The specificity of UVA-induced DNA damage in human melanocytes†‡§
Stéphane
Mouret
,
Anne
Forestier
and
Thierry
Douki
*
Laboratoire « Lésions des Acides Nucléiques », SCIB, UMR-E3 CEA/UJF-Grenoble 1, INAC, Grenoble, F-38054, France. E-mail: thierry.douki@cea.fr; Fax: (33) 4 38 78 50 90; Tel: (33) 4 38 78 31 91
First published on 10th October 2011
Abstract
Exposure to solar UV radiation is the origin of most skin cancers, including deadly melanomas. Melanomas are quite different from keratinocyte-derived tumours and exhibit a different mutation spectrum in the activated oncogenes, possibly arising from a different class of DNA damage. In addition, some data suggest a role for UVA radiation in melanomagenesis. To get further insight into the molecular mechanisms underlying induction of melanoma, we quantified a series of UV-induced DNA damage in primary cultures of normal human melanocytes. The results were compared with those obtained in keratinocytes from the same donors. In the UVB range, the frequency and the distribution of pyrimidine dimers was the same in melanocytes and keratinocytes. UVA was also found to produce thymine cyclobutane dimer as the major DNA lesion with an equal efficiency in both cell types. In contrast, following UVA-irradiation a large difference was found for the yield of 8-oxo-7,8-dihydroguanine; the level of this product was 2.2-fold higher in melanocytes than in keratinocytes. The comet assay showed that the induction of strand breaks was equally efficient in both cell types but that the yield of Fpg-sensitive sites was larger in melanocytes. Our data show that, upon UVA irradiation, oxidative lesions contribute to a larger extent to DNA damage in melanocytes than in keratinocytes. We also observed that the basal level of oxidative lesions was higher in the melanocytes, in agreement with a higher oxidative stress that may be due to the production of melanin. The bulk of these results, combined with qPCR and cell survival data, may explain some of the differences in mutation spectrum and target genes between melanomas and carcinomas arising from keratinocytes.
Introduction
Melanoma is the most deadly form of skin cancer because it often metastasizes and is poorly cured. These tumours, which originate from melanocytes, differ in several aspects from tumours derived from keratinocytes.1,2 Unlike squamous cell carcinomas, for which the frequency is related to the overall accumulation of sun exposure in life, melanomas seem to be more closely linked with episodes of sunburns in childhood.3 Melanomas also differ from other skin cancers regarding the most frequently mutated genes. While melanoma tumours often exhibit BRAF or N-ras mutations, other skin cancers are often associated with p53 mutations.4–6 Moreover, melanoma from sun-exposed and unexposed skin areas have different mutation spectra, supporting the existence of a distinct molecular pathway in melanomagenesis associated with UV exposure.4,7 A pending question is the respective role of UVA and UVB in the etiology of melanomas. Indeed, experiments using fish models suggested that UVA is more important than for other cancer types8 but these data were recently disputed.9 In contrast, studies involving genetically modified mice put forward a major involvement of UVB.10 The recent epidemiological data in humans using UVA-rich sunbeds11 emphasize a need for a better evaluation of the role of UVA.Identification of the physicochemical processes involved in the origin of melanomas may provide an explanation for the specific mutation spectrum observed in activated oncogenes in these skin tumours. In the case of basal and squamous cell carcinomas mutations in key genes, such as p53, are observed at bipyrimidine sites12,13 where dimerisation photoreactions can take place. The resulting DNA lesions, (6–4) photoproducts (64PPs) and, more likely, cyclobutane pyrimidine dimers (CPDs),14,15 are known to be mutagenic. Although this mutational signature involving bipyrimidine photoproducts is also frequent in melanoma,16–18 other types of mutations are observed that correspond to monomeric lesions, such as, for instance, the oxidized base 8-oxo-7,8-dihydroguanine (8-oxoGua).16,18 More importantly, mutations in activated oncogenes at the origin of the tumourigenesis process, such as the T to A transversion at position 1796 in BRAF, is not at a bipyrimidine site.17,19 These results raise the question of the involvement of non dimeric lesions and, in particular, of oxidative damage produced in the UVA range. Indeed, UVA photons are known to trigger photosensitized processes20 leading to the release of reactive oxygen species, which may then damage DNA by inducing strand breaks and oxidized bases. Interestingly, we and others have shown that UVA irradiation also leads to the formation of CPDs, in larger amounts than oxidative damage in human skin, in primary cultures of human cutaneous cells and in cultured rodent cells.21–25
In the present work, we investigated whether human melanocytes in primary culture exhibit a specific profile of DNA damage upon exposure to UVA. The results were compared with those obtained in keratinocytes from the same donors. Information on the effects of UVB in both cell types was also gathered for comparative purposes. In addition, insight into the DNA repair capacities of the two cell types was gained from RT-qPCR analysis.
Material and methods
Chemicals and enzymes
Keratinocyte-SFM, recombinant epidermal growth factor, bovine pituitary extract, Medium 254, human melanocyte growth supplement and phosphate-buffered saline (PBS) were purchased from Invitrogen (Cergy-Pontoise, France). Primocin was obtained from Cayla-Invivogen (Toulouse, France). Phosphodiesterase I, phosphodiesterase II, DNase II, alkaline phosphatase and nuclease P1 were purchased from Sigma (St Louis, MO).Cell culture
Normal human keratinocytes and melanocytes were isolated from the same skin tissue obtained after breast plastic surgery, from healthy donors with their informed consent (Department of “Chirurgie Plastique et Maxillo-faciale”, CHU Grenoble, France). All donors were Caucasian (16–62 year old) with a skin phototype between II and III according to the Fitzpatrick classification. Both cell types were isolated from the same skin explant and samples from at least three different donors were used for each experiment. The protocol for isolation of keratinocytes was reported previously24 and is based on growth in a serum free medium (Keratinocyte-SFM) supplemented with recombinant epidermal growth factor, bovine pituitary extract and primocin. Isolation and culture of melanocytes requires a different approach in order to prevent proliferation of excess keratinocytes. The skin, after washing with ethanol 70%, was rinsed 10 times in PBS containing 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 0.25 μg ml−1 amphotericin B and was finally immersed overnight in 0.25% trypsin at 4 °C. The action of trypsin was stopped by addition of Dulbecco's modified Eagle's medium with 10% fetal calf serum. Epidermis was detached from dermis and homogenized. After centrifugation, harvested cells were seeded in Medium 254 supplemented with human melanocyte growth supplement and primocin at a density of 5 × 106 cells in 25 cm2 flasks. Subsequently, melanocytes were separated from contaminating keratinocytes during the first 3 passages by limiting the duration of trypsin treatment that preferentially leads to the release of keratinocytes. Cells were cultured at 37 °C in a 5% CO2 atmosphere. In order to obtain sufficient numbers of cells, melanocytes were used between passage 5 and 8 while keratinocytes were used in passage 3.UV irradiations
The UVB source used was a VL 215 G irradiator (Bioblock Scientific, France) fitted with two 15 W tubes with a broad-spectrum distribution with a maximum emission at 312 nm. For UVA, a Waldman UVA 700 L irradiator fitted with a high pressure lamp MSR 700 (700 W) (Waldman, Germany) was used. The fluence rates of the UVB and UVA lamps were 0.05 and 4.4 J cm−2 min−1, respectively. The spectral distribution of these two sources is shown in Fig. S1.‡Keratinocytes and melanocytes were seeded at 2 × 105 cells in 60 mm in Petri dishes and grown to sub confluence for several days. Just before irradiation, the culture medium was removed and kept aside. Cells were rinsed twice with PBS. Irradiations were then performed in PBS with the lid removed, at room temperature for UVB or on ice for UVA. Immediately after UV irradiation, cells were trypsinized and recovered for DNA damage quantification or replaced in their previous culture medium for cell survival evaluation. All irradiations were performed in triplicate for each donor.
Cytotoxicity
Cell cytotoxicity was determined using the conversion of the tetrazolium salt, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) to formazan. Briefly, after UV irradiation, cells were incubated for 24 h in their reserved medium. Then, 200 μl of 5 mg ml−1 MTT were added in each dish and the cells were incubated for 2 h at 37 °C. DMSO was then added to dissolve the soluble blue formazan and the absorbance was read at 565 nm using a Multiskan RC microplate spectrophotometer (Labsystems, Finland). The results were expressed as the percentage of cell viability.HPLC-tandem mass spectrometry analyses
DNA was extracted from the two cell types by the use of a purification kit (DNEasy Blood and Tissue kit), involving Qiagen columns allowing the elimination of melanin. After extraction, DNA was solubilized in 50 μl of 0.1 mM deferoxiamine mesylate solution and then hydrolyzed by incubation with nuclease P1, DNase II and phosphodiesterase II (2 h, 37 °C, pH 6), followed by a second digestion step involving phosphodiesterase I and alkaline phosphatase (2 h, 37 °C, pH 8). This enzymatic treatment allowed the release of 8-oxo-7,8-dihydroguanine and unmodified bases as nucleosides while bipyrimidine photoproducts were released as modified dinucleoside monophosphates. An aliquot fraction of each sample was first analyzed for its content in 8-oxoGua using a previously reported assay26 involving positive ionization and ammonium formate-based HPLC elution buffer. The remaining part of the samples was used for the quantification of pyrimidine dimers performed in the negative ionization mode with triethylammonium acetate as a mobile phase, as previously described.27,28Comet assay
The alkaline single-cell gel electrophoresis assay was used to determine the presence of strand breaks and alkali-labile sites. Additional information on the level of oxidized bases was gathered from the quantification of formamidopyrimidine DNA glycosylase (Fpg)-sensitive sites using a modified version of the comet assay. The assay was essentially conducted as previously described.29 Briefly, after trypsin treatment, keratinocytes and melanocytes were embedded in low-melting point agarose at 37 °C (final concentration 0.6% in PBS) and rubbed on microscope slides coated with one dried layer of 1% normal agarose. Embedded cells were immediately irradiated with UVA (10 J cm−2). Then, all slides were immersed overnight in lysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% sodium lauroyl sarcosinate, 1% triton X-100, 10% DMSO, pH 10) in the dark at 4 °C. After lysis, slides were washed with neutralization buffer (0.4 M Tris-HCl, pH 8) and Fpg buffer (0.1 M KCl, 0.5 mM Na2EDTA, 0.04 M Tris, pH 8) before digestion (45 min at 37 °C) with Fpg (1.7 μg ml−1, 100 μl per slide) to convert purine base modifications into single strand breaks. The slides were then placed in a horizontal electrophoresis tank containing freshly alkaline buffer (1 mM EDTA, 300 mM NaOH) for 40 min at room temperature. Electrophoresis was performed at 25 V and 300 mA for 30 min. Then, the slides were rinsed in neutralization buffer and stained with ethidium bromide (10 μg ml−1). Finally, comet analysis was performed using the image analysis Comet IV software (Perceptive instrument, UK) and the quantification of DNA damage was performed using the percentage of tail DNA. For each sample, 50 comets of each slide were scored and the average tail intensity was determined from a triplicate experiment.Reverse Transcription (RT) and real time quantitative PCR (qPCR) analysis
Total RNA was extracted from three frozen pellets of melanocytes and keratinocytes from three distinct donors, using the GenElute mammalian total RNA miniprep kit (Sigma) according to the manufacturer's protocol. The quality of total RNA was assessed through a native agarose gel electrophoresis. We considered total RNA as intact when two acute bands corresponding to the 28S and 18S rRNA subunits were visualized. For both melanocytes and keratinocytes, 2 μg of total RNA were reverse-transcribed to cDNA (SuperscriptTM II Reverse Transcriptase, Invitrogen) in presence of 100 ng μl−1of random primers (Promega SARL, Charbonnières, France), dNTP mix (10 mM of each, Sigma), 5× first strand buffer, DTT (0.1 M) and Superscript II enzyme (Invitrogen). Twenty ng of each cDNA template was used in qPCR reactions with gene-specific primers. qPCR was performed in a MX3005p Multiplex Quantitative PCR system (Stratagene, La Jolla, CA) using MESA Blue qPCRTM Mastermix Plus for SYBR® Assay – Low Rox (Eurogentec SARL, Angers, France). At the end of each qPCR run, a dissociation curve analysis was performed to verify the integrity of primer-specific amplification, indicated by a single melt peak for each qPCR-product. Four housekeeping genes were tested to act as simultaneous endogenous controls in qPCR analysis. 18S ribosomal 1 (S18), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), cyclophilin (Cyclo) A and B were amplified in triplicate from both melanocytes and keratinocytes on a same qPCR run. The corresponding cycle threshold (Ct) values were exported to BestKeeper,30 an Excel-based pair-wise correlation tool that analyzes variability in the expression of individual genes among different conditions. Separately, S18, GAPDH, CycloA and CycloB exhibited a SD on Cts less than 1 between keratinocytes and melanocytes. This result showed that the reference genes were equally expressed in both cell types. In order to optimize the normalization, these four housekeeping genes were used for both melanocytes and keratinocytes of each donor to calculate the expression of targeted genes with respect of each of the references by using the expression R = 2(Ct(reference) − Ct(targeted gene)). The complete set of data for the four reference genes and the three donors was analysed for significance by ANOVA using the Statistica software. Seventeen genes were found to be significantly differently expressed in melanocytes and keratinocytes. Three of them were not further considered because modulation was observed only for one donor (polβ, NHTL1 and XPD). For all studied genes, ratios of expression in melanocytes to that in keratinocytes were calculated for the four reference genes and values were averaged for each donor (Supplementary information, Table S1 for the complete set of data‡).Results
Photosensitivity of melanocytes and keratinocytes
Melanocytes and keratinocytes were exposed to increasing fluences of either UVB or UVA radiation. Survival was then determined 24 h post-irradiation by using the MTT assay reflecting mitochondrial activity. In the UVB range, melanocytes were much more sensitive than keratinocytes (Fig. 1A), with an LD50 of 0.04 and 0.3 J cm−2, respectively. An opposite trend was observed in the UVA range (Fig. 1B) with a lower lethality for melanocytes than for keratinocytes. The fluences leading to 50% survival were 120 and 75 J cm−2, respectively. A similar comparison was previously reported and similar survival after UVB irradiation was found for both cell types.31,32 Yet these authors observed that melanocytes were much more resistant than keratinocytes to UVA. It may thus be concluded that, in relative values, the ratio between UVA and UVB survival is larger for melanocytes than keratinocytes.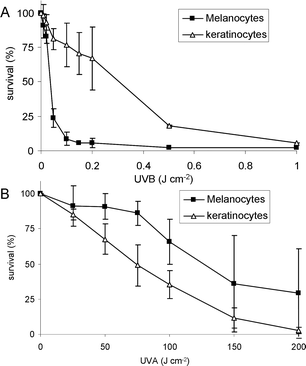 | ||
| Fig. 1 Cytotoxicity of UVB (upper panel) and UVA (lower panel) in primary culture of human melanocytes and keratinocytes. Cell viability was determined by the MTT assay. | ||
UV-induced pyrimidine dimers
The frequency of dimeric pyrimidine photoproducts was determined in cells exposed to either UVB or UVA radiation. In order to obtain individual values for all photoproducts involving thymine (T) and/or cytosine (C), a chromatographic assay was used. The assay is based on the extraction of DNA followed by release of the lesions as modified dinucleoside monophosphates which are subsequently quantified by HPLC coupled to tandem mass spectrometry (HPLC-MS/MS). Dose-dependent formation was studied for cultured keratinocytes and melanocytes from 3 or 4 donors.In the UVB range, a linear induction was observed for TT, TC, CT and CC CPDs, as well as for TT and TC 64PPs. Linear regression of these dose-dependent curves provided yields of formation, which were averaged among the three donors for both cell types (Fig. 2). Nearly identical yields were observed for all photoproducts between the two cell types. This similarity was also observed for each individual donor (data not shown). As previously reported for other cell types, UVA irradiation led to the sole formation of TT, TC and CT CPDs in melanocytes (Fig. 3). Like with UVB, the frequency and the relative yield of the different UVA-induced dimeric photoproducts were very similar in keratinocytes and melanocytes. For the TT CPD, the yields were 126.2 ± 27.4 and 121.6 ± 24.3 lesions/106 bases per kJ cm2 in keratinocytes and melanocytes, respectively (Fig. 4).
 | ||
| Fig. 2 Yield of formation of pyrimidine dimeric photoproducts upon UVB irradiation of keratinocytes and melanocytes. Photoproducts were quantified by HPLC-MS/MS. Differences in yield between the two cell types were not statistically significant. (n = 3). | ||
 | ||
| Fig. 3 Dose-dependent formation of CPDs in primary culture of human melanocytes exposed to UVA radiation. Photoproducts were quantified by HPLC-MS/MS. Data correspond to a representative donor. Irradiations and analyses were performed in triplicate. | ||
 | ||
| Fig. 4 Yield of formation of CPD and 8-oxoGua in melanocytes and keratinocytes exposed to UVA radiation. Modified bases were quantified by HPLC-MS/MS. No statistically significant difference was found for TT CPD while the p value was below 0.01 for 8-oxoGua (t test, n = 4). | ||
Oxidative DNA damage
We then determined the extent of oxidative lesions in cells exposed to UVA. The level of 8-oxoGua was measured by HPLC-MS/MS for four donors. In the absence of irradiation, a higher level was observed in melanocytes (12.7 ± 8.0 8-oxoGua per 106 bases) than in keratinocytes (5.8 ± 2.8 8-oxoGua per 106 bases). The formation of 8-oxoGua following exposure to 100 and 200 J cm−2 was also found to be larger in melanocytes than keratinocytes with a ratio of 3 to 1 (Fig. 4).The extent of oxidative DNA damage was then determined by the comet assay. This method permits the visualization of strand breaks and alkaline-sensitive sites. When combined with treatment with purified glycosylases it can also provide information on specific classes of modified bases. In the present work, bacterial formamidopyrimidine glycosylase was used to study oxidized purines, and in particular 8-oxoGua. Without irradiation, both strand breaks and Fpg-sensitive sites were more frequent in melanocytes than in keratinocytes (Table 1). Induction of lesions upon UVA irradiation followed the same trend, at least for oxidized bases. Indeed, a larger yield of Fpg-sensitive sites was observed in melanocytes, while formation of strand breaks was similar in both cell types.
| Background breaks* | Background Fpg** | UVA breaksb | UVA Fpgb*** | |
|---|---|---|---|---|
| a Results are expressed in % tail DNA and were obtained from 3 donors (n = 3, *: p < 0.05; **: p < 0.01. ***: p < 0.03). For each donor, experiments were performed in triplicate and results averaged. b The background values were subtracted from those determined after irradiation. | ||||
| keratinocytes | 10.7 ± 2.2 | 4.7 ± 3.2 | 16.6 ± 4.4 | 16.1 ± 3.0 |
| melanocytes | 27.5 ± 11.7 | 11.9 ± 2.7 | 14.6 ± 1.0 | 25.5 ± 0.3 |
DNA repair gene expression
The basal level of expression of a large series of DNA repair genes was compared in melanocytes and keratinocytes for three donors. Thirty-six genes were selected in the main repair pathways, including base excision repair (BER), nucleotide excision repair (NER), double strand breaks repair, and mismatch repair (MMR). Only little difference and rather large inter-individual variability was observed for more than half of the targeted genes (Table S1‡). Yet, several BER genes were significantly more expressed in melanocytes, namely UNG APE1 MYH XRCC1 LigIII and PARP1, while only APTX was less expressed (Fig. 5). In the nucleotide excision repair pathway, a rather higher expression of DDB1, XPA and PCNA was observed in melanocytes compared with keratinocytes, while CSB and ERCC1 (Fig. 5) were less expressed. No significant modulation was observed for the other repair pathways investigated with exception of polμ (double-strand breaks repair) and msh6 (MMR) that were respectively more and less expressed in melanocytes. | ||
| Fig. 5 Comparison of the expression of modulated DNA repair genes between melanocytes and keratinocytes. The upper panel shows data for BER while the lower one reports results for NER, double-strand breaks repair and MMR. Expression was determined by qPCR. Difference in gene expression was found to be statistically significant by ANOVA analysis (p < 0.01). | ||
Discussion
The possible contribution of UVA to the etiology of cutaneous melanoma has been a matter of discussion for many years.33 The question was raised by the observation that the influence of sun exposure and the nature of mutation was quite different from those of other skin cancers, at least in the relevant activated oncogenes.17,19 Because melanin, especially pheomelanin, was reported to exhibit oxidant properties,2,34 it was proposed that the nature of UV-induced DNA damage in melanocytes was different from those in other cell types.35 Evidence has thus been provided for a larger extent of oxidative lesions in this cell type.36,37 Yet, recent results show that UVA is also able to induce the formation of CPDs in higher yield than oxidative lesions in keratinocytes, fibroblasts and several cell lines.21,22,24 We therefore designed this study to compare the UV photochemistry of DNA between the primary cultures of melanocytes and keratinocytes. We further extended the work to the evaluation of DNA repair potency in the two cell types.Emphasis was first placed on the formation of bipyrimidine photoproducts. In the UVB range, no differences were observed in the yields of the four possible CPDs and of TT and TC 64PPs between melanocytes and keratinocytes. In addition, no Dewar valence isomer, the secondary photolysis product of 64PPs was detected (data not shown), in agreement with previous results obtained in similar, and rather low, dose ranges.22,28,38 Our observations of similar amounts of damage in the two cell types are in line with those reported for both cultured cells39,40 and whole skin.41
It was rather unexpected that melanocytes are not better protected against UV-induced CPDs since melanin is a good photoprotector in skin.42 This observation is not an experimental artefact since, in line with what has been reported by others,43 we observed that cultured melanocytes contained high amounts of melanin irrespective of the number of passages, as shown by their very heavy pigmentation. In contrast, primary cultures of keratinocytes were colourless. Yet, the yields of CPDs were the same between the two cell types. Interestingly, enhanced production of melanin was found to only marginally affect survival of UV-irradiated melanocytes.44,45 One explanation may be that melanin is mainly contained in melanosomes, present in the cytosol of the melanocytes 46 and therefore providing a poor protection to the nucleus. Such a hypothesis seems inconsistent with data obtained in whole skin where an inverse relationship between the amount of melanin and the yield of CPD is observed.47–49 It should, however, be remembered that these values mostly concern keratinocytes, which is the most frequent cell type in the epidermis. In skin, keratinocytes are loaded with melanin exhibiting perinuclear localization,46 thus likely efficiently shielding DNA. It would be interesting to determine whether the inverse correlation between skin type and CPD amounts induced in DNA holds for melanocytes in the skin. An alternative explanation to the apparent discrepancy between results in cultured cells and in skin may be that protection in the tissue is mostly performed by melanin released during differentiation of keratinocytes.
CPDs were also found to be produced in the DNA of melanocytes upon UVA irradiation, in agreement with previous observations in other cultured mammalian cells and in skin.21–23,25,50,51 No difference in efficiency was observed between keratinocytes and melanocytes. Again, melanin does not prevent formation of dimeric photoproducts. Interestingly, evidence is accumulating for a direct mechanism in UVA-induction of CPDs,52–54 resulting from a weak but significant absorption of DNA in this spectral region.52,55 It is thus not surprising that the ratio between the yields of CPDs in the UVA and UVB portion is not changed from one cell type to the other if the main parameter governing the frequency of damage is the amount of UV reaching DNA.
In contrast to dimeric photoproducts, significant differences were observed between melanocytes and keratinocytes in the case of UVA-induced oxidative lesions. This class of damage was not studied for UVB because it is produced in about two orders of magnitude lower yield than CPDs and 64PPs at these wavelengths.23,24 A first observation was that the basal level of endogenous oxidative lesions was higher in melanocytes. This was first observed on the level of 8-oxoGua measured by HPLC-MS/MS. Chromatographic measurements of 8-oxoGua have been shown in the past to be at risk of artifactual overestimation. Therefore, although an optimized extraction protocol was used, we applied the Comet assay to gather additional information. The trend in increased frequency of oxidative damage was confirmed for strand breaks and Fpg-sensitive sites. A likely explanation is that melanin synthesis is known to generate reactive oxygen species as by-products.56
In addition to this higher endogenous production of oxidative lesions, melanocytes were also found to accumulate more oxidation products than keratinocytes upon UVA irradiation, as previously reported.37,36 The ratio between the yields of CPDs and 8-oxoGua was 5.2 in keratinocytes, in agreement with previous results but decreased to 1.4 in melanocytes. This larger yield of 8-oxoGua was also observed for Fpg-sensitive sites in the Comet assay, which correspond mostly to 8-oxoGua. Interestingly, the yield of direct strand breaks was the same in the two cell types. This strongly suggests that singlet oxygen is the main mediator of the additional oxidative stress in melanocytes because this reactive oxygen species only induces 8-oxoGua, without damaging the DNA backbone.20,57
In line with these results, it may be reminded that melanin is a two-edge molecule that behaves as a photoprotector but also a pro-oxidant compound upon irradiation.58,59 Skin cells possess antioxidant defence system to protect against oxidative damage. However, it has been demonstrated that keratinocytes have higher antioxidant activities than melanocytes,60 which can also explain the higher background level of oxidative lesions in melanocytes. Moreover, in Caucasian melanocytes, the pheomelanin synthesis also consumes cysteine, which is the essential component of glutathione, and can thereby lower the capacity of antioxidant defence and increase the risk of oxidative damage in melanocytes.
The larger overall yield of DNA damage upon UVA irradiation of melanocytes with respect to keratinocytes suggested that DNA repair would be more efficient in the former cell line. This was the case for BER for which six genes were overexpressed among those studied; Only APTX was less expressed in melanocytes. Interestingly, the better adaptation of melanocytes to oxidative stress suggested by this increased expression of BER genes is in line with our observation that these cells were more resistant to UVA than keratinocytes. Yet, our repair genes expression data seems contradictory with recent published data showing reduced repair activity in melanocytes.37 Interestingly, the authors of this last work proposed that melanin exhibits an inhibitory effects on DNA repair. Thus, it is possible to envision an increased gene expression counteracted by a decrease of enzymatic activities.
A correlation between cell survival to UVB radiation and DNA repair of dimeric photoproducts was also observed. In contrast to UVA, melanocytes were found to be much more sensitive than keratinocytes in this wavelengths range. Accordingly, our gene expression profile showed that some key repair genes like CSB and ERCC1, important for the removal of CPDs by NER, are drastically less expressed in melanocytes than in keratinocytes. These results are in line with the recent data showing that repair of CPDs is less efficient in melanocytes.37 Interestingly, a clinical study recently highlighted a link between reduced DNA repair capacities and melanoma incidence.61
Conclusions
In summary, our results confirm that UVA induces CPDs in melanocytes with the same efficiency as in other cell types. This observation is another illustration of the biological relevance of this class of photoproducts over the all UV range and not only with UVB radiation. Yet, oxidative lesions were found to be more frequent in melanocytes than in keratinocytes, suggesting a higher UVA-induced oxidative stress likely resulting from the presence of melanin. The actual contribution of this genotoxic pathway to melanoma induction remains to be established.References
- G. Walker, Cutaneous melanoma: how does ultraviolet light contribute to melanocyte transformation?, Future Oncol., 2008, 4, 841–856 Search PubMed.
- N. Maddodi and V. Setaluri, Role of UV in cutaneous melanoma, Photochem. Photobiol., 2008, 84, 528–536 CrossRef CAS.
- D. C. Whiteman, C. A. Whiteman and A. C. Green, Childhood sun exposure as a risk factor for melanoma: a systematic review of epidemiologic studies, Cancer, Causes Control, 2001, 12, 69–82 CrossRef CAS.
- S. Jiveskog, B. Ragnarsson-Olding, A. Platz and U. Ringborg, N-ras mutations are common in melanomas from sun-exposed skin of humans but rare in mucosal membrane or unexposed skin, J. Invest. Dermatol., 1998, 111, 757–761 CrossRef CAS.
- S. Pavey, P. Johansson, L. Packer, J. Taylor, M. Stark, P. M. Pollock, G. J. Walker, G. M. Boyle, U. Harper, S. J. Cozzi, K. Hansen, L. Yudt, C. Schmidt, P. Hersey, K. A. Ellem, M. G. O'Rourke, P. G. Parsons, P. Meltzer, M. Ringner and N. K. Hayward, Microarray expression profiling in melanoma reveals a BRAF mutation signature, Oncogene, 2004, 23, 4060–4067 CrossRef CAS.
- P. M. Pollock, U. L. Harper, K. S. Hansen, L. M. Yudt, M. Stark, C. M. Robbins, T. Y. Moses, G. Hostetter, U. Wagner, J. Kakareka, G. Salem, T. Pohida, P. Heenan, P. Duray, O. Kallioniemi, N. K. Hayward, J. M. Trent and P. S. Meltzer, High frequency of BRAF mutations in nevi, Nat. Genet., 2003, 33, 19–20 CrossRef CAS.
- J. A. Curtin, J. Fridlyand, T. Kageshita, H. N. Patel, K. J. Busam, H. Kutzner, K. H. Cho, S. Aiba, E. B. Brocker, P. E. LeBoit, D. Pinkel and B. C. Bastian, Distinct sets of genetic alterations in melanoma, N. Engl. J. Med., 2005, 353, 2135–2147 CrossRef CAS.
- R. B. Setlow, Spectral regions contributing to melanoma: a personal view, J. Invest. Dermatol. Symp. Proc., 1999, 4, 46–49 Search PubMed.
- D. L. Mitchell, A. A. Fernandez, R. S. Nairn, R. Garcia, L. Paniker, D. Trono, H. D. Thames and I. Gimenez-Conti, Ultraviolet A does not induce melanomas in a Xiphophorus hybrid fish model, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9329–9334 CrossRef CAS.
- E. C. De Fabo, F. P. Noonan, T. Fears and G. Merlino, Ultraviolet B but not ultraviolet A radiation initiates melanoma, Cancer Res., 2004, 64, 6372–6376 CAS.
- S. G. Coelho and V. J. Hearing, UVA tanning is involved in the increased incidence of skin cancers in fair-skinned young women, Pigm. Cell Melanoma Res., 2010, 23, 57–63 Search PubMed.
- D. E. Brash, J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin and J. Ponten, A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10124–10128 CAS.
- A. Ziegler, D. J. Leffel, S. Kunala, H. W. Sharma, P. E. Shapiro, A. E. Bale and D. E. Brash, Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4216–4220 CAS.
- J. Jans, W. Schul, Y. G. Sert, Y. Rijksen, H. Rebel, A. P. Eker, S. Nakajima, H. van Steeg, F. R. de Gruijl, A. Yasui, J. H. Hoeijmakers and G. T. van der Horst, Powerful skin cancer protection by a CPD-photolyase transgene, Curr. Biol., 2005, 15, 105–115 CrossRef CAS.
- Y. H. You, D. H. Lee, J. H. Yoon, S. Nakajima, A. Yasui and G. P. Pfeifer, Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells, J. Biol. Chem., 2001, 276, 44688–44694 CrossRef CAS.
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. L. Lin, G. R. Ordonez, G. R. Bignell, K. Ye, J. Alipaz, M. J. Bauer, D. Beare, A. Butler, R. J. Carter, L. N. Chen, A. J. Cox, S. Edkins, P. I. Kokko-Gonzales, N. A. Gormley, R. J. Grocock, C. D. Haudenschild, M. M. Hims, T. James, M. M. Jia, Z. Kingsbury, C. Leroy, J. Marshall, A. Menzies, L. J. Mudie, Z. M. Ning, T. Royce, O. B. Schulz-Trieglaff, A. Spiridou, L. A. Stebbings, L. Szajkowski, J. Teague, D. Williamson, L. Chin, M. T. Ross, P. J. Campbell, D. R. Bentley, P. A. Futreal and M. R. Stratton, A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2010, 463, 191–197 CrossRef CAS.
- T. Hocker and H. S. Tsao, Ultraviolet radiation and melanoma: A systematic review and analysis of reported sequence variants, Hum. Mutat., 2007, 28, 578–588 CrossRef CAS.
- Y. Wang, J. J. DiGiovanna, J. B. Stern, T. J. Hornyak, M. Raffeld, S. G. Khan, K. S. Oh, M. C. Hollander, P. A. Dennis and K. H. Kraemer, Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 6279–6284 CrossRef CAS.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woffendin, M. J. Garnett, W. Bottomley, N. Davis, N. Dicks, R. Ewing, Y. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. C. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Siegler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Futreal, Mutations of the BRAF gene in human cancer, Nature, 2002, 417, 949–954 CrossRef CAS.
- J. Cadet, T. Douki, J. L. Ravanat and P. Di Mascio, Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation, Photochem. Photobiol. Sci., 2009, 8, 903–911 RSC.
- S. Courdavault, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Larger yield of cyclobutane dimers than 8 oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2004, 556, 135–142 CrossRef CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- C. Kielbassa, L. Roza and B. Epe, Wavelength dependence of oxidative DNA damage induced by UV and visible light, Carcinogenesis, 1997, 18, 811–816 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- D. Perdiz, P. Grof, M. Mezzina, O. Nikaido, E. Moustacchi and E. Sage, Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis, J. Biol. Chem., 2000, 275, 26732–26742 CAS.
- S. Frelon, T. Douki, J.-L. Ravanat, C. Tornabene and J. Cadet, High performance liquid chromatography-tandem mass spectrometry measurement of radiation-induced base damage to isolated and cellular DNA, Chem. Res. Toxicol., 2000, 13, 1002–1010 CrossRef CAS.
- T. Douki and J. Cadet, Individual determination of the yield of the main-UV induced dimeric pyrimidine photoproducts in DNA suggests a high mutagenicity of CC photolesions, Biochemistry, 2001, 40, 2495–2501 CrossRef CAS.
- T. Douki, M. Court, S. Sauvaigo, F. Odin and J. Cadet, Formation of the main UV-induced thymine dimeric lesions within isolated and cellular DNA as measured by high performance liquid chromatography-tandem mass spectrometry, J. Biol. Chem., 2000, 275, 11678–11685 CrossRef CAS.
- S. Mouret, S. Sauvaigo, A. Peinnequin, A. Favier, J. C. Beani and M. T. Leccia, E6*oncoprotein expression of human papillomavirus type-16 determines different ultraviolet sensitivity related to glutathione and glutathione peroxidase antioxidant defence, Exp. Dermatol., 2005, 14, 401–410 CrossRef CAS.
- M. W. Pfaffl, G. W. Horgan and L. Dempfle, Relative expression software tool (REST (c)) for group-wise comparison and statistical analysis of relative expression results in real-time PCR, Nucleic Acids Res., 2002, 30, e36 CrossRef.
- S. M. Deleeuw, S. Janssen, J. Simons, P. H. M. Lohman, B. J. Vermeer and A. A. Schothorst, The UV action spectra for the clone-forming ability of cultured human melanocytes and keratinocytes, Photochem. Photobiol., 1994, 59, 430–436 CrossRef CAS.
- P. Larsson, E. Andersson, U. Johansson, K. Ollinger and I. Rosdahl, Ultraviolet A and B affect human melanocytes and keratinocytes differently. A study of oxidative alterations and apoptosis, Exp. Dermatol., 2005, 14, 117–123 CrossRef CAS.
- L. P. Lund and G. S. Timmins, Melanoma, long wavelength ultraviolet and sunscreens: Controversies and potential resolutions, Pharmacol. Ther., 2007, 114, 198–207 CrossRef CAS.
- M. A. Cotter, J. Thomas, P. Cassidy, K. Robinette, N. Jenkins, S. R. Florell, S. Leachman, W. E. Samlowski and D. Grossman, N-acetylcysteine protects melanocytes against oxidative stress/damage and delays onset of ultraviolet induced melanoma in mice, Clin. Cancer Res., 2007, 13, 5952–5958 CrossRef CAS.
- S. Q. Wang, R. Setlow, M. Berwick, D. Polsky, A. A. Marghoob, A. W. Kopf and R. S. Bart, Ultraviolet A and melanoma: A review, J. Am. Acad. Dermatol., 2001, 44, 837–846 CrossRef CAS.
- X. Z. Song, N. Mosby, J. Yang, A. Xu, Z. Abdel-Malek and A. L. Kadekaro, alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes, Pigm. Cell Melanoma Res., 2009, 22, 809–818 Search PubMed.
- H. T. Wang, B. Choi and M. S. Tang, Melanocytes are deficient in repair of oxidative DNA damage and UV-induced photoproducts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12180–12185 CrossRef CAS.
- S. Courdavault, C. Baudouin, M. Charveron, B. Canghilem, A. Favier, J. Cadet and T. Douki, Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations, DNA Repair, 2005, 4, 836–844 CrossRef CAS.
- S. M. Deleeuw, W. I. M. Simons, B. J. Vermeer and A. A. Schothorst, Comparison of melanocytes and keratinocytes in ultraviolet-induced DNA-damage per minimum erythema dose sunlight - Applicability of ultraviolet action spectra for risk estimates, J. Invest. Dermatol., 1995, 105, 259–263 CrossRef CAS.
- A. A. Schothorst, L. M. Evers, K. C. Noz, R. Filon and A. A. Vanzeeland, Pyrimidine dimer induction and repair in cultured human skin keratinocytes or melanocytes after irradiation with monochromatic ultraviolet-radiation, J. Invest. Dermatol., 1991, 96, 916–920 CrossRef CAS.
- A. R. Young, C. S. Potten, O. Nikaido, P. G. Parsons, J. Boenders, J. M. Ramsden and C. A. Chadmick, Human melanocytes and keratinocytes exposed to UVB or UVA in vivo show comparable levels of thymine dimers, J. Invest. Dermatol., 1998, 111, 936–940 CrossRef CAS.
- M. Brenner and V. J. Hearing, The protective role of melanin against UV damage in human skin, Photochem. Photobiol., 2008, 84, 539–549 CrossRef CAS.
- S. M. De Leeuw, N. P. M. Smit, M. Van Veldhoven, E. M. Pennings, S. Pavel, J. Simons and A. A. Schothorst, Melanin content of cultured human melanocytes and UV-induced cytotoxicity, J. Photochem. Photobiol., B, 2001, 61, 106–113 CrossRef CAS.
- C. Kowalczuk, M. Priestner, C. Baller, A. Pearson, N. Cridland, R. Saunders, K. Wakamatsu and S. Ito, Effect of increased intracellular melanin concentration on survival of human melanoma cells exposed to different wavelengths of UV radiation, Int. J. Radiat. Biol., 2001, 77, 883–889 CrossRef CAS.
- H. Z. Hill, G. J. Hill, K. Cieszka, P. M. Plonka, D. L. Mitchell, M. F. Meyenhofer, P. T. Xin and R. E. Boissy, Comparative action spectrum for ultraviolet light killing of mouse melanocytes from different genetic coat color backgrounds, Photochem. Photobiol., 1997, 65, 983–989 CrossRef CAS.
- H. Y. Park, M. Kosmadaki, M. Yaar and B. A. Gilchrest, Cellular mechanisms regulating human melanogenesis, Cell. Mol. Life Sci., 2009, 66, 1493–1506 CrossRef CAS.
- S. Mouret, M.-T. Leccia, J.-L. Bourrain, T. Douki, J.-C. Beani and Individual, photosensitivity of human skin and UVA-induced pyrimidine dimers in DNA, J. Invest. Dermatol., 2011, 131, 1539–1546 CrossRef CAS.
- T. Tadokoro, N. Kobayashi, B. Z. Zmudzka, S. Ito, K. Wakamatsu, Y. Yamaguchi, K. S. Korossy, S. A. Miller, J. Z. Beer and V. J. Hearing, UV-induced DNA damage and melanin content in human skin differing in racial/ethnic origin, FASEB J., 2003, 17, 1177–1179 CAS.
- Y. Yamaguchi, K. Takahashi, B. Z. Zmudzka, A. Kornhauser, S. A. Miller, T. Tadokoro, W. Berens, J. Z. Beer and V. J. Hearing, Human skin responses to UV radiation: pigment in the upper epidermis protects against DNA damage in the lower epidermis and facilitates apoptosis, FASEB J., 2006, 20, 1486–1488 CrossRef CAS.
- S. E. Freeman, H. Hacham, R. W. Gange, D. J. Maytum, J. C. Sutherland and B. M. Sutherland, Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 5605–5609 CrossRef CAS.
- F. E. Quaite, B. M. Sutherland and J. C. Sutherland, Action spectrum for DNA damage in alfalfa lowers predicted impact of ozone depletion, Nature, 1992, 358, 576–578 CrossRef CAS.
- S. Mouret, C. Philippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8, 1706–1711 RSC.
- Y. Jiang, M. Rabbi, M. Kim, C. H. Ke, W. Lee, R. L. Clark, P. A. Mieczkowski and P. E. Marszalek, UVA generates pyrimidine dimers in DNA directly, Biophys. J., 2009, 96, 1151–1158 CrossRef CAS.
- Z. Kuluncsics, D. Perdiz, E. Brulay, B. Muel and E. Sage, Wavelength dependence of ultraviolet-induced DNA damage distribution: involvement of direct or indirect mechanisms and possible artefacts, J. Photochem. Photobiol., B, 1999, 49, 71–80 CrossRef CAS.
- J. C. Sutherland and K. P. Griffin, Absorption spectrum of DNA for wavelengths greater than 300 nm, Radiat. Res., 1981, 86, 399–409 CrossRef CAS.
- F. L. Meyskens, P. Farmer and J. P. Fruehauf, Redox regulation in human melanocytes and melanoma, Pigm. Cell Res., 2001, 14, 148–154 Search PubMed.
- J.-L. Ravanat, C. Saint-Pierre, P. Di Mascio, G. R. Martinez, M. H. Medeiros and J. Cadet, Damage to isolated DNA mediated by singlet oxygen, Helv. Chim. Acta, 2001, 84, 3702–3709 CrossRef CAS.
- S. Gidanian, M. Mentelle, F. L. Meyskens and P. J. Farmer, Melanosomal damage in normal human melanocytes induced by UVB and metal uptake - A basis for the pro-oxidant state of melanoma, Photochem. Photobiol., 2008, 84, 556–564 CrossRef CAS.
- L. Marrot, J. P. Belaidi, C. Jones, P. Perez and J. R. Meunier, Molecular responses to stress induced in normal human Caucasian melanocytes in culture by exposure to simulated solar UV, Photochem. Photobiol., 2005, 81, 367–375 CrossRef CAS.
- J. J. Yohn, D. A. Norris, D. G. Yrastorza, I. J. Buno, J. A. Leff, S. S. Hake and J. E. Repine, Disparate antioxidant enzyme activities in cultured human cutaneous fibroblasts, keratinocytes, and melanocytes, J. Invest. Dermatol., 1991, 97, 405–409 CrossRef CAS.
- Q. Y. Wei, J. E. Lee, J. E. Gershenwald, M. I. Ross, P. F. Mansfield, S. S. Strom, L. E. Wang, Z. Z. Guo, Y. W. Qiao, C. I. Amos, M. R. Spitz and M. Duvic, Repair of UV light-induced DNA damage and risk of cutaneous malignant melanoma, J. Natl. Cancer Inst., 2003, 95, 308–315 CrossRef CAS.
Footnotes |
| † Contribution to the themed issue on the biology of UVA. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1: emission spectra of the UV lamps and Table S1: list and expression of studied DNA repair genes.. See DOI: 10.1039/c1pp05185g |
| § This work was supported by a grant from the French “Agence National pour la Recherche” (ANR-07-PCVI-0004-01). |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |