3K4me3 occupancy (reduction in open, transcriptional active chromatin), a substantial increase of the methylation of ∼50% (10 weeks) and ∼70% (15 weeks) was also detected at the promoter of P16INK4a for chronically UVA-irradiated cells (Fig. 3). The P16INK4a promoter of the unirradiated HaCaT control cells remained virtually unmethylated (Fig. 3).
 | ||
| Fig. 3 Sustained promoter hypermethylation of P16 INK4a after chronic UVA-irradiation (200 kJ m −2 week−1) of HaCaT human skin keratinocytes. Quantitative methylation specific PCR (qMSP) revealed a strong increase of P16INK4a promoter methylation from ∼0% to ∼50% (10 weeks UVA) and to ∼70% (15 weeks UVA). Error bars indicate standard deviations. N ≥ 3. **: p < 0.01. | ||
In order to investigate whether these epigenetic changes would have any impact on the gene, we further analyzed the expression patterns of P16INK4a. Indeed, a drastic decrease of the mRNA expression with 20-fold (for 10 weeks) and 40-fold (for 15 weeks) reduction was detected for P16INK4a. A slight decrease with ∼3-fold reduction of the P21WAF1/CIP1 transcription was seen for both 10 weeks and 15 weeks UVA-treated cells. No distinct deviation for KLF4 was detected (Fig. 4). Transcription of epigenetic factors DNMT1 (DNA methylase) and HDAC1 (histone deacetylase) was not altered in chronically UVA-treated cells.
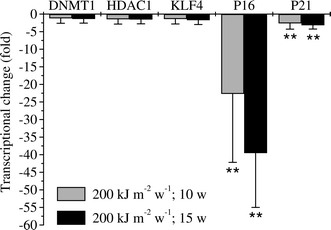 | ||
| Fig. 4 Sustained transcriptional changes of P16 INK4a and P21 WAF1 / CIP1 after chronic UVA-irradiation (200 kJ m −2 week −1 ) of HaCaT human skin keratinocytes . P16INK4a transcription was persistently decreased (∼20-fold after 10 weeks UVA and ∼40-fold after 15 weeks UVA) upon chronic UVA-irradiation. P21WAF1/CIP1 showed a decrease of transcription of about 3-fold after 10 weeks and 15 weeks chronic UVA-irradiation. Transcription for KLF4, DNMT1 and hTERT was not altered. HPRT1 (hypoxanthine phosphoribosyltransferase 1) and TBP (TATA box binding protein) were used as house keeping genes for normalisation. Error bars indicate standard deviations. N ≥ 3. Fold-change of the transcription upon UVA was obtained by setting the control as one-fold. Two-fold threshold was applied as criterion of altered transcriptional response. **: p < 0.01. | ||
Epigenetic alterations at P16INK4a in tumor-derived cells
As UVA-radiation is able to induce epigenetic changes in human skin cells (see above) it is interesting to analyse if the same or different epigenetic modification can be found in skin tumors. We, therefore, examined the histone patterns and the mRNA expression of the same panel of genes (see above) in re-cultivated tumor cells (from 4 tumors of 4 mice) originating from chronically UVA-irradiated HaCaT skin keratinocytes (200 kJ m−2 week−1, 15 weeks;48). All 4 cell samples exhibited consistent epigenetic and transcriptional changes at least over 3 passages. A striking increase of the heterochromatin mark H3K9me3 (∼200%) and the repressive mark H3K27me3 (∼100%) could be measured (Fig. 5). | ||
| Fig. 5 Changes in the histone methylation patterns at the KLF4 , NANOG , P16 INK4a , P21WAF1/CIP1and hTERT promoter regions in tumor-derived cells (from 4 tumors of 4 mice) originating from chronically UVA-irradiated HaCaT skin keratinocytes (200 kJ m−2week−1, 15 weeks). (a) An increase of H3K4me3 was obtained for NANOG (100%) and hTERT (200%) in the tumor-derived cells. (b) A striking increase (∼200%) of the H3K9me3 mark at the P16INK4a promoter region was found in the tumor-derived cells. The occupancy of H3K9me3 at the KLF4 and P21WIF1/CIP1 promoters was slightly reduced (∼50%). (c) An increase of the H3K27me3 mark (100%) for P16INK4a was also obvious in the tumor-derived cells. Error bars indicate standard deviations. N ≥ 3. *: p < 0.05; **: p < 0.01. | ||
The permissive mark H3K4me3 at P16INK4a was slightly altered (∼40% reduced in tumor-derived cells, Fig. 5), however not significant (p > 0.05). An increase of the H3K4me3 mark for hTERT (∼200%) and for NANOG (∼100%) was also detected in the tumor-derived cells (Fig. 5). Reduction of the H3K9me3 mark (∼50%) on the promoter of both KLF4 and P21WAF1/CIP1 was seen. In addition, an increase of the H3K4me3 mark (200%) for KLF4 and a reduction of H3K9me3 mark for both NANOG (∼60%) and hTERT (∼50%) were also detected in the tumor-derived cells; however, significance did not reach the p < 0.05 level in these cases (Fig. 5). In accordance with the increased H3K9me3 and H3K27me3 occupancy (and a slight reduction of H3K4me3), a hypermethylation was also detected at the promoter of P16INK4a (∼0% in the control cellsversus 85% in the tumor-derived cells, Fig. 6).
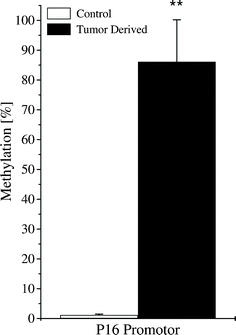 | ||
| Fig. 6 Sustained promoter hypermethylation of P16 INK4a in tumor-derived cells . Quantitative methylation specific PCR (qMSP) revealed a strong increase of P16INK4a promoter methylation from ∼0% in the control cells to ∼85% in the tumor-derived cells. Error bars indicate standard deviations. N ≥ 3. **: p < 0.01. | ||
Pronounced transcriptional repression of P16INK4a (∼35-fold) could be obtained (Fig. 7). A slight increase of the mRNA expression (2-fold) has been detected for KLF4 and P14ARF, though with p > 0.05 (Fig. 7). Transcription level of HIC1, LIN28 remained unchanged.
 | ||
| Fig. 7 Sustained transcriptional repression of P16 INK4a in the tumor-derived cells . P16INK4a transcription was persistently decreased (∼35-fold) in the tumor-derived cells. Transcription of HIC1, KLF4, LIN28 and P14ARF was not significantly altered. HPRT1 (hypoxanthine phosphoribosyltransferase 1) and TBP (TATA box binding protein) were used as house keeping genes for normalisation. Fold-change of the transcription upon UVA was obtained by setting the control as one-fold. Error bars indicate standard deviations. N ≥ 3. Two-fold threshold was applied as criterion of altered transcriptional response. **: p < 0.01. | ||
Cell cycle distribution after UVA-irradiation
To access a possible change in cell cycle regulation upon DNA damage by UVA in chronically UVA-irradiated HaCaT cells a single UVA-irradiation (400 kJ m−2) was applied to the cells. Twenty two hours after irradiation, cell cycle distribution was analysed by flow cytometry. In HaCaT control cells G1-fraction increased as a result of UVA-irradiation from 46% to 62%. A similar UVA-induced increase of the G1-fraction (from 40% to 60%) was obtained in the chronically UVA-treated HaCaT cells (data not shown). A concomitant decrease (∼15%) of the S-phase fraction was detected for both control and the chronically UVA-irradiated HaCaT cells (data not shown). Thus, despite of a down regulated P16INK4a transcription, a UVA-induced G1-arrest seems to be retained in chronically UVA-irradiated HaCaT cells. The sub-G1 phase indicative of apoptosis has been measured viaflow cytometry, too. In the control cells only a small fraction (2%) of the sub-G1 phase was identified as apoptotic 22 h post UVA-irradiation (400 kJ m−2). The same small amount (2%) of UVA-induced apoptotic cells was found for the chronically UVA-irradiated HaCaT cells (data not shown).Discussion
UV-radiation is meanwhile accepted to be the main environmental (and artificial) risk factor for development of non-melanocytic skin cancer (basal cell carcinoma, BCC; and squamous cell carcinoma, SCC) as well as malignant melanoma (MM).1–6 The International Agency for the Research on Cancer (IARC) has therefore grouped solar UV (UVB = 280–315 nm and UVA = 315–400 nm) as well as UV-radiation used in sunbeds in category 1a (“carcinogenic to humans”).65 This has been reasoned by the overwhelming evidence coming from epidemiological data and in vitro and in vivo experiments that prove a causal connection between UV and skin cancer. In particular, the UV-dependent induction of specific signature mutations in the genome of UV-irradiated human skin and human skin cell lines has strongly contributed to this scientifically accepted view. However, the question of which cellular pathways are involved in skin cancer development and progression and how UV-radiation (UVB, UVA or UVA + UVB) influences certain pathways is still a matter of ongoing research. Particularly, the effects of UV-radiation on epigenetic regulatory networks, which control gene expression and epidermal (stem) cell fate has not been studied in detail, although it's known that epigenetic mechanisms seem to play an important role in processes leading to and maintaining skin tumors.We have recently reported48 that defined treatment regimes (1 × 20 or 4 × 5 J cm−2 per week) of environmental highly relevant UVA doses cause tumorigenic conversions of HaCaT skin keratinocytes, allowing them to form well-differentiated SCCs in nude mice. This has been considered to be an interesting finding because it clearly shows that reasonable doses of UVA (which comprises 95% of solar UV-radiation and is the main spectral UV-component in sunbeds) possess tumorigenic potential (a dose of 20 J cm−2 = 200 kJ m−2 of UVA can be accumulated by human skin on a sunny day in 1 h at noon in central Europe48). When tumor cells derived from mice were re-cultivated, tumorigenicity was characterized by distinct chromosomal aberrations most probably introduced by UVA-induced DNA single strand and double strand breaks, which have been detected in UVA-irradiated HaCaT cells and primary human epidermal keratinocytes.48
To further elucidate the mechanisms of UV-induced photocarcinogenesis, we investigated in our present work whether UVA-radiation is able to induce epigenetic changes in UVA-exposed human keratinocytes and whether epigenetic changes can be found in cells re-cultivated from SCCs that have been induced in mouse skin by transplanted human UVA-irradiated skin keratinocytes.
UVA-induced epigenetic changes in the P16INK4a gene
Our results show that UVA-radiation is able to alter epigenetic marks in a number of selected genes that are involved in regulation of (epidermal) stem cell fate (NANOG, KLF4), tumor suppression (P16INK4a, P21WAF1/CIP1) and telomere maintenance (hTERT). The most significant changes have been found for P16INK4a after UVA-irradiation. Tri-methylation of histone H3 at Lsy4 (H3K4me3) a permissive mark for open, transcriptionally active chromatin was reduced by a factor of 4 after 10 weeks and by a factor of 9 after 15 weeks chronic irradiation (200 kJ m−2 per week) of HaCaT cells with UVA (Fig. 2). This goes along with a drastic increase in methylation of the P16INK4a promoter region (Fig. 3). Together, both UVA-induced alterations of the methylation patterns result in a pronounced repression of P16INK4a (Fig. 4). These findings are in agreement with very recent results of chronically UVB-irradiated mice,47 showing promoter hypermethylation of P16INK4a and reduced expression of P16INK4a in irradiated mouse skin as well as in SCCs, which developed in mouse skin. Our results show that these effects can also be found in human skin keratinocytes after chronic UVA-irradiation and that, additionally, epigenetic histone modifications are involved in the repression of P16INK4a. Furthermore, our results demonstrate that UVA-radiation does not induce a single, common epigenetic change throughout the genome (or at least in the genes selected for our investigation). Rather, UVA-induced histone modifications seem to be promoter-/gene-specific. Whereas 10 weeks or 15 weeks UVA-irradiation increases e.g.H3K4me3 in the KLF4 promoter (see Fig. 2), the same irradiation protocol leads to a drastic decrease in H3K4me3 in the P16INK4a promoter. This indicates that UV-induced epigenetic modifications render the expression profile gene-specifically (probably depending on the gene's function) in order to create a coordinated cellular “UV-response”, which determines future cell fate after UV-irradiation.Epigenetic alterations at P16INK4a in tumor-derived cells
Because UV-radiation is a known risk factor for skin cancer induction, UVA seems to alter epigenetic patterns (see above) and epigenetic modifications are involved in skin carcinogenesis, we were interested to see whether epigenetic changes comparable with those found in UVA-irradiated HaCaT cells could be detected in tumor-derived cells.Our results show that cells re-cultivated from squamous cell carcinoma which developed in mouse skin after transplantation of chronically UVA-irradiated HaCaT cells48 were characterized by a drastic repression of P16INK4a (see Fig. 7). This repression was introduced again by DNA hypermethylation of the promoter region (see Fig. 6). However, contrary to our findings in HaCaT cells (see above), repression via promoter hypermethylation was not supported by a decrease in permissive histone H3K4me3 levels. Rather, we detected an increase of heterochromatin marker H3K9me3 (transcriptionally silenced chromatin) as well as an increase in repressive marker H3K27me3 (see Fig. 2 and 5).
This indicates that UVA-induced epigenetic silencing of P16INK4a expression, which has been introduced early after UVA-irradiation of human keratinocytes (“UV-response”), might persist and can be detected again, later in the tumor (SCC). However, early and late silencing seems to be supported by different epigenetic histone modifications (H3K4me3 decrease compared to H3K9me3 and H3K27me3 increase). Whether H3K9me3 (and H3K27me3)-supported (late) silencing of P16INK4a (in the tumor) is still a consequence of the early, UVA-induced H3K4me3-related repression of P16INK4a or represents a distinct (new) epigenetic modification that is needed to facilitate e.g. tumor growth and progression has, however, still to be investigated.
Our results are supported by the recent findings in chronically UVB-irradiated mice, which also show that P16INK4a is epigenetically silenced in UVB-irradiated skin and UVB-induced tumors in mouse skin.47 These changes occur as a consequence of DNA promoter hypermethylation, which could be attributed to increased expression of maintenance DNA methyltransferase 1 (DNMT1) and de novo methyltransferases DNMT3a and DNMT3b. Increased expression of DNA methyltransferases was accompanied by epigenetic histone modifications involved in tumor suppressor gene silencing.47
In a first, yet incomplete, analysis of expression levels of mediators of epigenetic modifications, like DNMT1 or HDAC1 (which might influence DNA methylationvia the known cross-talk between histone deacetylation and DNA methylation66,67), we have not been able to confirm the findings of Nandakumar et al.47DNMT1 as well as HDAC1 expression did not change after chronic UVA-irradiation of HaCaT cells (see Fig. 4). Furthermore, first experiments in our group, dealing with expression levels of DNMT3a and DNMT3b after UVA-irradiation did not indicate any differences in comparison to unirradiated controls (data not shown). Whether this discrepancy between results published by Nandakumar47 and our findings is a consequence of the different irradiation qualities (UVBvs.UVA) or whether it reflects differences in the cellular systems used (mice vs. human) has still to be elucidated in future investigations.
In the present investigation we also detected altered epigenetic histone modifications after UVA-irradiation for certain other genes in the selected panel of genes studied (e.g.H3K4me3-, H3K9me3- and H3K27me3-levels in KLF4, and H3K9me3-level in P21WAF1/CIP1, see Fig. 2), which reach statistical significance. These modifications might be interesting too, because the important role of KLF4 in transcriptional control of epidermal specification, differentiation and skin cancer, involving P21WAF1/CIP1 is known.68–71 The prominent role of KLF4 in reprogramming somatic cells into induced pluripotent stem cells (iPS cells) is well documented.72 A detailed analysis of the first results presented here will be given in further publications where we are dealing with the effects of UV-radiation on the epigenetic control of epidermal stem cell fate (R. Greinert, in preparation). Other epigenetic changes (e.g.H3K9me3 in NANOG) could be detected, however, not on a reasonable statistical level. Further investigation is needed to clarify these points.
In this investigation we focused on epigenetic changes introduced by UVA in P16INK4a. To our knowledge we were able to show for the first time that UVA is able to induce sustained epigenetic modifications, which lead to a repression of P16INK4a in chronically irradiated HaCaT cells and in tumor (SCC) derived, re-cultivated cells. This might have important implications. First, UVA is the main spectral component of solar UV-radiation (95%) and of artificial UV used in sunbeds. The detection of any epigenetic change introduced by this radiation quality therefore adds new arguments to the risk assessment of UVA. Second, because it's known that certain epigenetic modifications can be interpreted as “hallmarks of cancer”,41 our finding that UVA-radiation is able to epigenetically repress e.g.P16INK4a and possibly other genes extends our knowledge about the mechanisms of skin cancer induction, development and progression.
Epigenetic modifications in P16INK4a have been demonstrated in a variety of diseases, including skin cancer.28 Reduced expression levels for P16INK4a have been reported in BCC samples compared to the surrounding normal tissue,24 although other investigations point in an opposite direction. Svensson et al.73 reported that increased P16INK4a expression has been detected in a highly invasive BCC subtype with infiltrative growth pattern, followed by ceased proliferation. On the contrary, Consciene et al. did not find any difference in P16INK4a expression among different histological types of BCCs and suggested that P16INK4a expression is not correlated with the degree of proliferation and malignancy. Rather, P16INK4a overexpression was significantly associated with tumor location on sun exposed areas.74
In the case of squamous cell carcinoma, several reports described hypermethylation of P16INK4a and P14ARF.16,75,76 Brown et al.16 have been able to demonstrate, that P16INK4a and P14ARF were silenced by DNA methylation in 36% and 42% of human primary SCCs, respectively. However, these findings have only been insufficiently supported by another study (only 7% P16INK4a methylation).75 The results of our investigation might indicate that down regulation of P16INK4a expression in SCCs16,75,76 and certain BCCs24 is caused by UVA-induced epigenetic silencing of P16INK4a.
A number of other genes have been reported to be transcriptionally down regulated by promoter hypermethylation in human primary BCCs and SCCs. 14-3-3sigma, a P53 dependent inhibitor of cell cycle progression and a prominent regulator of senescence and clonal evolution of human keratinocytes27 has been shown to be promoter-hypermethylated at high frequency (68%) in BCCs.77 Furthermore, T-cadherin (involved in cell adhesion) was silenced by promoter-specific hypermethylation in primary BCCs (24%) and SCCs (43%).78 E-cadherin, another cell adhesion gene, was even silenced in 67% of human primary SCCs.79 In addition, Fraga et al. reported about a number of other genes, which have been transcriptionally down regulated by promoter hypermethylation in primary human SCCs or SCC cell lines, including cysteine- and glycine-rich protein 2, insulin-like growth factor binding protein-3 and CXCR4, among others.80
All together these data indicate, that epigenetic regulation on the DNA-level (promoter hypermethylation) and on the histone-level play important roles in the development and progression of non-melanocytic skin cancer. Our data presented in this work are indicative for an involvement of UVA-radiation in these changes, especially in squameous cell carcinoma (SCC).
Biophysical and molecular mechanisms
The possible biophysical and molecular mechanisms by which UVA-radiation induces the epigenetic changes, described in this investigation, are still not known. In our investigation UVA-radiation seems to induce a G1-block in the cell cycle, despite the fact that P16INK4a, known to be involved in cell cycle regulation, is epigenetically repressed in the chronically UVA-irradiated HaCaT cells. No significant amounts of apoptotic cells have been found in UVA-irradiated HaCaT cells or tumor derived cells (see results). These preliminary data seem to indicate that a UVA dependent epigenetic repression of P16INK4a neither affects a UVA-induced G1-block nor influences apoptotic pathways (under the experimental conditions chosen in this investigation).It is clear that UV-radiation induces specific types of damage in DNA directly (e.g.CPDs and 6-4 PP)81 and/or produces a variety of reactive oxygen species (ROS) via complex photophysical and/or photochemical interactions with photosensitive molecules (e.g.flavins) within the cell.82 ROS could be responsible for further cellular damage (DNA base-modifications, DNA double strand and single strand breaks, etc8). Therefore, it might be reasonable to speculate that UV-induced DNA-damage and/or it's enzymatic repair triggers certain signal cascades responsible for the activation of epigenetic “mediators” like DNA-methyltransferases, histone methyltransferases, histone acetyltransferases (HAC), histone deacetylases (HDAC) and others, that bring about epigenetic changes in response to UV-radiation.
However, the mechanistical interpretation for these epigenetic changes still awaits scientific explanation. In hairless mice UV-irradiation is associated with progressive global hypomethylation of DNA in skin cells.83 Furthermore, a mechanistic link between P53 (which is often inactivated due to mutagenic effects of UV-radiation in skin cancer) and DNA methylation has been proposed. Peterson et al. found that P53 is able to repress transcription of DNA methyltransferase DNMT1 directly. Mutational inactivation of P53 (by UV) might therefore lead to activation of DNMT1 transcription and expression resulting in increased DNA methylation activity.84
All together, the data of our investigation show for the first time that chronic exposure of human skin cells to UVA-radiation is able to induce specific epigenetic changes in the genome. In particular, the tumor suppressor P16INK4a seems to be epigenetically silenced in UVA-irradiated human keratinocytes and cells derived from squamous cell carcinoma. This sheds new light on the etiology of photocarcinogenesis and might even have implications for new therapy options because epigenetic alterations are principally revertible.
Acknowledgements
The work was supported by BMBF-02NUK003D (Bundesministerium für Bildung und Forschung, Germany). Critical reading of the manuscript by Dr Kohelia Choudhury is gratefully acknowledged.References
- B. K. Armstrong and A. Kricker, The epidemiology of UV induced skin cancer, J. Photochem. Photobiol., B, 2001, 63, 8–18 CrossRef CAS.
- J. E. Cleaver and E. Crowley, UV damage, DNA repair and skin carcinogenesis, Front. Biosci., 2002, 7, d1024–1043 CrossRef CAS.
- J. Ramos, J. Villa, A. Ruiz, R. Armstrong and J. Matta, UV dose determines key characteristics of nonmelanoma skin cancer, Cancer Epidemiol., Biomarkers Prev., 2004, 13, 2006–2011 CAS.
- M. Norval, A. P. Cullen, F. R. de Gruijl, J. Longstreth, Y. Takizawa, R. M. Lucas, F. P. Noonan and J. C. van der Leun, The effects on human health from stratospheric ozone depletion and its interactions with climate change, Photochem. Photobiol. Sci., 2007, 6, 232–251 CAS.
- U. Leiter and C. Garbe, Epidemiology of melanoma and nonmelanoma skin cancer–the role of sunlight, Adv. Exp. Med. Biol., 2008, 624, 89–103 CrossRef.
- R. Greinert, Skin cancer: new markers for better prevention, Pathobiology, 2009, 76, 64–81 CrossRef.
- H. Ikehata and T. Ono, The mechanisms of UV mutagenesis, J. Radiat. Res., 2011, 52, 115–125 CrossRef CAS.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 3–17 CrossRef CAS.
- P. J. Rochette, J. P. Therrien, R. Drouin, D. Perdiz, N. Bastien, E. A. Drobetsky and E. Sage, UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells, Nucleic Acids Res., 2003, 31, 2786–2794 CrossRef CAS.
- S. Mouret, M. Charveron, A. Favier, J. Cadet and T. Douki, Differential repair of UVB-induced cyclobutane pyrimidine dimers in cultured human skin cells and whole human skin, DNA Repair, 2008, 7, 704–712 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. L. Lin, G. R. Ordonez, G. R. Bignell, K. Ye, J. Alipaz, M. J. Bauer, D. Beare, A. Butler, R. J. Carter, L. Chen, A. J. Cox, S. Edkins, P. I. Kokko-Gonzales, N. A. Gormley, R. J. Grocock, C. D. Haudenschild, M. M. Hims, T. James, M. Jia, Z. Kingsbury, C. Leroy, J. Marshall, A. Menzies, L. J. Mudie, Z. Ning, T. Royce, O. B. Schulz-Trieglaff, A. Spiridou, L. A. Stebbings, L. Szajkowski, J. Teague, D. Williamson, L. Chin, M. T. Ross, P. J. Campbell, D. R. Bentley, P. A. Futreal and M. R. Stratton, A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2010, 463, 191–196 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- S. Mouret, C. Philippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8, 1706–1711 CAS.
- P. Boukamp, Non-melanoma skin cancer: what drives tumor development and progression?, Carcinogenesis, 2005, 26, 1657–1667 CrossRef CAS.
- V. L. Brown, C. A. Harwood, T. Crook, J. G. Cronin, D. P. Kelsell and C. M. Proby, p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma, J. Invest. Dermatol., 2004, 122, 1284–1292 CrossRef CAS.
- M. Serrano, G. J. Hannon and D. Beach, A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4, Nature, 1993, 366, 704–707 CrossRef CAS.
- L. Sigalotti, A. Covre, E. Fratta, G. Parisi, F. Colizzi, A. Rizzo, R. Danielli, H. J. Nicolay, S. Coral and M. Maio, Epigenetics of human cutaneous melanoma: setting the stage for new therapeutic strategies, J. Transl. Med., 2010, 8, 56 CrossRef.
- D. E. Freedberg, S. H. Rigas, J. Russak, W. Gai, M. Kaplow, I. Osman, F. Turner, J. A. Randerson-Moor, A. Houghton, K. Busam, D. Timothy Bishop, B. C. Bastian, J. A. Newton-Bishop and D. Polsky, Frequent p16-independent inactivation of p14ARF in human melanoma, J. Natl. Cancer Inst., 2008, 100, 784–795 CrossRef CAS.
- K. Ranade, C. J. Hussussian, R. S. Sikorski, H. E. Varmus, A. M. Goldstein, M. A. Tucker, M. Serrano, G. J. Hannon, D. Beach and N. C. Dracopoli, Mutations associated with familial melanoma impair p16INK4 function, Nat. Genet., 1995, 10, 114–116 CrossRef CAS.
- J. W. Rocco and D. Sidransky, p16(MTS-1/CDKN2/INK4a) in cancer progression, Exp. Cell Res., 2001, 264, 42–55 CrossRef CAS.
- J. Campisi, Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors, Cell, 2005, 120, 513–522 CrossRef CAS.
- C. J. Collins and J. M. Sedivy, Involvement of the INK4a/Arf gene locus in senescence, Aging Cell, 2003, 2, 145–150 CrossRef CAS.
- P. Kanellou, A. Zaravinos, M. Zioga and D. A. Spandidos, Deregulation of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in basal cell carcinoma, Br. J. Dermatol., 2009, 160, 1215–1221 CrossRef CAS.
- A. Pacifico and G. Leone, Role of p53 and CDKN2A inactivation in human squamous cell carcinomas, J. Biomed. Biotechnol., 2007, 2007, 43418 CrossRef.
- N. Soufir, J. P. Moles, C. Vilmer, C. Moch, O. Verola, J. Rivet, A. Tesniere, L. Dubertret and N. Basset-Seguin, P16 UV mutations in human skin epithelial tumors, Oncogene, 1999, 18, 5477–5481 CrossRef CAS.
- R. van Doorn, N. A. Gruis, R. Willemze, P. A. van der Velden and C. P. Tensen, Aberrant DNA methylation in cutaneous malignancies, Semin. Oncol., 2005, 32, 479–487 CrossRef CAS.
- N. Popov and J. Gil, Epigenetic regulation of the INK4b-ARF-INK4a locus: in sickness and in health, Epigenetics, 2010, 5, 685–690 CrossRef CAS.
- G. Palmieri, M. Capone, M. L. Ascierto, G. Gentilcore, D. F. Stroncek, M. Casula, M. C. Sini, M. Palla, N. Mozzillo and P. A. Ascierto, Main roads to melanoma, J. Transl. Med., 2009, 7, 86 CrossRef.
- E. Gronniger, B. Weber, O. Heil, N. Peters, F. Stab, H. Wenck, B. Korn, M. Winnefeld and F. Lyko, Aging and chronic sun exposure cause distinct epigenetic changes in human skin, PLoS Genet., 2010, 6, e1000971 Search PubMed.
- U. G. Sathyanarayana, A. Y. Moore, L. Li, A. Padar, K. Majmudar, V. Stastny, P. Makarla, M. Suzuki, J. D. Minna, Z. Feng and A. F. Gazdar, Sun exposure related methylation in malignant and non-malignant skin lesions, Cancer Lett., 2007, 245, 112–120 CrossRef CAS.
- M. Kulis and M. Esteller, DNA methylation and cancer, Adv. Genet., 2010, 70, 27–56 CrossRef.
- M. Berdasco and M. Esteller, Aberrant epigenetic landscape in cancer: how cellular identity goes awry, Dev. Cell, 2010, 19, 698–711 CrossRef CAS.
- V. Davalos and M. Esteller, MicroRNAs and cancer epigenetics: a macrorevolution, Curr. Opin. Oncol., 2010, 22, 35–45 CrossRef CAS.
- A. Portela and M. Esteller, Epigenetic modifications and human disease, Nat. Biotechnol., 2010, 28, 1057–1068 CrossRef CAS.
- S. S. Oliver and J. M. Denu, Dynamic interplay between histone H3 modifications and protein interpreters: emerging evidence for a “histone language”, ChemBioChem, 2011, 12, 299–307 CrossRef CAS.
- T. Kouzarides, Chromatin modifications and their function, Cell, 2007, 128, 693–705 CrossRef CAS.
- A. Barski, S. Cuddapah, K. Cui, T. Y. Roh, D. E. Schones, Z. Wang, G. Wei, I. Chepelev and K. Zhao, High-resolution profiling of histone methylations in the human genome, Cell, 2007, 129, 823–837 CrossRef CAS.
- C. Sawan and Z. Herceg, Histone modifications and cancer, Adv. Genet., 2010, 70, 57–85 CrossRef CAS.
- H. Hashimoto, P. M. Vertino and X. Cheng, Molecular coupling of DNA methylation and histone methylation, Epigenomics, 2010, 2, 657–669 CrossRef CAS.
- P. W. Laird, Cancer epigenetics, Hum. Mol. Genet., 2005, 14 Spec No 1, R65–76 CrossRef.
- A. Sharonov, T. Gustavsson, S. Marguet and D. Markovitsi, Photophysical properties of 5-methylcytidine, Photochem. Photobiol. Sci., 2003, 2, 362–364 CAS.
- G. P. Pfeifer, p53 mutational spectra and the role of methylated CpG sequences, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2000, 450, 155–166 CrossRef CAS.
- G. P. Pfeifer, Mutagenesis at methylated CpG sequences, Curr. Top. Microbiol. Immunol., 2006, 301, 259–281 CrossRef CAS.
- M. F. Denissenko, J. X. Chen, M. S. Tang and G. P. Pfeifer, Cytosine methylation determines hot spots of DNA damage in the human P53 gene, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3893–3898 CrossRef CAS.
- Y. H. You and G. P. Pfeifer, Similarities in sunlight-induced mutational spectra of CpG-methylated transgenes and the p53 gene in skin cancer point to an important role of 5-methylcytosine residues in solar UV mutagenesis, J. Mol. Biol., 2001, 305, 389–399 CrossRef CAS.
- V. Nandakumar, M. Vaid, T. O. Tollefsbol and S. K. Katiyar, Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice, Carcinogenesis, 2011, 32, 597–604 CrossRef CAS.
- K. Wischermann, S. Popp, S. Moshir, K. Scharfetter-Kochanek, M. Wlaschek, F. de Gruijl, W. Hartschuh, R. Greinert, B. Volkmer, A. Faust, A. Rapp, P. Schmezer and P. Boukamp, UVA radiation causes DNA strand breaks, chromosomal aberrations and tumorigenic transformation in HaCaT skin keratinocytes, Oncogene, 2008, 27, 4269–4280 CrossRef CAS.
- M. LeBoeuf, A. Terrell, S. Trivedi, S. Sinha, J. A. Epstein, E. N. Olson, E. E. Morrisey and S. E. Millar, Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells, Dev. Cell, 2010, 19, 807–818 CrossRef CAS.
- T. Abbas and A. Dutta, p21 in cancer: intricate networks and multiple activities, Nat. Rev. Cancer, 2009, 9, 400–414 CrossRef CAS.
- J. C. Heng, Y. L. Orlov and H. H. Ng, Transcription factors for the modulation of pluripotency and reprogramming, Cold Spring Harbor Symp. Quant. Biol., 2010, 75, 237–244 CrossRef.
- P. Zhang, R. Andrianakos, Y. Yang, C. Liu and W. Lu, Kruppel-like factor 4 (Klf4) prevents embryonic stem (ES) cell differentiation by regulating Nanog gene expression, J. Biol. Chem., 2010, 285, 9180–9189 CrossRef CAS.
- J. L. Cox, S. K. Mallanna, X. Luo and A. Rizzino, Sox2 uses multiple domains to associate with proteins present in Sox2-protein complexes, PLoS One, 2010, 5, e15486 Search PubMed.
- S. R. Viswanathan, J. T. Powers, W. Einhorn, Y. Hoshida, T. L. Ng, S. Toffanin, M. O'Sullivan, J. Lu, L. A. Phillips, V. L. Lockhart, S. P. Shah, P. S. Tanwar, C. H. Mermel, R. Beroukhim, M. Azam, J. Teixeira, M. Meyerson, T. P. Hughes, J. M. Llovet, J. Radich, C. G. Mullighan, T. R. Golub, P. H. Sorensen and G. Q. Daley, Lin28 promotes transformation and is associated with advanced human malignancies, Nat. Genet., 2009, 41, 843–848 CrossRef CAS.
- S. R. Kinney and S. Pradhan, Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells, Prog. Mol. Biol. Transl. Sci., 2011, 101, 311–333 CrossRef CAS.
- G. L. Sen, J. A. Reuter, D. E. Webster, L. Zhu and P. A. Khavari, DNMT1 maintains progenitor function in self-renewing somatic tissue, Nature, 2010, 463, 563–567 CrossRef CAS.
- M. Frye, A. G. Fisher and F. M. Watt, Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation, PLoS One, 2007, 2, e763 Search PubMed.
- M. G. Lee, C. Wynder, D. A. Bochar, M. A. Hakimi, N. Cooch and R. Shiekhattar, Functional interplay between histone demethylase and deacetylase enzymes, Mol. Cell. Biol., 2006, 26, 6395–6402 CrossRef CAS.
- V. Dehennaut and D. Leprince, Implication of HIC1 (Hypermethylated In Cancer 1) in the DNA damage response, Bull. Cancer, 2009, 96, E66–72 Search PubMed.
- S. B. Baylin and J. E. Ohm, Epigenetic gene silencing in cancer – a mechanism for early oncogenic pathway addiction?, Nat. Rev. Cancer, 2006, 6, 107–116 CrossRef CAS.
- R. L. Zinn, K. Pruitt, S. Eguchi, S. B. Baylin and J. G. Herman, hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site, Cancer Res., 2007, 67, 194–201 CrossRef CAS.
- E. A. Attia, L. S. Seada, M. H. El-Sayed and S. M. El-Shiemy, Study of telomerase reverse transcriptase (hTERT) expression in normal, aged, and photo-aged skin, Int. J. Dermatol., 2010, 49, 886–893 CrossRef.
- J. A. Dahl and P. Collas, A rapid micro chromatin immunoprecipitation assay (microChIP), Nat. Protoc., 2008, 3, 1032–1045 CrossRef CAS.
- J. Vandesompele, K. De Preter, F. Pattyn, B. Poppe, N. Van Roy, A. De Paepe and F. Speleman, Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes, GenomeBiology, 2002, 3, RESEARCH0034 CrossRef.
- F. El Ghissassi, R. Baan, K. Straif, Y. Grosse, B. Secretan, V. Bouvard, L. Benbrahim-Tallaa, N. Guha, C. Freeman, L. Galichet and V. Cogliano, A review of human carcinogens–part D: radiation, Lancet Oncol., 2009, 10, 751–752 CrossRef.
- F. Fuks, W. A. Burgers, A. Brehm, L. Hughes-Davies and T. Kouzarides, DNA methyltransferase Dnmt1 associates with histone deacetylase activity, Nat. Genet., 2000, 24, 88–91 CrossRef CAS.
- J. S. You, J. K. Kang, E. K. Lee, J. C. Lee, S. H. Lee, Y. J. Jeon, D. H. Koh, S. H. Ahn, D. W. Seo, H. Y. Lee, E. J. Cho and J. W. Han, Histone deacetylase inhibitor apicidin downregulates DNA methyltransferase 1 expression and induces repressive histone modifications via recruitment of corepressor complex to promoter region in human cervix cancer cells, Oncogene, 2008, 27, 1376–1386 CrossRef CAS.
- J. A. Segre, Epidermal barrier formation and recovery in skin disorders, J. Clin. Invest., 2006, 116, 1150–1158 CrossRef CAS.
- J. A. Segre, C. Bauer and E. Fuchs, Klf4 is a transcription factor required for establishing the barrier function of the skin, Nat. Genet., 1999, 22, 356–360 CrossRef CAS.
- Y. C. Chew, G. Adhikary, G. M. Wilson, E. A. Reece and R. L. Eckert, Protein Kinase C (PKC) {delta} Suppresses Keratinocyte Proliferation by Increasing p21Cip1 Level by a KLF4 Transcription Factor-dependent Mechanism, J. Biol. Chem., 2011, 286, 28772–28782 CrossRef CAS.
- W. J. Choi, S. H. Youn, J. H. Back, S. Park, E. J. Park, K. J. Kim, H. R. Park, A. L. Kim and K. H. Kim, The role of KLF4 in UVB-induced murine skin tumor development and its correlation with cyclin D1, p53, and p21(Waf1/Cip1) in epithelial tumors of the human skin, Arch. Dermatol. Res., 2011, 303, 191–200 CrossRef CAS.
- A. Banito, S. T. Rashid, J. C. Acosta, S. Li, C. F. Pereira, I. Geti, S. Pinho, J. C. Silva, V. Azuara, M. Walsh, L. Vallier and J. Gil, Senescence impairs successful reprogramming to pluripotent stem cells, Genes Dev., 2009, 23, 2134–2139 CrossRef CAS.
- S. Svensson, K. Nilsson, A. Ringberg and G. Landberg, Invade or proliferate? Two contrasting events in malignant behavior governed by p16(INK4a) and an intact Rb pathway illustrated by a model system of basal cell carcinoma, Cancer Res., 2003, 63, 1737–1742 CAS.
- I. Conscience, N. Jovenin, C. Coissard, M. Lorenzato, A. Durlach, F. Grange, P. Birembaut, C. Clavel and P. Bernard, P16 is overexpressed in cutaneous carcinomas located on sun-exposed areas, Eur. J. Dermatol., 2006, 16, 518–522 CAS.
- L. N. Tyler, L. Ai, C. Zuo, C. Y. Fan and B. R. Smoller, Analysis of promoter hypermethylation of death-associated protein kinase and p16 tumor suppressor genes in actinic keratoses and squamous cell carcinomas of the skin, Mod. Pathol., 2003, 16, 660–664 CrossRef.
- J. L. Arbiser, C. Y. Fan, X. Su, B. O. Van Emburgh, F. Cerimele, M. S. Miller, J. Harvell and M. P. Marinkovich, Involvement of p53 and p16 tumor suppressor genes in recessive dystrophic epidermolysis bullosa-associated squamous cell carcinoma, J. Invest. Dermatol., 2004, 123, 788–790 CrossRef CAS.
- D. Lodygin, A. S. Yazdi, C. A. Sander, T. Herzinger and H. Hermeking, Analysis of 14-3-3sigma expression in hyperproliferative skin diseases reveals selective loss associated with CpG-methylation in basal cell carcinoma, Oncogene, 2003, 22, 5519–5524 CrossRef CAS.
- T. Takeuchi, S. B. Liang, N. Matsuyoshi, S. Zhou, Y. Miyachi, H. Sonobe and Y. Ohtsuki, Loss of T-cadherin (CDH13, H-cadherin) expression in cutaneous squamous cell carcinoma, Lab. Invest., 2002, 82, 1023–1029 CAS.
- M. C. Chiles, L. Ai, C. Zuo, C. Y. Fan and B. R. Smoller, E-cadherin promoter hypermethylation in preneoplastic and neoplastic skin lesions, Mod. Pathol., 2003, 16, 1014–1018 CrossRef.
- M. F. Fraga, M. Herranz, J. Espada, E. Ballestar, M. F. Paz, S. Ropero, E. Erkek, O. Bozdogan, H. Peinado, A. Niveleau, J. H. Mao, A. Balmain, A. Cano and M. Esteller, A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors, Cancer Res., 2004, 64, 5527–5534 CrossRef CAS.
- D. L. Mitchell, Effects of cytosine methylation on pyrimidine dimer formation in DNA, Photochem. Photobiol., 2000, 71, 162–165 CrossRef CAS.
- D. Dardalhon, A. R. Angelin, G. Baldacci, E. Sage and S. Francesconi, Unconventional effects of UVA radiation on cell cycle progression in S. pombe, Cell Cycle, 2008, 7, 611–622 CrossRef CAS.
- A. Mittal, C. Piyathilake, Y. Hara and S. K. Katiyar, Exceptionally high protection of photocarcinogenesis by topical application of (−)-epigallocatechin-3-gallate in hydrophilic cream in SKH-1 hairless mouse model: relationship to inhibition of UVB-induced global DNA hypomethylation, Neoplasia, 2003, 5, 555–565 CAS.
- E. J. Peterson, O. Bogler and S. M. Taylor, p53-mediated repression of DNA methyltransferase 1 expression by specific DNA binding, Cancer Res., 2003, 63, 6579–6582 CAS.
- J. G. Herman, J. R. Graff, S. Myohanen, B. D. Nelkin and S. B. Baylin, Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9821–9826 CrossRef CAS.
- T. Valenta, J. Lukas, L. Doubravska, B. Fafilek and V. Korinek, HIC1 attenuates Wnt signaling by recruitment of TCF-4 and beta-catenin to the nuclear bodies, EMBO J., 2006, 25, 2326–2337 CrossRef CAS.
- J. M. Martinez, C. A. Afshari, P. R. Bushel, A. Masuda, T. Takahashi and N. J. Walker, Differential toxicogenomic responses to 2,3,7,8-tetrachlorodibenzo-p-dioxin in malignant and nonmalignant human airway epithelial cells, Toxicol. Sci., 2002, 69, 409–423 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |