4-thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy†
Olivier
Reelfs
*a,
Peter
Karran
b and
Antony R.
Young
a
aKing's College London, King's College London School of Medicine, Division of Genetics and Molecular Medicine, St John's Institute of Dermatology, London, U.K SE19RT. * Corresponding author. E-mail: olivier.reelfs@kcl.ac.uk; Fax: +44 (0) 207 188 8050; Tel: +44 (0) 207 188 7209
bCancer Research UK London Research Institute, Clare Hall Laboratories, South Mimms, Herts, U.K. EN6 3LD
First published on 2nd November 2011
Abstract
Photochemotherapy, in which ultraviolet radiation (UVR: 280–400 nm) or visible light is combined with a photosensitizing drug to produce a therapeutic effect that neither drug or radiation can achieve alone, is a proven therapeutic strategy for a number of non-malignant hyperproliferative skin conditions and various cancers. Examples are psoralen plus UVA (320–400 nm) radiation (PUVA) and photodynamic therapy (PDT). All existing photochemotherapies have drawbacks – for example the association of PUVA with the development of skin cancer, and pain that is often associated with PDT treatment of skin lesions. There is a clear need to develop alternative approaches that involve lower radiation doses and/or improved selectivity for target cells. In this review, we explore the possibility to address this need by exploiting thionucleoside-mediated DNA photosensitisation to low, non toxic doses of UVA radiation.
 Olivier Reelfs | Olivier Reelfs obtained his PhD from the Swiss Institute for Experimental Cancer Research (ISREC) on the role of iron in solar UVA radiation-induced signal transduction. He has since then continued to investigate the effects of UVA on biomolecules in the context of skin cancer, notably at Cancer Research UK. He has recently focused his research on the study of novel compounds with potential in photochemotherapy. He is currently a senior research fellow at King's College London, with a major interest in DNA damage and photobiology. |
Introduction
Photochemotherapy (Fig. 1) for the treatment of skin disorders has made significant advances in the past three decades. For a long time psoralen plus UVA (320–400 nm) radiation (PUVA) was the treatment of choice for psoriasis but its association with a significant increase of squamous cell carcinoma (SCC) has resulted in a marked decline in its use;1 an increase in melanoma has also been reported.2 The most widely used psoralen in PUVA is 8-methoxypsoralen (8-MOP), which photobinds to DNA resulting in monoadducts and crosslinks that initiate therapeutic responses by immunological mechanisms.3,4 The carcinogenic properties of PUVA could have been predicted from its in vitro photomutagenic properties5 and its photocarcinogenic properties in mice.6 Nevertheless, extra-corporeal PUVA is still widely used, notably for the treatment of cutaneous T-cell lymphoma (CTCL).7 Another example of photochemotherapy is photodynamic therapy (PDT) that, with topical application of the sensitizer combined with visible radiation, is widely used for the treatment of non-melanoma skin cancer.8,9PDT is also increasingly used to treat a range of internal malignancies9,10 and its clinical effects are mediated by the generation of reactive oxygen species (ROS). There is no evidence that PDT is carcinogenic but its use in skin is often associated with severe pain.11,12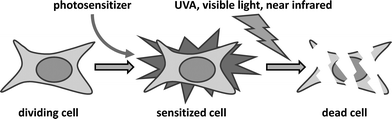 | ||
| Fig. 1 Photochemotherapy. The principle involves the incorporation by target cells, of a molecule (photosensitizer) which sensitizes them to killing by subsequent irradiation with a waveband (UV, visible, or near-infrared) that ideally matches the optical properties of the photosensitizer. | ||
In photochemotherapy, neither drug nor radiation should have any effect on its own. The photosensitizers employed are often not specific for the target tissue. Nevertheless, when combined with the appropriate radiation wavelength and the possibility of localized irradiation (e.g. with optic fibers), photochemotherapy becomes an extremely targeted treatment modality. The demise of PUVA in dermatology means that PDT is rapidly becoming the only photochemotherapeutic option, albeit for different clinical indications. PDT has disadvantages, however. These include pain and adverse reactions, such as persistent photosensitization and a strict dependence on oxygen, which is not always present in tumour tissue.13,14 In view of these drawbacks, it is apparent that there would be advantages in developing new photochemotherapy options. Here we review the photochemical properties of 4-thiothymidine (S4TdR) and the potential exploitation of S4TdR plus UVA in photochemotherapy.
Optical properties of thiobase/nucleosides
The canonical DNA/RNA bases absorb short wavelength UVC (100–280 nm) and UVB (280–320 nm) but have no significant absorbance in the UVA (320–400 nm) region. As a result, DNA is a weak UVA chromophore and is largely insensitive to direct UVA-induced damage. Skin, for example, is about 1000 times less sensitive to UVA than to UVB radiation at a molecular and clinical level.15 Some base analogs do absorb UVA radiation, however, and their incorporation into DNA sensitizes it to direct damage by UVA. The thiobases/nucleosides: 6-thioguanine, 6-mercaptopurine, S4-thymidine (S4TdR), and S4-bromouracil are all UVA chromophores with absorbance maxima around 340 nm and illustrate the general principle that introduction of a sulphur atom into purines or pyrimidines shifts their absorbance maximum into the UVA region. (Fig. 2).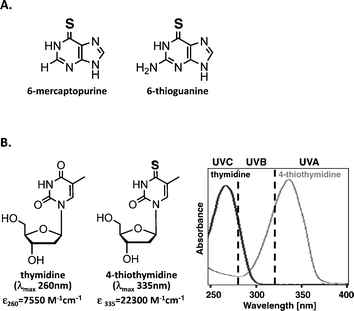 | ||
| Fig. 2 A. Major thiopurines used in clinics as immunosuppressants. B. Thymidine (TdR) and 4-thiothymidine (S4TdR) molecules and their absorption spectra. Replacement of the oxygen atom by a sulphur in position 4 of thymidine results in a bathochromic shift of λmax from UVC (260 nm) to UVA (335 nm), allowing DNA to become a UVA chromophore if S4TdR gets incorporated into its chain. Extinction coefficients of the two molecules at their λmax are indicated. | ||
Metabolism of S4TdR
The introduction of base analogs into DNAviapurine or pyrimidine nucleoside salvage pathways is a common therapeutic strategy. Thymidine and its analogues, such as S4TdR are salvaged by the thymidine kinase (TK) pathway (Fig. 3). All proliferating cells express TK. Its activity is tightly coupled to the cell cycle and increases significantly in S phase. S4TdR can be introduced into the DNA of cultured cells simply by including the thiodeoxynucleoside in the growth medium. TK expression is essential for S4TdR utilization.16 Incorporation of exogenous thymidine analogues is influenced by both the intracellular pool of thymidine nucleotides produced by the de novo synthesis and the extracellular concentration of competitor TdR. The calf serum used for cell culture may contain high and variable concentrations of TdR, and to attain maximal and consistent S4TdR utilization, competing serum TdR is removed by dialysis.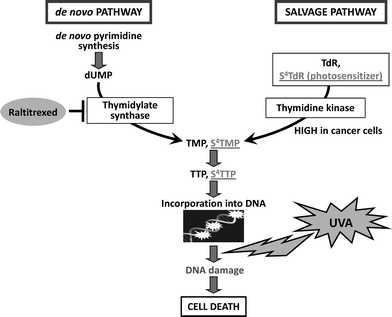 | ||
| Fig. 3 Metabolism of 4-thiothymidine S4TdR is incorporated into DNAvia the salvage pathway. This process is strictly dependent on phosphorylation of the thionucleoside by the enzyme thymidine kinase (TK), which is highly active in cancer cells. | ||
S4TdR incorporation into DNA of cultured cells is generally linear with respect to the external thionucleoside concentration up to around 100 μM. The extent of DNA S4TdR substitution varies among cell lines with maximum levels in cultured human, rat or hamster cells lying between 0.1–2% TdR replacement.16–19
Potential ways to increase S4TdR/UVA toxicity: thymidylate synthase inhibitors
De novo TdR synthesis (Fig. 3) is abrogated by treatment with thymidylate synthase (TS) inhibitors. By forcing cells to rely on salvage for the supply of thymidine nucleotides, this represents a possible strategy to facilitate the incorporation of S4TdR into DNA and further enhancing its effects. The TS inhibitor Tomudex (Raltitrexed or ZD1694, Astra-Zeneca) is currently used to treat advanced colorectal and breast cancers.20In vitro, Raltitrexed enhanced S4TdR incorporation into the DNA of bladder cancer cell lines by a factor of 3–10-fold.18 This promising result warrants further investigation to address efficiency of this drug combination in vivo in animal models. Investigation on the possible use of TS inhibitors, such as 5-fluorouracil (5-FU or Efudix®, Adrucil®) or the folate antimetabolite pemetrexed (Alimta®, Eli Lilly and Company) should also be considered.Cytotoxicity and mutagenicity in the absence of UVA
One important property of S4TdR that distinguishes it from the thiopurine analog 6-TG is its extremely low toxicity. At comparable levels of DNA substitution, 6-TG is highly toxic21 whereas S4TdR is without detectable effect. This has been verified in numerous human cell lines, including Raji Burkitt's lymphoma, HeLa, A2780 ovarian carcinoma, HT1080 fibrosarcoma, RTS3b squamous cell carcinoma, in primary and SV40-transformed human fibroblasts, human keratinocytes and rat bladder carcinoma cells (e.g. MYU-3L). In all cases, growth for at least two cell cycles at concentrations up to 300 μM S4TdR caused no significant growth impairment.16,18,22This lack of cytotoxicity reflects the coding properties of S4TdR and of its methylated derivative S4meTdR. The potential lethality of DNA 6-TG stems from its infrequent non-enzymatic S-methylation by S-adenosylmethionine to form DNA S6-thiomethylguanine (S6meG). Following its replication, this ambiguously coding base engages mismatch repair, an event that results in cell death. DNA S4TdR can also undergo facile S-methylation but this does not provoke processing by mismatch repair and so S4TdR is non-toxic. Massey et al. (2002) suggested that the reason for this lack of cytotoxicity is that neither DNA S4T nor S4meT codes ambiguously.17 Helix thermal stability measurements and studies of coding preferences by primer extension indicated that the highly preferred S4T:A and S4meT:G base pairings retain the thermodynamic and structural stability of canonical T:A and C:G pairs. Formation of S4T:G or S4meT:A during replication (when mismatch repair operates) is extremely improbable and this absence of coding ambiguity is responsible for the inability of DNA S4T or S4meT to engage mismatch repair. This, in turn, underlies the ability of cells to tolerate high levels of DNA S4TdR.
S4TdR is a poor mutagen. Growth of hamster CHO-D422 cells in 100 μM S4TdR increased the frequency of mutation at the APRT gene by only 1.5 fold16 and the absence of mutagenicity has been confirmed in a more recent extensive study.19 It seems likely that this weak mutagenicity reflects the normal coding properties of DNA S4T.
Synergistic toxicity of S4TdR and UVA radiation
S4TdR is a powerful UVA photosensitizer. The ability of cells to proliferate indefinitely in medium containing S4TdR contrasts dramatically to the profound cytotoxicity of S4TdR combined with UVA radiation of wavelength around 340 nm. Maximal DNA substitution achieved by several days growth of MRC5VA human fibroblasts in the presence of S4TdR, reduced the D37 (the dose required to introduce one lethal event per cell) for UVA from 70 kJ m−2 to 0.5 kJ m−2 – a sensitization factor of >100-fold.16 This dramatic UVA sensitization, which has been reproduced in numerous cell lines (Table 1), is dependent on both the UVA dose and on the level of DNA substitution.| Cell name | Cell type | Experimental Condition | Approx.% survival (by colony forming assay) | Reference |
|---|---|---|---|---|
| NHF | primary skin fibroblasts | 100uM S4TdR/10kJ m−2UVA | 10 | 22 |
| NHK | primary skin keratinocytes | idem | 10 | 22 |
| HT1080 | fibrosarcoma | idem | 1 | 22 |
| RTS3b | spontaneously immortalized keratinocytes | idem | 1 | 22 |
| HaCaT | spontaneously immortalized keratinocytes | idem | 2 | 19 |
| MRC5VA | SV40-transformed lung fibroblasts | idem | 0.03 | 16 |
| CHO-D422 | Chinese Hamster Ovary cells | idem | 0.001 | 19 |
| XP12RO | XPA fibroblasts | 100uM S4TdR/0.1kJ m−2UVA | 8 | 16 |
| GM04429f | XPA fibroblasts | idem | 25 | 16 |
| GM08437b | XPF fibroblasts | idem | 25 | 16 |
This synergistic S4TdR/UVA toxicity has an absolute requirement for incorporation of S4TdR into DNA and a TK negative variant of Raji cells is completely insensitive to S4TdR/UVA treatment.16Thymidine kinase activity is up-regulated by a factor of about 10-fold during S-phase23 and, importantly, elevated TK activity is a common property of malignant cells.24 This differential TK activity suggests that S4TdR treatment might selectively photosensitize rapidly dividing cells (e.g. tumour cells) while leaving normal cells relatively unaffected. This possibility is supported by our findings that two established skin-related cell lines with high proliferating fraction (54–57%) were 10 to 100-fold more susceptible to killing by S4TdR/UVA than primary keratinocytes and fibroblasts with a lower proliferative fraction (3–20%)(Fig. 4).
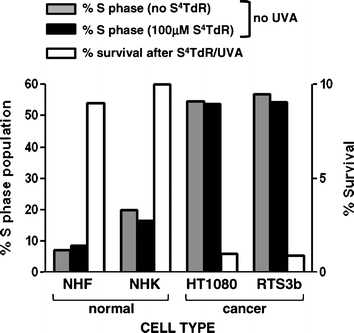 | ||
| Fig. 4 Specific inactivation of fast-dividing cells by S4TdR/UVA S4TdR is incorporated into the DNA during replication and is not cytotoxic on its own. Normal cells have a low population of S phase cells, contrarily to cancer cells. In the presence of UVA, normal cells incubated with S4TdR have a higher survival in contrast to very poor survival of cancer cells. | ||
Photosensitizing mechanism
The UVA energy absorbed by thiobase-substituted DNA can cause damage by Type I photosensitization or it may be transferred to molecular oxygen to generate reactive oxygen species (ROS) in a Type II photosensitizing reaction (Fig. 5).25 Some of these reactions occur when the thiobases are present in DNA. For DNA 6-TG, UVA sensitization is predominantly by an oxygen-dependent Type II reaction involving the formation of singlet oxygen, which has deleterious effects on DNA and proteins26–28 (Attard & Karran, this issue of PPS53).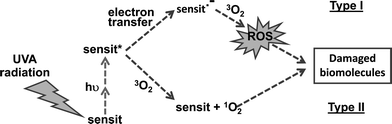 | ||
| Fig. 5 Type I versus Type II photosensitization. | ||
There is currently a paucity of information regarding the mechanism by which DNA S4TdR increases UVA sensitivity. In solution, in the presence of molecular oxygen, 355 nm laser photolysis of S4TdR generates singlet oxygen with a quantum yield of 0.5, suggesting that a Type II mechanism is possible – at least in this experimental context.29 However, UVA irradiation of S4T-containing oligonucleotides generated photolesions more consistent with a ROS-independent mechanism.19,30 In agreement with this possibility, UVA activation of DNA S4TdR in cells did not generate measurable ROS.19 As a consequence, the DNA damage S4TdR/UVA produces appears to be qualitatively different to that caused by high dose UVA alone. Clearly the context and chemical environment of S4TdR plays an important role in determining its photochemical fate. Identification of the DNA S4T photoproducts will help to clarify the mechanism of photosensitization.
Mechanism of cytotoxicity of S4TdR plus UVA
Cells from nucleotide excision repair (NER)-defective xeroderma pigmentosum (XP) patients are hypersensitive to UVC and UVB radiation, both of which cause crosslinking between adjacent DNA pyrimidines to form (6–4) pyrimidine:pyrimidone [(6–4)PP] adducts and cyclobutane pyrimidine dimers (CPD). These cells are generally slightly sensitive to high doses of UVA alone, most likely due to the formation of some CPD by these wavelengths.31,32 Massey et al. (2001) showed that XP complementation group A (XPA) fibroblasts were about 30-fold more sensitive to S4TdR/UVA compared to NER-proficient MRC5VA cells. A rather poorly characterized XPA ‘revertant’ cell line (XP129), acknowledged to have regained UVC resistance together with the ability to excise UVC-induced (6–4)PP, but not CPD33 exhibited almost wild-type S4TdR/UVA sensitivity.16 These findings suggested that intrastrand DNA lesions that resemble (6–4)PP and are excised by NER are significant contributors to S4TdR/UVA-induced cell death. Consistent with this possibility, early chemical studies showed that UVA irradiation of oligodeoxynucleotides containing a S4T placed 3′ to a T generated a thietane photoproduct, which bears a strong resemblance to UVB-type (6–4)PP adduct. There are, however, important differences between these photoproducts (Fig. 6).30,34 The equilibrium between the ring-closed thietane and its open ring (6–4) isomer (S5-(6–4)T:T) favours the ring-closed structure,35–37 whereas its oxygen-containing oxetane homolog is overwhelmingly in the ring open form.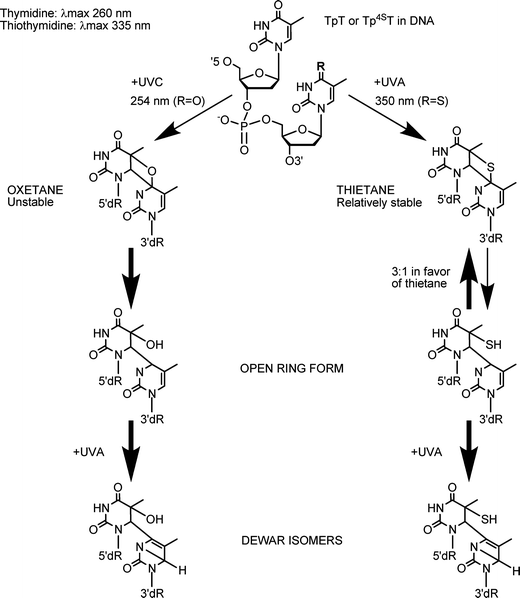 | ||
Fig. 6 Photochemistry of TpT or Tp4ST dimers upon UV irradiation (adapted from ref. 30,36,37). Irradiation of TpT or Tp4ST dimers with UVC and UVA radiation generates either an oxetane or a thietane molecule, respectively. Whereas the oxetane is highly unstable and spontaneously converts into its ring-open form, the thietane is relatively stable and exists in an equilibrium with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in favour of the thietane to its ring-open form. Further irradiation of the ring-open forms of (6–4)T:T and S5-(6–4)T:T with 300 nm converts them into their respective Dewar isomers. :1 ratio in favour of the thietane to its ring-open form. Further irradiation of the ring-open forms of (6–4)T:T and S5-(6–4)T:T with 300 nm converts them into their respective Dewar isomers. | ||
The thietane exhibits fluorescence (Ex. 320 nm, Em. 370 nm) and a fluorescent photoproduct with the expected properties of S5-(6–4)T:T was recently identified in DNA from human cells treated with S4TdR and UVA.19HaCaT keratinocytes excised this photoproduct efficiently. Nevertheless, it was more persistent than the canonical (6–4) Py:Py lesions. Removal of S5-(6–4)T:T adducts was even slower in CCRF-CEM leukemia cells in which most of them persisted for up to 24 h. Rapid repair of (6–4) Py:Py lesions reflects the significant DNA distortion they create. We hypothesize that the relative persistence of S5-(6–4)T:T reflects a less pronounced DNA distortion. This, in turn, may be a consequence of the predominance of a ring-closed form.
Both ring-open oxetane and the thietane forms readily convert into their Dewar valence photoisomer (Dewar-PP) following further exposure to UVA radiation. The UVA of sunlight is the waveband range responsible for photoisomerisation of the canonical (6–4)PP in cells.38 Interestingly, canonical Dewar-PP are not repaired at the same rates in different cells and/or species: In hamster CHO cells, the rate were slow and similar to repair of CPD,38 while in human fibroblasts the repair was slightly faster39 and in human keratinocytes, comparable to (6–4)PP repair.40 The fate of DNA thietane-related Dewar-PP photoproducts in cells remains to be determined.
The extreme sensitivity of NER-deficient cells and cells with defects in homologous recombination (HR) or in the processing of interstrand crosslinks (ICL)19 indicates that the cytotoxicity of S4TdR/UVA depends on its ability to induce bulky DNA lesions that are severe impediments to DNA replication. We identified two possible candidate DNA lesions: ICL and S5-(6–4)T:T adducts, but the formation of additional potentially cytotoxic DNA lesions has not been ruled out. DNA–protein crosslinks are formed following UVA irradiation of S4TdR-containing oligonucleotides41,42 and DNA from S4TdR/UVA-treated bladder cancer cells.18 Although little is known about how DNA–protein crosslinks are repaired, there are suggestions that relatively small adducts can be removed by a mechanism that is partially dependent on NER.43,44 Double strand breaks (DSB) were generated in S4TdR/UVA-treated HaCaT cells,19 consistent with previous observations (B. Montaner, P. Karran, unpublished) that DNA S4TdR photoactivation strongly inhibits DNA replication. Of note, inhibition of DNA replication by other thio-containing photoproducts has also been reported.45 Despite acting as a UVA sensitizer, DNA S4TdR did not detectably increase the yield of DNA single strand breaks, oxidized guanine and CPD in HaCaT cells even under conditions that are supralethal and result in <1% cell survival.19
A better understanding of the precise mechanism of S4TdR/UVA-mediated toxicity and its possible repair awaits direct chemical characterization and quantitation of S4T DNA photoproducts.
S4TdR/UVA-induced cell death
In experiments with human fibrosarcoma cells and spontaneously immortalized keratinocytes, S4TdR/UVA induced cell death within 24 h. The associated morphologic changes, membrane blebbing and cell fragmentation were consistent with death by apoptosis, as was the appearance of Annexin V positive cells and cleaved caspase 3.22Death occurred to similar extent and kinetics in different cell types. Levels of the proapoptotic proteins Bax and Bak were not detectably changed by S4TdR/UVA treatment, although a conformational conversion of the Bak protein into an active form – a mark of apoptosis induction,46 occurred in a majority of cells. None of these molecular changes occurred when cells were treated with S4TdR or UVA alone, therefore confirming that S4TdR/UVA toxicity is strictly dependent on the photoactivation of the thionucleoside in DNA. Further investigation is needed to elucidate the exact mechanism of S4TdR/UVA-induced apoptosis, and to determine whether additional/alternative pathways of cell death (e.g. necrosis) are induced.The same study revealed that S4TdR/UVA treatment caused p53 stabilization and induced downstream components of the p53 DNA damage response. Nevertheless, S4TdR/UVA-induced toxicity was largely p53-independent and isogenic cell lines expressing either E6 protein from oncogenic human papilloma virus (HPV) 18, known to target p53 for destruction, or HPV5, which does not promote p53 degradation, exhibited similar sensitivity.22 This property of inducing cell death in a p53-independent manner represents a key advantage for a treatment aimed at UVA-accessible cancers, such as non-melanoma skin cancer (NMSC), in which p53 is often mutated or absent.47,48
Animal models
Investigations to date indicate that S4TdR/UVA is weakly mutagenic in comparison to PUVA.19,49 The combination of high toxicity, low mutagenicity and selectivity for dividing cells suggests that S4Td/UVA might be an effective and safe treatment for cancers that are associated with relatively rapid proliferation. Encouraging data has recently emerged from a preliminary investigation using a rat bladder carcinoma model.18 In these experiments cancer cells are instilled directly into the bladder where they normally form tumours over the course of five days. Intravenous administration of S4TdR resulted in a measurable incorporation of S4TdR into the tumor DNA. Significantly, this was around 3-fold higher than the incorporation into normal bladder epithelium. When intravenous (iv) S4TdR was combined with UVA delivered to the bladder via a fibre optic device inserted through the urethra, tumour growth was prevented in 3/5 animals. This compares to tumor growth in 20/20 control animals.With respect to potential post-treatment tissue photosensitivity associated with systemic S4TdR treatment, it is noteworthy that two hours after intravenous administration of S4TdR, DNA substitution in rat skin and eye was at least 10-fold lower than the levels in other normal tissues. This had decreased to undetectable levels by 20 h. This short persistence of S4TdR in the DNA of tissues naturally exposed to solar UV, if replicated in humans, would compare favourably with the several week-long persistent photosensitivity associated with PDT treatment. This study represents the first use of S4TdR/UVA in an animal model of cancer and the encouraging findings pave the way to further investigations on the possible use of this treatment for other types of hyperproliferative conditions, including cancer.
Conclusions
In vitro and animal studies suggest that S4TdR has potential as a photochemotherapeutic drug. Its specific incorporation into the DNA of replicating cells suggests a highly targeted destruction of tumour cells, with minimal collateral damage to normal cells. The low mutagenicity of combined S4TdR/UVA compared to PUVA indicates that it may not represent a great risk for unwanted therapy-related late effects. Although the animal studies must be regarded as preliminary, they are encouraging enough to warrant confirmation with larger groups of animals and extension to other cancer models.From a practical point of view, the development of S4TdR/UVA photochemotherapy should include the development of custom-made UVA sources, taking into account possible changes in optical properties of S4TdR when it gets incorporated into the DNA. Indeed, the emission spectrum of any source used in photochemotherapy should, ideally, match the absorption spectrum of the active molecule or any complexes it forms with biomolecules. This principle has not been applied in the empirical development of PUVA, which has used sources peaking at about 360 nm when action spectrum studies would indicate peak activity at about 330 nm.50,51 Similarly, for PDT, a wide range of sources are used and it is clear that they are often optically sub-optimal.52 For S4TdR, typically, the use of sources that peak at about 340 nm but extend into the longer wavelength UVA1 region, which penetrates deep into normal dermis (Tewari et al., this issue of PPS54) will have to be investigated.
Acknowledgements
We are grateful to Prof Alan Boddy and Dr Simon Pridgeon from the Northern Institute for Cancer Research (Newcastle upon Tyne, U.K.) for sharing their results on their in vivo animal model, prior to publication. We thank the British Skin Foundation and Cancer Research UK for supporting this work which was also supported by the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's & St Thomas’ NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust.References
- R. S. Stern and E. J. Lunder, Risk of squamous cell carcinoma and methoxsalen (psoralen) and UV-A radiation (PUVA) - A meta-analysis, Arch. Dermatol., 1998, 134, 1582–1585 Search PubMed.
- R. S. Stern and P. F. U. Study, The risk of melanoma in association with long-term exposure to PUVA, J. Am. Acad. Dermatol., 2001, 44, 755–761 CrossRef CAS.
- R. S. Stern, Psoralen and ultraviolet a light therapy for psoriasis, N. Engl. J. Med., 2007, 357, 682–690 Search PubMed.
- T. P. Singh, B. Huettner, H. Koefeler, G. Mayer, I. Bambach, K. Wallbrecht, M. P. Schon and P. Wolf, Platelet-Activating Factor Blockade Inhibits the T-Helper Type 17 Cell Pathway and Suppresses Psoriasis-Like Skin Disease in K5.hTGF-beta 1 Transgenic Mice, Am. J. Pathol., 2011, 178, 699–708 Search PubMed.
- D. Papadopoulo, F. Sagliocco and D. Averbeck, Mutagenic Effects of 3-Carbethoxypsoralen and 8-Methoxypsoralen Plus 365-nm Irradiation in Mammalian-Cells, Mutat. Res., Genet. Toxicol., 1983, 124, 287–297 Search PubMed.
- A. R. Young, I. A. Magnus, A. C. Davies and N. P. Smith, A Comparison of the Phototumorigenic Potential of 8-MOP and 5-MOP in Hairless Albino Mice Exposed to Solar Simulated Radiation, Br. J. Dermatol., 1983, 108, 507–518 Search PubMed.
- R. Knobler, M. L. Barr, D. R. Couriel, J. L. M. Ferrara, L. E. French, P. Jaksch, W. Reinisch, A. H. Rook, T. Schwarz and H. Greinix, Extracorporeal photopheresis: Past, present, and future, J. Am. Acad. Dermatol., 2009, 61, 652–665 Search PubMed.
- P. Babilas, S. Schreml, M. Landthaler and R. M. Szeimies, Photodynamic therapy in dermatology: state-of-the-art, Photodermatol., Photoimmunol. Photomed., 2010, 26, 118–132 Search PubMed.
- D. Fayter, M. Corbett, M. Heirs, D. Fox and A. Eastwood, A systematic review of photodynamic therapy in the treatment of pre-cancerous skin conditions, Barrett's oesophagus and cancers of the biliary tract, brain, head and neck, lung, oesophagus and skin, Health Technol. Assess., 2010, 14, 1–288 Search PubMed.
- J. H. Pinthus, A. Bogaards, R. Weersink, B. C. Wilson and J. Trachtenberg, Photodynamic therapy for urological malignancies: Past to current approaches, J. Urol., 2006, 175, 1201–1207 Search PubMed.
- S. K. Attili, R. Dawe and S. Ibbotson, A review of pain experienced during topical photodynamic therapy – Our experience in Dundee, Photodiagn. Photodyn. Ther., 2011, 8, 53–57 Search PubMed.
- S. H. Ibbotson, Adverse effects of topical photodynamic therapy, Photodermatol., Photoimmunol. Photomed., 2011, 27, 116–130 Search PubMed.
- A. M. Bugaj, Targeted photodynamic therapy - a promising strategy of tumor treatment, Photochem. Photobiol. Sci., 2011, 10, 1097–1109 RSC.
- M. Verhille, P. Couleaud, R. Vanderesse, D. Brault, M. Barberi-Heyob and C. Frochot, Modulation of Photosensitization Processes for an Improved Targeted Photodynamic Therapy, Curr. Med. Chem., 2010, 17, 3925–3943 CrossRef CAS.
- A. R. Young, C. A. Chadwick, G. I. Harrison, O. Nikaido, J. Ramsden and C. S. Potten, The similarity of action spectra for thymine dimers in human epidermis and erythema suggests that DNA is the chromophore for erythema, J. Invest. Dermatol., 1998, 111, 982–988 CrossRef CAS.
- A. Massey, Y.-Z. Xu and P. Karran, Photoactivation of DNA thiobases as a potential novel therapeutic option, Curr. Biol., 2001, 11, 1142–1146 CrossRef CAS.
- A. Massey, Y. Z. Xu and P. Karran, Ambiguous coding is required for the lethal interaction between methylated DNA bases and DNA mismatch repair, DNA Repair, 2002, 1, 275–286 Search PubMed.
- S. W. Pridgeon, R. Heer, G. A. Taylor, D. R. Newell, K. O'Toole, M. Robinson, Y. Z. Xu, P. Karran and A. V. Boddy, Thiothymidine combined with UVA as a potential novel therapy for bladder cancer, Br. J. Cancer, 2011, 104, 1869–1876 Search PubMed.
- O. Reelfs, P. MacPherson, X. Ren, Y.-Z. Xu, P. Karran and A. Young, Identification of potentially cytotoxic lesions induced by UVA photoactivation of DNA 4-thiothymidine in human cells, Nucleic Acids Res., 2011 DOI:10.1093/nar/gkr674.
- D. Cunningham, J. R. Zalcberg, U. Rath, I. Olver, E. Van Cutsem, C. Svensson, J. F. Seitz, P. Harper, D. Kerr and G. Perez-Manga et al., ‘Tomudex’ (ZD1694): results of a randomised trial in advanced colorectal cancer demonstrate efficacy and reduced mucositis and leucopenia. The ‘Tomudex’ Colorectal Cancer Study Group, Eur. J. Cancer, 1995, 31A, 1945–1954 Search PubMed.
- P. F. Swann, T. R. Waters, D. C. Moulton, Y. Z. Xu, Q. Zheng, M. Edwards and R. Mace, Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine, Science, 1996, 273, 1109–1111 Search PubMed.
- O Reelfs, Y. Z. Xu, A. Massey, P. Karran and A. Storey, Thiothymidine plus low-dose UVA kills hyperproliferative human skin cells independently of their human papilloma virus status, Mol. Cancer Ther., 2007, 6, 2487–2495 CrossRef.
- J. L. Sherley and T. J. Kelly, Regulation of human thymidine kinase during the cell cycle, J. Biol. Chem., 1988, 263, 8350–8358 CAS.
- M. Hengstschlager, M. Pfeilstocker and E. Wawra, Thymidine kinase expression. A marker for malignant cells, Adv. Exp. Med. Biol., 1998, 431, 455–460 Search PubMed.
- J. Cadet, T. Douki, J. L. Ravanat and P. Di Mascio, Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation, Photochem. Photobiol. Sci., 2009, 8, 903–911 RSC.
- P. O'Donovan, C. M. Perrett, X. Zhang, B. Montaner, Y. Z. Xu, C. A. Harwood, J. M. McGregor, S. L. Walker, F. Hanaoka and P. Karran, Azathioprine and UVA light generate mutagenic oxidative DNA damage, Science, 2005, 309, 1871–1874 CrossRef CAS.
- B. Montaner, P. O'Donovan, O. Reelfs, C. M. Perrett, X. Zhang, Y. Z. Xu, X. Ren, P. Macpherson, D. Frith and P. Karran, Reactive oxygen-mediated damage to a human DNA replication and repair protein, EMBO Rep., 2007, 8, 1074–1079 CrossRef CAS.
- X. Zhang, G. Jeffs, X. Ren, P. O'Donovan, B. Montaner, C. M. Perrett, P. Karran and Y. Z. Xu, Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light, DNA Repair, 2007, 6, 344–354 CrossRef CAS.
- Y. Harada, T. Suzuki, T. Ichimura and Y. Z. Xu, Triplet formation of 4-thiothymidine and its photosensitization to oxygen studied by time-resolved thermal lensing technique, J. Phys. Chem. B, 2007, 111, 5518–5524 CrossRef CAS.
- M. A. Warren, J. B. Murray and B. A. Connolly, Synthesis and characterisation of oligodeoxynucleotides containing thio analogues of (6-4) pyrimidine-pyrimidinone photo-dimers, J. Mol. Biol., 1998, 279, 89–100 CrossRef CAS.
- C. Kielbassa and B. Epe, DNA damage induced by ultraviolet and visible light and its wavelength dependence, Methods Enzymol., 2000, 319, 436–445 CAS.
- T. Douki, A. Reynaud-Angelin, J. Cadet and E. Sage, Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42, 9221–9226 CrossRef CAS.
- J. E. Cleaver, F. Cortes, L. H. Lutze, W. F. Morgan, A. N. Player and D. L. Mitchell, Unique DNA repair properties of a xeroderma pigmentosum revertant, Mol. Cell Biol., 1987, 7, 3353–3357 Search PubMed.
- A. Favre, C. Saintome, J. L. Fourrey, P. Clivio and P. Laugaa, Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid-protein interactions, J. Photochem. Photobiol., B, 1998, 42, 109–124 CrossRef CAS.
- R. O. Rahn and J. L. Hosszu, Photochemical Studies of Thymine in Ice, Photochem. Photobiol., 1969, 10, 131–137 CrossRef CAS.
- P. Clivio, J. L. Fourrey, J. Gasche and A. Favre, DNA Photodamage Mechanistic Studies - Characterization of a Thietane Intermediate in a Model Reaction Relevant to 6-4 Lesions, J. Am. Chem. Soc., 1991, 113, 5481–5483 CrossRef CAS.
- P. Clivio, J. L. Fourrey, J. Gasche and A. Favre, Novel Insight into the Stereochemical Pathway Leading to (6-4) Pyrimidine-Pyrimidone Photoproducts in DNA, Tetrahedron Lett., 1992, 33, 1615–1618 CrossRef CAS.
- D. Perdiz, P. Grof, M. Mezzina, O. Nikaido, E. Moustacchi and E. Sage, Distribution and repair of bipyrimidine photoproducts in solar UV-irradiated mammalian cells. Possible role of Dewar photoproducts in solar mutagenesis, J. Biol. Chem., 2000, 275, 26732–26742 CAS.
- B. S. Rosenstein and D. L. Mitchell, The repair of DNA damages induced in normal human skin fibroblasts exposed to simulated sunlight, Radiat. Res., 1991, 126, 338–342 Search PubMed.
- S. Courdavault, C. Baudouin, M. Charveron, B. Canguilhem, A. Favier, J. Cadet and T. Douki, Repair of the three main types of bipyrimidine DNA photoproducts in human keratinocytes exposed to UVB and UVA radiations, DNA Repair, 2005, 4, 836–844 CrossRef CAS.
- B. Bartholomew, B. R. Braun, G. A. Kassavetis and E. P. Geiduschek, Probing close DNA contacts of RNA polymerase III transcription complexes with the photoactive nucleoside 4-thiodeoxythymidine, J. Biol. Chem., 1994, 269, 18090–18095 Search PubMed.
- T. T. Nikiforov and B. A. Connolly, Oligodeoxynucleotides containing 4-thiothymidine and 6-thiodeoxyguanosine as affinity labels for the Eco RV restriction endonuclease and modification methylase, Nucleic Acids Res., 1992, 20, 1209–1214 CAS.
- J. T. Reardon and A. Sancar, Repair of DNA-polypeptide crosslinks by human excision nuclease, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4056–4061 Search PubMed.
- T. Nakano, A. Katafuchi, M. Matsubara, H. Terato, T. Tsuboi, T. Masuda, T. Tatsumoto, S. P. Pack, K. Makino, D. L. Croteau, B. Van Houten, K. Iijima, H. Tauchi and H. Ide, Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells, J. Biol. Chem., 2009, 284, 27065–27076 Search PubMed.
- A. Ito, F. T. Robb, J. G. Peak and M. J. Peak, Base-specific damage induced by 4-thiouridine photosensitization with 334-nm radiation in M13 phage DNA, Photochem. Photobiol., 1988, 47, 231–240 CAS.
- M. C. Wei, T. Lindsten, V. K. Mootha, S. Weiler, A. Gross, M. Ashiya, C. B. Thompson and S. J. Korsmeyer, tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c, Genes Dev., 2000, 14, 2060–2071 CAS.
- D. E. Brash, J. A. Rudolph, J. A. Simon, A. Lin, G. J. McKenna, H. P. Baden, A. J. Halperin and J. Ponten, A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 10124–10128 CAS.
- A. Ziegler, D. J. Leffell, S. Kunala, H. W. Sharma, M. Gailani, J. A. Simon, A. J. Halperin, H. P. Baden, P. E. Shapiro and A. E. Bale et al., Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4216–4220 CAS.
- E. Sage, E. A. Drobetsky and E. Moustacchi, 8-Methoxypsoralen induced mutations are highly targeted at crosslinkable sites of photoaddition on the non-transcribed strand of a mammalian chromosomal gene, EMBO J., 1993, 12, 397–402 Search PubMed.
- A. R. Young and I. A. Magnus, An Action Spectrum for 8-MOP Induced Sunburn Cells in Mammalian Epidermis, Br. J. Dermatol., 1981, 104, 541–548 CAS.
- K. H. Kaidbey, An Action Spectrum for 8-Methoxypsoralen-Sensitized Inhibition of DNA-Synthesis In vivo, J. Invest. Dermatol., 1985, 85, 98–101 Search PubMed.
- R. M. Sayre, J. C. Dowdy and R. W. Gottschalk, Comparative effectiveness of clinically used light sources for cutaneous protoporphyrin IX-based photodynamic therapy, J. Cosmetic Laser Ther., 2011, 13, 63–68 Search PubMed.
- N. R. Attard and P. Karran, UVA photosensitization of thiopurines and skin cancer in organ transplant recipients, Photochem. Photobiol. Sci., 2012 10.1039/C1PP05194F.
- A. Tewari, C. Lahmann, R. Sarkany, J. Bergemann and A. R. Young, Human erythema and matrix metalloproteinase-1 mRNA induction, in vivo, share an action spectrum which suggests common chromophores, Photochem. Photobiol. Sci., 2012 10.1039/C1PP05243H.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |