Autophagic-lysosomal dysregulation downstream of cathepsin B inactivation in human skin fibroblasts exposed to UVA†
Sarah D.
Lamore
and
Georg T.
Wondrak
*
Department of Pharmacology and Toxicology, College of Pharmacy & University of Arizona, Arizona Cancer Center, 1515 North Campbell Avenue, Tucson, AZ 85724, USA. E-mail: wondrak@pharmacy.arizona.edu; Fax: +1 520-626-3797; Tel: +1 520-626-9017
First published on 20th July 2011
Abstract
Recently, using 2D-DIGE proteomics we have identified cathepsin B as a novel target of UVA in human Hs27 skin fibroblasts. In response to chronic exposure to noncytotoxic doses of UVA (9.9 J cm−2, twice a week, 3 weeks), photooxidative impairment of cathepsin B enzymatic activity occurred with accumulation of autofluorescent aggregates colocalizing with lysosomes, an effect mimicked by pharmacological antagonism of cathepsin B using the selective inhibitor CA074Me. Here, we have further explored the mechanistic involvement of cathepsin B inactivation in UVA-induced autophagic-lysosomal alterations using autophagy-directed PCR expression array analysis as a discovery tool. Consistent with lysosomal expansion, UVA upregulated cellular protein levels of the lysosomal marker glycoprotein Lamp-1, and increased levels of the lipidated autophagosomal membrane constituent LC3-II were detected. UVA did not alter expression of beclin 1 (BECN1), an essential factor for initiation of autophagy, but upregulation of p62 (sequestosome 1, SQSTM1), a selective autophagy substrate, and α-synuclein (SNCA), an autophagic protein substrate and aggresome component, was observed at the mRNA and protein level. Moreover, UVA downregulated transglutaminase-2 (TGM2), an essential enzyme involved in autophagolysosome maturation. Strikingly, UVA effects on Lamp-1, LC3-II, beclin 1, p62, α-synuclein, and transglutaminase-2 were mimicked by CA074Me treatment. Taken together, our data suggest that UVA-induced autophagic-lysosomal alterations occur as a consequence of impaired autophagic flux downstream of cathepsin B inactivation, a novel molecular mechanism potentially involved in UVA-induced skin photodamage.
Introduction
Cutaneous exposure to solar UVA radiation (320–400 nm), representing most (>95%) of the solar UV energy incident on human skin, has been identified as a causative factor involved in skin photodamage. Even though solar UVA radiation results in little photoexcitation of DNA directly, it may be involved in cutaneous photoaging and photocarcinogenesis through induction of photooxidative stress mediated by reactive oxygen species (ROS).1–7 However, in spite of the emerging role of UVA in cutaneous photooxidative stress, the identity of specific molecular targets modulated by UVA and causatively involved in solar skin cell damage remains largely undefined.Expression of cathepsins, a family of proteases involved in lysosomal protein degradation and numerous other intra- and extracellular activities, plays an important role in skin structure and function including hair follicle morphogenesis, epidermal differentiation, wound healing, and MHC-II-mediated antigen presentation.8–10 Importantly, molecular changes affecting cathepsins have been identified as causative factors in various skin pathologies such as tumorigenesis and inflammatory dysregulation.11–14
Recent research has focused on the mechanistic involvement of cathepsins in UV-induced cutaneous alterations and photodamage. Specifically, a role of cathepsin K in solar elastosis has been substantiated in human skin fibroblasts where an age-related decline in cathepsin K maturation was shown to compromise the process of orderly intracellular elastin degradation leading to subsequent accumulation of elastin in the extracellular space.15 Stimulation of cathepsin G production in response to UVA was observed in dermal fibroblasts potentially contributing to the remodeling of elastotic areas in sun-damaged skin.16 Moreover, UVA exposure causes alternate trafficking of cathepsin L in dermal fibroblasts leading to extracellular release, an effect that has been linked to the anti-fibrotic activity of repetitive cutaneous UVA exposure.17
Recently, we have identified the lysosomal cysteine protease cathepsin B as a novel target of UVA-induced photooxidative stress in cultured human Hs27 skin fibroblasts, a finding consistent with other research suggesting a mechanistic involvement of UVA-induced cathepsin B impairment in skin photoaging.18–20 In fibroblasts chronically exposed to non-cytotoxic doses of solar simulated UVA, our 2D-DIGE (differential-in-gel-electrophoresis)/mass spectrometric analysis identified cathepsin B as the protein displaying the most pronounced downregulation.18 UVA-induced loss of intracellular specific enzymatic activity of cathepsin B was accompanied by extensive lysosomal accumulation of lipofuscin-like autofluorescence, molecular changes that were suppressed in part by antioxidant treatment during UVA exposure. Interestingly, pharmacological inhibition of cathepsin B mimicked UVA-induced cellular changes including lysosomal expansion with accumulation of autofluorescence and deficient cathepsin B protein maturation.
Here we have further explored the molecular changes associated with UVA-induced lysosomal impairment using autophagy-focused gene expression array analysis followed by immunoblot detection of key proteins mechanistically involved in the lysosomal-autophagic pathway. Our expression array data confirm a pronounced similarity between the molecular effects caused by UVA exposure and pharmacological inhibition of cathepsin B, suggesting that functional impairment of cathepsin B is a heretofore unrecognized causative factor in cutaneous UVA photodamage that operates upstream of autophagic-lysosomal dysregulation.
Materials and methods
Chemicals
CA074Me was purchased from Enzo Life Sciences (Plymouth Meeting, PA), 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI), and LysoTracker Red™ were purchased from Invitrogen (Carlsbad, CA). All other chemicals were from Sigma Chemical Co. (St. Louis, MO).Cell culture
Dermal neonatal foreskin Hs27 fibroblasts from ATCC (Manassas, VA) were cultured in DMEM containing 10% bovine calf serum. Cells were maintained at 37 °C in 5% CO2, 95% air in a humidified chamber.Irradiation with solar simulated UVA
A KW large area light source solar simulator, model 91293, from Oriel Corporation (Stratford, CT) was used, equipped with a 1000 W xenon arc lamp power supply, model 68920, and a VIS-IR bandpass blocking filter plus UVB and C blocking filter (output 320–400 nm plus residual 650–800 nm). The output was quantified using a dosimeter from International Light Inc. (Newburyport, MA), model IL1700, with a SED033 detector for UVA (range 315–390 nm, peak 365 nm). Using the UVB/C blocking filter, the dose at 365 mm from the source, which was used for all experiments, was 5.39 mJ cm−2 s−1UVA radiation with a residual UVB dose of 3.16 μJ cm−2 s−1.For chronic UVA treatment, an exposure regimen was selected that delivered a physiologically relevant dose of UVA without causing compromised cell viability or altered proliferative rate after reseeding:
‘Three week’ UVA regimen: Cells were exposed to 9.9 J cm−2UVA (30 min exposure time) twice a week for a total of 18 days (59.4 J cm−2 total UVA dose).
‘One week’ UVA regimen: Cells were exposed to 9.9 J cm−2UVA (30 min exposure time) for four consecutive days (39.6 J cm−2 total UVA dose).
For UVB, a bank of two FS20T12 UVB bulbs (National Biological Corp., Beachwood, OH) was used, fitted with Kodacel filters (Eastman Kodak, Rochester, NY) to eliminate any residual UVC. The output was quantified using a UVX radiometer with a detector for UVB (UVP, Inc., Upland, CA) as 0.47 mJ cm−2 s−1UVB radiation. ‘One week’ UVB regimen: Cells were exposed to 25 mJ cm−2UVB for four consecutive days (100 mJ cm−2 total UVB dose).
For all regimens, cells were seeded at 5 × 105cells per 100 mm dish and incubated overnight prior to the first UV exposure. Before each irradiation, cells were first washed with PBS and then irradiated under PBS. After irradiation, PBS was removed and fresh culture medium was added. For all analyses, cells were harvested 1 h after the last irradiation.
Cathepsin B enzymatic activity
Cathepsin B activity was measured using a fluorimetric cathepsin B assay kit employing the cathepsin B substrate Ac-Arg-Arg-AFC (200 μM final concentration; 1 h at 37 °C; BioVision, Inc., Mountain View, CA) according to manufacturer's instructions as published recently.18 As a negative control, analysis was performed in the presence of the cathepsin B inhibitor Z-Phe-Phe-FMK (200 μM final concentration). The release of free amino-4-trifluoromethylcoumarin (AFC) was measured using a fluorescence plate reader (λex 400 nm, λem 505 nm; SpectraMax Gemini, Molecular Devices, Sunnyvale, CA). Protein concentration of cell lysates was determined using the Pierce™ BCA Protein Assay Kit (ThermoScientific, Rockford, IL), and cathepsin B activity was normalized to protein concentration.Flow cytometric analysis of cell viability
Cell viability was determined using flow cytometric analysis of annexinV-PI stained cells using an apoptosis detection kit (APO-AF, Sigma, St. Louis, MO) according to the manufacturer's specifications.21Detection of intracellular oxidative stress by flow cytometric analysis
Induction of intracellular oxidative stress by photosensitization was analyzed by flow cytometry using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) according to a published standard procedure.21 One hour after the last irradiation, DCFH-DA (5 μg mL−1 concentration) was added to the culture medium and cells were incubated for 1 h in the dark (37 C, 5% CO2). Cells were harvested, washed with PBS, resuspended in 300 μL PBS and immediately analyzed by flow cytometry.Flow cytometric quantification of cellular autofluorescence
One hour after the last irradiation or CA074Me treatment, cells were harvested, washed with PBS, resuspended in 300 μL PBS, and immediately analyzed by flow cytometry (λex 488 nm, λem 530 ± 15 nm).Confocal fluorescence microscopy
Cells were trypsinized, reseeded on glass bottom 35 mm dishes (MatTek Corp., Ashland, MA) at 1 × 105cells per dish and cultured overnight. Prior to live imaging, cells were incubated in LysoTracker Red DND-99 (75 nM in growth medium) for 1 h at 37 °C/5% CO2. Medium was removed and cells were incubated in DAPI (3 μM in HBSS) for 30 min. Cells were then washed several times and kept in HBSS for fluorescence microscopy. Using a SP5 spectral confocal microscopy system equipped with a Leica DMI6000 inverted microscope (Wetzlar, Germany), DAPI was detected between 450–550 nm with excitation at 405 nm (UV laser source). Autofluorescence and LysoTracker Red were visualized using an argon laser (λex 488 nm), and a spectral scan (560–700 nm with 10 nm increments) was performed. Image analyses were performed using Leica Confocal Imaging software and distinction between fluorescent signals was accomplished by spectral separation.Transmission electron microscopy
Cells were trypsinized, reseeded and cultured for 4 h. Cells were fixed in situ with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4), postfixed in 1% osmium tetroxide in cacodylate buffer, washed, scraped and pelleted. Cells were then stained in 2% aqueous uranyl acetate, dehydrated through a graded series (50, 70, 90 and 100%) of ethanol and infiltrated with Spurr's resin, then allowed to polymerize overnight at 60 °C. Sections (50 nm) were cut, mounted onto uncoated 150 mesh copper grids, and stained with 2% lead citrate. Sections were examined in a CM12 Transmission Electron Microscope (FEI, Hillsboro, OR) operated at 80 kV with digital image collection (AMT, Danvers, MA).Gene expression array analysis
Total cellular RNA (5 × 106cells) was prepared using the RNeasy kit from Qiagen (Valencia, California). Reverse transcription was performed using the RT2 First Strand kit (SA Biosciences, Frederick, MD) and 1 μg total RNA. Expression array analysis using the Human Autophagy RT2 Profiler™ PCR Expression Array or the Human Stress and Toxicity RT2 Profiler™ PCR Expression Array (SA Biosciences), each profiling the expression of 84 genes, was performed as published recently.22 In addition, LAMP1, not contained in either array was measured individually with each PCR reaction consisting of 3.75 μl of cDNA added to 12.5 μl of TaqMan Universal PCR Master Mix (Roche Molecular Systems), 1.25 μl of gene-specific primer/probe mix [Applied Biosystems: LAMP1 (assay ID Hs00174766_m1), GAPDH(assay ID Hs99999905_m1)] and 7.5 μl of PCR water. All reactions were run using the following PCR conditions: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s alternating with 60 °C for 1 min (Applied Biosystems 7000 SDS, Foster City, CA). Gene-specific product was normalized to GAPDH and quantified using the comparative (ΔΔCt) Ct method as described in the ABI Prism 7000 sequence detection system user guide.Immunoblot detection
Cells were lysed in 1× SDS-PAGE sample buffer and heated for 3 min at 95 °C. Samples were separated by 12% SDS-PAGE followed by transfer to nitrocellulose membranes (Optitran, Whatman, Piscataway, NJ). Membranes were incubated with primary antibody in 5% milk-PBST overnight at 4 °C. HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibody (Jackson Immunological Research, West Grove, PA) was used at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20000 in 5% milk-PBST followed by visualization using enhanced chemiluminescence detection reagents. Equal protein loading was examined by β-actin detection. The following primary antibodies were used: rabbit anti-Lamp-1 monoclonal antibody, 1:1000 (Cell Signaling Technology, Danvers, MA); mouse anti-beclin 1 monoclonal antibody, 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-sequestosome 1 (p62) monoclonal antibody, 1:200 (Santa Cruz Biotechnology); rabbit anti-α-synuclein monoclonal antibody, 1:1000 (Cell Signaling Technology); rabbit anti-LC3 polyclonal antibody, 1:500 (Novus Biologics, Littleton, CO); rabbit anti-transglutaminase 2 monoclonal antibody, 1:2000 (Cell Signaling Technology); mouse anti-actin monoclonal antibody, 1:1500 (Sigma).
:20000 in 5% milk-PBST followed by visualization using enhanced chemiluminescence detection reagents. Equal protein loading was examined by β-actin detection. The following primary antibodies were used: rabbit anti-Lamp-1 monoclonal antibody, 1:1000 (Cell Signaling Technology, Danvers, MA); mouse anti-beclin 1 monoclonal antibody, 1:1000 (Santa Cruz Biotechnology, Santa Cruz, CA); mouse anti-sequestosome 1 (p62) monoclonal antibody, 1:200 (Santa Cruz Biotechnology); rabbit anti-α-synuclein monoclonal antibody, 1:1000 (Cell Signaling Technology); rabbit anti-LC3 polyclonal antibody, 1:500 (Novus Biologics, Littleton, CO); rabbit anti-transglutaminase 2 monoclonal antibody, 1:2000 (Cell Signaling Technology); mouse anti-actin monoclonal antibody, 1:1500 (Sigma).
Statistical analysis
The results are presented as means (±SD) of at least three independent experiments. All data were analyzed employing one-way analysis of variance (ANOVA) with Tukey's post hoc test using the Prism 4.0 software unless specified otherwise. Differences were considered significant at p < 0.05 (*p < 0.05; **p < 0.01; ***p < 0.001).Results
UVA-induced lysosomal alterations are mimicked by pharmacological inhibition of cathepsin B
First, UV-induced impairment of cathepsin B specific enzymatic activity was examined in total cellular extracts prepared from human skin fibroblasts exposed to non-cytotoxic doses of chronic UVA (‘1 week regimen’: 39.6 J cm−2 total dose; ‘3 week regimen’: 59.4 J cm−2 total dose) that did not diminish cellular viability (Fig. 1A and D) but were associated with induction of cellular oxidative stress (Fig. 1B). As a positive control, the cathepsin B inhibitor CA074Me (1μM, q.d., four consecutive days) was used causing complete loss of cathepsin B specific enzymatic activity without causing cell death. As published recently, enzymatic activity was greatly diminished in response to UVA exposure.18 However, no effects were observed in response to UVB exposure administered at sublethal doses (1 week regimen: 100 mJ cm−2 total dose). Quantitative assessment of cellular autofluorescence using flow cytometry and subsequent visualization by confocal fluorescence microscopy revealed pronounced accumulation of autofluorescent material (λex 488 nm/λem 553–611 nm) that occurred with a punctate cytosolic staining pattern in response to UVA and was similarly induced by pharmacological inhibition of cathepsin B (Fig. 1C and E). Electron microscopy indicated accumulation of cytosolic membraneous vesicles containing osmiophilic material that occurred in the absence of lysosomal desintegration or membrane permeabilization in response to either UVA or pharmacological inhibition of cathepsin B (Fig. 1F). These dramatic changes indicative of lysosomal expansion were then substantiated by confocal microscopy using the lysosomal stain lysotracker Red and immunodetection of Lamp-1, a lysosomal marker protein (Fig. 1G–H).23 Confocal imaging revealed a punctate pattern of intact lysotracker Red-positive vesicles that was strongly increased in response to either UVA- or CA074Me-treatment, a finding also substantiated by quantitative flow cytometry (data not shown). Accumulation of Lamp-1 protein occurred in response to both UVA exposure regimens (‘1 week’ and ‘3 week’) and CA074Me treatment. Lamp-1 accumulation occurred at the level of the extensively glycosylated form of this lysosomal transmembrane glycoprotein detected at an apparent size range between 80 and 100 kDa, and the nonglycosylated form running at about 40 kDa was also observed. Neither treatment affected Lamp-1 expression at the transcriptional level as determined by quantitative RT-PCR assessing mRNA levels of LAMP1, the gene encoding Lamp-1 (Fig. 1I).![UVA-induced lysosomal changes in human Hs27 skin fibroblasts are mimicked by pharmacological inhibition of cathepsin B. (A) Cathepsin B specific enzymatic activity in Hs27 cells exposed to UVA (‘1 week’ regimen: 39.6 J cm−2 total dose; ‘3 week’ regimen: 59.4 J cm−2 total dose), UVB (1 week regimen: 100 mJ cm−2 total dose), or CA074Me (1μM, q.d., 4 consecutive days) (n ≥ 3, mean ± SD). (B) Cellular oxidative stress as determined by flow cytometric analysis of DCF-fluorescence in UVA-exposed (‘1 week’ regimen) and CA074Me-treated (as specified in A) cells (n ≥ 3, mean ± SD). (C) Cellular autofluorescence intensity as determined by flow cytometric analysis ((groups as in B; n ≥ 3, mean ± SD). (D–G) After exposure to chronic UVA (‘3 week’ regimen), CA074Me (as specified in A), or mock treatment, cells were analyzed for: (D) viability as examined by flow cytometric analysis of annexin V-propidium iodide-stained cells; (E) cellular autofluorescence as visualized by confocal microscopy; (F) occurrence of osmiophilic vesicles as visualized by transmission electron microscopy [L (osmiophilic vesicles indicative of lysosomal lipofuscin accumulation); M (mitochondrion); N (nucleus)]. (G) lysosomal staining (Lysotracker Red) as visualized by confocal microscopy; nuclear counterstain: DAPI. (H) Cellular Lamp-1 protein levels in response to CA074Me (as specified in A) or UVA exposure (‘1 week’ and ‘3 week’ regimen) as examined by immunoblot analysis. (I) LAMP-1mRNA levels in Hs27 cells exposed to UVA (‘3 week’ regimen) or CA074Me as determined by real time RT-PCR analysis (mean ± SD, n = 3).](/image/article/2012/PP/c1pp05131h/c1pp05131h-f1.gif) | ||
| Fig. 1 UVA-induced lysosomal changes in human Hs27 skin fibroblasts are mimicked by pharmacological inhibition of cathepsin B. (A) Cathepsin B specific enzymatic activity in Hs27 cells exposed to UVA (‘1 week’ regimen: 39.6 J cm−2 total dose; ‘3 week’ regimen: 59.4 J cm−2 total dose), UVB (1 week regimen: 100 mJ cm−2 total dose), or CA074Me (1μM, q.d., 4 consecutive days) (n ≥ 3, mean ± SD). (B) Cellular oxidative stress as determined by flow cytometric analysis of DCF-fluorescence in UVA-exposed (‘1 week’ regimen) and CA074Me-treated (as specified in A) cells (n ≥ 3, mean ± SD). (C) Cellular autofluorescence intensity as determined by flow cytometric analysis ((groups as in B; n ≥ 3, mean ± SD). (D–G) After exposure to chronic UVA (‘3 week’ regimen), CA074Me (as specified in A), or mock treatment, cells were analyzed for: (D) viability as examined by flow cytometric analysis of annexin V-propidium iodide-stained cells; (E) cellular autofluorescence as visualized by confocal microscopy; (F) occurrence of osmiophilic vesicles as visualized by transmission electron microscopy [L (osmiophilic vesicles indicative of lysosomal lipofuscin accumulation); M (mitochondrion); N (nucleus)]. (G) lysosomal staining (Lysotracker Red) as visualized by confocal microscopy; nuclear counterstain: DAPI. (H) Cellular Lamp-1 protein levels in response to CA074Me (as specified in A) or UVA exposure (‘1 week’ and ‘3 week’ regimen) as examined by immunoblot analysis. (I) LAMP-1mRNA levels in Hs27 cells exposed to UVA (‘3 week’ regimen) or CA074Me as determined by real time RT-PCR analysis (mean ± SD, n = 3). | ||
RT2 Profiler™ Autophagy PCR array analysis identifies UVA-induced alteration of SQSTM1, PRKAA2, SNCA, and TGM2 expression that is mimicked by pharmacological inhibition of cathepsin B
To further explore the nature of UVA-induced lysosomal-autophagic alterations at the gene expression level we then employed the RT2 Profiler™ Autophagy PCR array platform (Table 1 and Fig. 2) that allows quantitative assessment of treatment-induced transcriptional changes of 84 autophagy-related genes. In human Hs27 dermal fibroblasts, chronic UVA exposure (‘3 week’ regimen) altered expression levels of eight genes on the array by at least two-fold. The gene displaying the most pronounced upregulation in response to UVA was identified as SQSTM1 (sequestosome 1; 3.5-fold) encoding p62, a multidomain scaffold/adaptor protein involved in autophagic degradation of ubiquitinated proteins and organelles that serves as a selective autophagic substrate itself.24,25 Moderate UVA-induction at the mRNA level was also observed with PRKAA2 (encoding the catalytic subunit of 5′ adenosine monophosphate-activated protein kinase, a sensor of cellular energy status) and SNCA (encoding α-synuclein, an autophagic protein substrate and aggresome component).26,27 Pronounced downregulation in response to chronic UVA occurred with TGM2 encoding transglutaminase 2, an essential enzymatic factor involved in autophagolysosome maturation.28,29| Gene Symbol | Gene Name | Fold change | |
|---|---|---|---|
| UVA | CA074Me | ||
| (A) | |||
| SQSTM1 (NM_003900) | Sequestosome 1 | 3.5* | 4.6* |
| PRKAA2 (NM_006252) | Protein kinase, AMP-activated, alpha 2 catalytic subunit | 2.2* | 2.3* |
| SNCA (NM_000345) | Synuclein, alpha (non A4 component of amyloid precursor) | 2.0* | 6.7* |
| TGM2 (NM_004613) | Transglutaminase 2 | −6.3* | −4.4* |
| (B) | |||
| MAP1LC3B (NM_022818) | Microtubule-associated protein 1 light chain 3 beta | 2.4* | 1.4 |
| HSP90AA1 (NM_001017963) | Heat shock protein 90 kDa alpha (cytosolic), class A member 1 | 2.1* | −1.0 |
| EIF2AK3 (NM_004836) | Eukaryotic translation initiation factor2-alpha kinase 3 | 2.0* | −1.2 |
| HSPA8 (NM_006597) | Heat shock 70 kDa protein 8 | 2.0* | −1.0 |
| CLN3 (NM_000086) | Ceroid-lipofuscinosis, neuronal 3 | 1.5 | 2.2* |
| CTSS (NM_004079) | Cathepsin S | 1.4 | 2.8* |
| IGF1 (NM_000618) | Insulin-like growth factor 1 (somatomedin C) | −1.8 | −2.6* |
| (C) | |||
| AKT1 (1MM_005163), AMBRA1 (NM_017749), APP (NM_000484), ARSA (NM_000487), ATG10 (NM_031482), ATG12 (NM_004707), ATG16L1 (NM_017974), ATG16L1 (NM_017974), ATG16L2 (NM_033388), ATG3 (NM_022488), ATG4A (NM_052936), ATG4B (NM_178326), ATG4C (NM_178221), ATG4D (NM_032885), ATG5 (NM_004849), ATG7 (NM_006395), ATG9A (NM_024085), ATG9B (NM_173681), BAD (NM_004322), BAK1 (NM_001188), BAX (NM_004324), BCL2 (NM_000633), BCL2L1 (NM_138578), BECN1 (NM_003766), BID (NM_001196), BNIP3 (NM_004052), CASP3 (NM_004346), CASP8 (NM_001228), CDKN1B (NM_004064), CDKN2A (NM_000077), CTSB (NM_001908), CXCR4 (NM_003467), DAPK1 (NM_004938), DRAM (NM_018370), EIF4G1 (NM_182917), ESR1 (NM_000125), FADD (NM_003824), FAS (NM_000043), GAA (NM_000152), GABARAP (NM_007278), GABARAPL1 (NM_031412), GABARAPL2 (NM_007285), HDAC1 (NM_004964), HGS (NM_004712), HTT (NM_002111), IFNA2 (NM_000605), IFNA4 (NM_021068), IFNG (NM_000619), INS (NM_000207), IRGM (XR_040407), MAP1LC3A (NIVM81509), MAPK14 (NM_001315), MAPK8 (NM_002750), NFKB1 (NM_003998), PIK3C3 (NM_002647), PIK3CG (NM_002649), PIK3R4 (NM_014602), PRKAA1 (NM_006251), PTEN (NM_000314), RAB24 (NM_130781), RB1 (NM_000321), RGS19 (NM_005873), RPS6KB1 (NM_003161), TGFB1 (NM_000660), FAM176A (NM_032181), TMEM74 (NIVM53015), TMEM77 (NM_178454), TNF (NM_000594), TNFSF10 (NM_003810), TP53 (NM_000546), TP73 (NM_005427), ULK1 (NM_003565), ULK2 (NM_014683), UVRAG (NM_003369) | |||
 | ||
| Fig. 2 Gene expression changes affecting the autophagic-lysosomal pathway in human skin fibroblasts induced by UVA exposure or pharmacological inhibition of cathepsin B. (A) Scatter blot of differential gene expression in response to chronic UVA exposure (‘3 week’ regimen) or CA074Me treatment (1 μM, q.d., 4 consecutive days) versus mock treatment as analyzed using the RT2 Human Autophagy™ PCR Expression Array (as summarized in Table 1). Upper and lower lines represent the cut-off indicating two-fold up- or down-regulated expression, respectively. Arrows mark genes displaying similar expression changes in response to both treatments. (B) Comparative analysis of autophagic-lysosomal- and heat shock-related gene expression changes induced by UVA- versusCA074Me-treatment from combined RT2 Human Autophagy™ and Stress and Toxicity PathwayFinder™ PCR Expression arrays (n = 3, mean ± SD; only where applicable: * denotes statistically significant differences (p < 0.05) between treated and untreated control; # denotes statistically significant differences (p < 0.05) between UVA- and CA074Me-treated samples). | ||
Unexpectedly, pharmacological inhibition of cathepsin B closely mimicked this specific pattern of UVA-induced expression changes affecting autophagy-related genes (Fig. 2 and Table 1). CA074Me-treatment (1 μM, q.d., 4 subsequent days) caused gene upregulation (SQSTM1, PRKAA2, SNCA) and downregulation (TGM2) as observed with UVA.
In order to further explore the unexpected similarities between expression changes caused by UVA and CA074Me we expanded the array of genes interrogated for transcriptional changes combining data derived from RT2 Human Autophagy™ and Stress PathwayFinder™ PCR Expression analysis (Fig. 2B). Consistent with data published earlier,18 chronic UVA exposure caused a pronounced upregulation of the cellular heat shock response, but these dramatic changes were not induced by CA074Me treatment. Comparative assessment of expression changes induced by UVA- versusCA074Me-treatment revealed statistically significant differences affecting heat shock protein-encoding genes including HSPA1A (11-fold), HSPA6 (1041-fold), HSPA8 (2-fold), and HSPCA/HSP90AA1 (2.1-fold) all of which with the exception of HMOX1 (UVA: 18-fold versusCA074Me: 4.1-fold) were only upregulated in response to UVA (n = 3, p < 0.05; Fig. 2B). Moreover, upregulated expression of EIF2AK3 (encoding eukryotic translation initiation factor2-alpha kinase 3, also called PERK, a regulator of the unfolded protein response) and MAP1LC3B (encoding microtubuli-associated protein 1 light chain 3 beta, also called LC3, a protein involved in microtubuli assembly and autophagosome formation) only occurred in UVA-exposed fibroblasts. In contrast, transcriptional changes exclusively observed in CA074Me-treated fibroblasts included CLN3 (2.2-fold upregulation) encoding the lysosomal protein CLN3 impaired in juvenile ceroid lipofuscinosis, CTSS (2.8-fold upregulation) encoding the lysosomal cysteine-protease cathepsin S, and IGF1 (2.6-fold downregulation) encoding insulin-like growth factor 1.
Immunoblot detection of Lamp-1, beclin 1, LC3-II, p62, α-synuclein, and transglutaminase 2 confirms UVA-induced autophagic-lysosomal dysregulation that is mimicked by pharmacological inhibition of cathepsin B
We further examined the occurrence of UVA- and CA074Me-induced alterations indicative of autophagic-lysosomal dysfunction at the protein level (Fig. 3). After detecting upregulation of the lysosomal glycoprotein Lamp-1 at the protein but not at the transcript level (Fig. 1H–I), we focused on immunodetection of LC3-II, a lipidated autophagosomal component and established indicator of autophagosome accumulation.25 Indeed, pronounced accumulation of LC3-II occurred in response to both UVA and CA074Me treatment (Fig. 3A). However, protein levels of beclin1, a key initiator and established hallmark of autophagy known to be upregulated in response to various autophagy-inducing stimuli, remained unchanged in response to either treatment (Fig. 3A).25,30 In contrast, pronounced accumulation of p62 (sequestosome 1, encoded by SQSTM1), a selective autophagy substrate known to undergo depletion during active autophagy, occurred at the protein level in response to either treatment (Fig. 3B).24,25 Further evidence in support of impaired autophagic-lysosomal protein turnover was obtained from immunodetection of α-synuclein (encoded by SNCA) (Fig. 3B). In response to either treatment we observed cellular accumulation of α-synuclein, a protein known to undergo autophagosomal degradation and primarly implicated in cytoplasmic inclusion body and aggresome formation.26,27 In contrast, a pronounced decrease of cellular levels of transglutaminase 2 (TG2 encoded by TGM2), an essential enzyme involved in autophagosome maturation, was observed.28,29 | ||
| Fig. 3 Protein changes affecting the autophagic-lysosomal pathway in human skin fibroblasts induced by UVA exposure or pharmacological inhibition of cathepsin B. After exposure to CA074Me (1μM, q.d., 4 consecutive days) or UVA (3 week regimen: 59.4 J cm−2 total dose) protein levels were determined by Western blot analysis using β-actin detection as a loading control. Panel A: beclin 1, LC3-I, LC3-II; panel B: p62, α-synuclein, transglutaminase 2 (TG2). | ||
Discussion
The mechanistic involvement of the lysosomal cysteine protease cathepsin B in UV-induced cutaneous alterations and photodamage has attracted considerable research interest.1,3,18–20,31,32 Earlier work has shown that in human fibroblasts exposed to cytotoxic doses of UVA, photooxidative rupture of lysosomal membranes was followed by cytoplasmic release of proteases including cathepsin B that degrade ferritin with mobilization of redox-active iron representing a key factor in UVA-induced fibroblast photodamage.1,3,31 Similarly, lysosomal release of cathepsin B was identified as a causative factor involved in melanocyte cell death that occurred in response to exposure to apoptogenic doses of UVA/B significantly higher than the ones used in our model of chronic UVA exposure.32More recently, we have demonstrated that chronic UVA exposure (at doses that neither interfere with proliferation rate nor cause rupture of lysosomal membranes or necrotic cell death) targets cathepsin B through a photooxidative mechanism.18 UVA-induced loss of cathepsin B activity caused impaired cathepsin B maturation (that occurred in the absence of changes affecting transcription of CTSB, the gene encoding cathepsin B), possibly from interference with autoproteolytic cleavage, effects also observed upon pharmacological inhibition using CA074Me. These changes were associated with accumulation of lipofuscin-like autofluorescent material displaying lysosomal colocalization, a change suggestive of lysosomal impairment.
In the current follow up study, we present evidence suggesting that UVA-induced inhibition of cathepsin B enzymatic activity is the causative factor operating upstream of lysosomal-autophagic impairment, a finding largely based on the identification of a similar pattern of changes affecting cellular phenotype and gene expression at the mRNA and protein level, observed in response to both UVA-treatment and pharmacological inhibition of cathepsin B. In addition, recent research has confirmed cathepsin B enzymatic activity as an important regulator of autophagic flux further supporting the hypothesis that cathepsin B inactivation is sufficient to serve as a causative mediator of UVA-induced alterations.33 Indeed, in macrophages derived from CTSB-knockout mice or wildtype macrophages exposed to CA074Me, autophagic flux was significantly impaired leading to the accumulation and stabilization of LC3-II containing autophagosomal and autophagolysosomal vesicles. However, the molecular mechanism underlying cathepsin B involvement in the regulation of autophagic flux is largely unknown.
In our experiments, gene array analysis revealed expression changes consistent with UVA-induced autophagic-lysosomal dysregulation affecting SQSTM1, PRKAA2, SNCA, TGM2, MAP1LC3B, HSP90AA1, EIF2AK3, and HSPA8 by at least two-fold (Table 1A and B; Fig. 2). Strikingly, comparative gene expression analysis indicated that the most pronounced changes (SQSTM1, PRKAA2, SNCA, TGM2) also occurred upon pharmacological inhibition of cathepsin B enzymatic activity using CA074Me, but no changes affecting CTSB expression occurred in response to either treatment (Table 1 and Fig. 2B).18 Furthermore, a compelling similarity between the UVA- and CA074Me-induced expression pattern was detected at the protein level affecting Lamp-1, LC3-II, beclin-1, p62, α-synuclein, and transglutaminase-2 (Fig. 1H and 3).
Lysosomal expansion [as already suggested by accumulation of cytosolic autofluorescent and osmiophilic vesicles, accompanied by extensive lysotracker Red staining (Fig. 1E–G)] was further substantiated by detection of UVA- and CA074Me-induced accumulation of the lysosomal marker protein Lamp-1 (Fig. 1H), an extensively N-glycosylated transmembrane glycoprotein involved in lysosomal motility during vesicular fusion and trafficking (Fig. 1I).23
Importantly, in response to UVA exposure as well as selective pharmacological inhibition of cathepsin B, pronounced upregulation of the selective autophagy substrate p62 (sequestosome 1, encoded by SQSTM1), a cargo receptor for autophagic degradation of specific ubiquitinated target proteins, occurred at the mRNA and protein level (Table 1; Fig. 2 and 3B).34 The multidomain scaffold/adaptor protein p62 plays an important role in protein aggregate formation, cell survival, and apoptosis that involves binding of polyubiquitinated TRAF6 (involved in activation of the transcription factor NFκB) and caspase-8 (involved in apoptotic execution), respectively.25,34,35 Importantly, p62 also binds the autophagy regulator Atg8/LC3via its LIR region, and p62 has been proposed to regulate the packing and delivery of polyubiquitinated misfolded or aggregated proteins and dysfunctional organelles for clearance through autophagy36 It is now established that cellular levels of p62 are regulated through autophagy with p62 serving as a selective autophagy protein substrate.24,25 Indeed, increased autophagy is associated with reduction of cellular p62 levels, whereas inhibition of autophagy has been shown to upregulate cellular p62 levels, a finding further substantiated by the observation that p62 accumulates in autophagy-deficient mice.25,37 Therefore, pronounced upregulation of p62 protein levels observed equally in response to UVA exposure or CA074Me treatment is consistent with the occurrence of autophagic blockade downstream of cathepsin B inhibition (Fig. 3B).
Consistent with impaired turnover of the autophagy substrate p62 and accumulation of the lysosomal membrane protein Lamp-1, pronounced upregulation of α-synuclein (encoded by SNCA), another autophagy substrate, occurred in response to either treatment in dermal fibroblasts (Table 1; Fig. 2 and 3B). Indeed, recent research has shown that α-synuclein is degraded at least partly by chaperone-mediated autophagy involving cathepsins.26,27 Interestingly, α-synuclein constitutes the major protein in cytoplasmic Lewy bodies that accumulate in nigrostriatal neurons during progression of Parkinson's disease where SNCA mutations may cause early-onset of this neurodegenerative disease.38 Important functions of α-synuclein include modulation of lipid vesicle dynamics and mitochondrial energy production through complex I binding, but little is know about its specific function in cutaneous cells where expression in melanocytes has been documented.39
Consistent with autophagic-lysosomal dysregulation and blockade, our study also identified downregulation of transglutaminase 2 (encoded by TGM2), an important factor in autophagosome maturation, at the mRNA and protein level (Table 1; Fig. 2 and 3B).28 Specifically, it has been shown that genetic ablation of transglutaminase 2 resulted in accumulation of LC3-II, the lipidated autophagosomal membrane component, on pre-autophagic vesicles.29 In contrast, subsequent formation of the acidic vesicular organelles in the same cells was suppressed, suggesting an impairment of the final maturation of autophagolysosomes associated with LC3-II accumulation that occurs in response to transglutaminase 2 downregulation.
Indeed, we observed pronounced formation of LC3-II in response to UVA and CA074Me exposure (Fig. 3A). LC3, the mammalian homologue of yeast Atg8 is an essential factor for autophagosome formation that relocalizes to and participates in the formation of the autophagosomal membrane after C-terminal proteolytic processing and posttranslational phospholipid-conjugation.25 Therefore, after relocalization of LC3-I to newly-formed vesicles a more rapidly migrating lipidated form (LC3-II) is detectable by SDS-PAGE (Fig. 3A). Importantly, recent evidence suggests that lysosomal cathepsins including cathepsin B and D are involved in LC3-II turnover, and pharmacological inhibition of cathepsin B was shown to induce accumulation of LC3-II in macrophages.33,40
In the context of autophagic alterations, it is important to note that the above changes of autophagic-lysosomal components occurred in the absence of altered expression at the mRNA or protein level affecting beclin 1 (encoded by BECN1) (Table 1; Fig. 2 and 3A). Indeed, beclin 1 is a critical component in the class III PI3 kinase complex (PI3KC3) involved in autophagosome formation.25,30 Due to its role in the initiation of autophagy, upregulation of beclin I is an established hallmark indicative of autophagic induction.
It is also important to note that upregulation of Lamp-1 protein levels was not accompanied by changes at the transcriptional level (LAMP1) (Fig. 1H and I), yet expression changes affecting SQSTM1 (p62), SNCA (α-synuclein), and TGM2 (transglutaminase 2) at the mRNA level paralleled changes at the protein level (Table 1 and Fig. 2). Interestingly, it has been demonstrated recently that SQSTM1 is under transcriptional control of Nrf2, and that p62 activates Nrf2-dependent transcription by binding and inactivating the Nrf2-antagonistic factor Keap1, representing a positive feedback loop of p62-dependent transcriptional activation of SQSTM1 expression.41,42 In the context of autophagic impairment and p62 accumulation, it is therefore tempting to speculate that SQSTM1 upregulation observed at the mRNA level occurs as a consequence of p62 protein accumulation downstream of cathepsin B inhibition. However, the mechanistic basis underlying UVA- and CA074Me-induced transcriptional changes affecting SNCA and TGM2 remains unknown, awaiting further experimental interrogation.
Taken together these data demonstrate for the first time that chronic exposure to UVA induces autophagic-lysosomal dysregulation in human dermal fibroblasts. Moreover, we provide compelling evidence that functional impairment of cathepsin B activity is a heretofore unrecognized causative factor in cutaneous UVA photodamage operating upstream of autophagic-lysosomal alterations as summarized in Fig. 4. This model proposes that inactivation of cathepsin B as a consequence of either UVA-induced photooxidative stress or direct pharmacological inhibition causes dynamic changes that are most consistent with a blockade of autophagic flux leading to lysosomal expansion with accumulation of lipofuscin-like material as evident from increases in autofluorescent (Fig. 1E), lysotracker-positive (Fig. 1G), and osmiophilic vesicles (Fig. 1F). Supporting this molecular mechanism, recent research has demonstrated that inhibition of cathepsin B enzymatic activity is sufficient to cause delay of autophagic flux in macrophages.33 Consistent with this finding, in cathepsin D-deficient and cathepsins B and L double-deficient mice, abnormal vacuolar structures resembling autophagosomes accumulate in neurons of the brain.40 It has also been shown earlier that primary lysosomal dysfunction may impair autophagic flux as observed in genetic lysosomal storage disorders (LSD).43,44 Moreover, in cells from LSD mice, an impairment of the autophagic pathway may also be associated with the inefficient degradation of aggregate-prone proteins such as mutated α-synuclein. It is therefore tempting to speculate that a deficiency in lysosomal cathepsin B enzymatic activity caused by UVA-photooxidative inactivation impairs autophagic-lysosomal function in a similar way. Since various autophagic pathways including microautophagy, macroautophagy, and chaperone-mediated autophagy all depend on lysosomal function, the specific molecular consequences associated with UVA-induced autophagic-lysosomal dysfunction downstream of cathepsin B inactivation remain to be elucidated.25
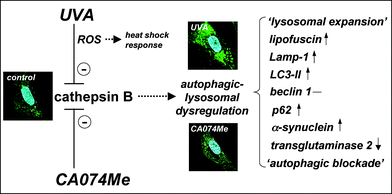 | ||
| Fig. 4 Cathepsin B inactivation as a causative factor in cutaneous UVA photodamage operating upstream of autophagic-lysosomal alterations. The model proposes that inactivation of cathepsin B as a consequence of either UVA-induced photooxidative stress (mediated by ROS) or direct pharmacological antagonism (by the specific inhibitor CA074Me) causes authophagic-lysosomal dysregulation. Effects of chronic UVA exposure on phenotypic markers including cellular autofluorescence (not shown) and lysotracker Red staining (confocal microscopy images, panels as in Fig. 1G) the expression pattern of the autophagic-lysosomal factors Lamp-1, LC3-II, beclin 1, p62, α-synuclein, and transglutaminase 2 are mimicked by CA074Me treatment indicating that UVA exposure causes autophagic-lysosomal dysregulation downstream of cathepsin B inactivation, a novel molecular mechanism potentially involved in UVA-induced skin photodamage. Differential stress response gene expression (including heat shock protein encoding genes) occurs in response to UVA but not CA074Me due to the causative involvement of ROS in UVA-induced cathepsin B inhibition that are not generated as a result of direct pharmacological antagonism by CA074Me. | ||
According to our model, ROS are mechanistically involved in cathepsin B inactivation only in UVA-treated cells and are not required for the causation of CA074Me-induced alterations. Indeed, ROS formation was only observed in response to UVA exposure (Fig. 1B), and significant differences in stress response gene expression were observed as evident from upregulation of heat shock protein encoding genes that occurred primarily in response to UVA but not CA074Me (Fig. 2B), a difference also substantiated at the protein level by immunodetection of Hsp70 as published recently.18
Apart from UVA-induced cathepsin B inactivation observed by us and other,18–20 earlier research has demonstrated that UVA exposure may also interfere with proteasomal protein degradation, another mechanism potentially underlying pathologically altered protein turnover in the context of skin photodamage.45,46 Moreover, lipofuscin-dependent proteasomal inhibition has been demonstrated in human dermal fibroblasts representing another mechanism potentially exacerbating dysfunctional protein turnover under UV stress.47 Future experimentation will therefore address the question if UVA-induced cathepsin B inhibition and autophagic-lysosomal alteration synergizes and interfaces with UVA-induced proteasomal dysfunction.
Further evidence in support of a causative role of cathepsin B in UVA photodamage and our current model of autophagic-lysosomal alterations will be obtained from ongoing studies in our laboratory that involve murine embryonal fibroblasts derived from CTSB-deficient mice (MEF T−/−) representing a stringent genetic model of cathepsin B ablation.48 It will also be interesting to investigate if cathepsin B inhibition induced by either genetic or pharmacological antagonism sensitizes skin cells to UVA-induced damage, a scenario that would be compatible with fluorescent lipofuscin-epitopes acting as endogenous UVA-photosensitizers that further enhance UVA-photooxidative stress, a mechanism suggested by our recent studies demonstrating pronounced UVA-photosensitizer activity of the lipid peroxidation-derived lipofuscin-epitope DHP identified in human skin.21
In addition to further substantiating the emerging role of cathepsin B as a novel target in skin photodamage and autophagic-lysosomal dysfunction, it will be fascinating to elucidate the functional implications of these changes for skin photocarcinogenesis and photoaging.
Abbreviations
| CTSB | cathepsin B (gene) |
| DAPI | 4′,6-diamidino-2-phenylindole dihydrochloride |
| DCFH-DA | 2′,7′-dichlorodihydrofluorescein diacetate |
| 2D-DIGE | two-dimensional difference gel electrophoresis |
| DMEM | Dulbecco's modified Eagle's medium |
| FITC | fluorescein isothiocyanate |
| HBSS | Hank's balanced salt solution |
| HMOX1 | heme oxygenase 1 (gene) |
| PBS | phosphate buffered saline |
| PI | propidium iodide |
| q.d. | each day (quaque die) |
| ROS | reactive oxygen species |
| TG2 | transglutaminase 2 |
| UVA | ultraviolet A |
Acknowledgements
Flow cytometric analysis was performed at the Arizona Cancer Center flow cytometry core facility. Supported in part by grants from the National Institutes of Health [R01CA122484, ES007091, ES006694, Arizona Cancer Center Support Grant CA023074].References
- C. Pourzand, R. D. Watkin, J. E. Brown and R. M. Tyrrell, Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: the role of ferritin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6751–6756 CrossRef CAS.
- A. Valencia and I. E. Kochevar, Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes, J. Invest. Dermatol., 2007, 128, 214–222 CrossRef.
- S. Basu-Modak, D. Ali, M. Gordon, T. Polte, A. Yiakouvaki, C. Pourzand, C. Rice-Evans and R. M. Tyrrell, Suppression of UVA-mediated release of labile iron by epicatechin-a link to lysosomal protection, Free Radical Biol. Med., 2006, 41, 1197–1204 CrossRef CAS.
- K. Scharffetter-Kochanek, M. Wlaschek, P. Brenneisen, M. Schauen, R. Blaudschun and J. Wenk, UV-induced reactive oxygen species in photocarcinogenesis and photoaging, Biol. Chem., 1997, 378, 1247–1257 CAS.
- G. T. Wondrak, M. K. Jacobson and E. L. Jacobson, Endogenous UVA-photosensitizers: mediators of skin photodamage and novel targets for skin photoprotection, Photochem. Photobiol. Sci., 2006, 5, 215–237 CAS.
- J. Cadet, T. Douki, J. L. Ravanat and P. Di Mascio, Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation, Photochem. Photobiol. Sci., 2009, 8, 903–911 CAS.
- A. W. Girotti, Photosensitized oxidation of membrane lipids: reaction pathways, cytotoxic effects, and cytoprotective mechanisms, J. Photochem. Photobiol., B, 2001, 63, 103–113 CrossRef CAS.
- P. L. Zeeuwen, Epidermal differentiation: the role of proteases and their inhibitors, Eur. J. Cell Biol., 2004, 83, 761–773 CrossRef CAS.
- H. Buth, P. Luigi Buttigieg, R. Ostafe, M. Rehders, S. R. Dannenmann, N. Schaschke, H. J. Stark, P. Boukamp and K. Brix, Cathepsin B is essential for regeneration of scratch-wounded normal human epidermal keratinocytes, Eur. J. Cell Biol., 2007, 86, 747–761 CrossRef.
- O. Vasiljeva, T. Reinheckel, C. Peters, D. Turk, V. Turk and B. Turk, Emerging roles of cysteine cathepsins in disease and their potential as drug targets, Curr. Pharm. Des., 2007, 13, 387–403 CrossRef CAS.
- E. Frohlich, B. Schlagenhauff, M. Mohrle, E. Weber, C. Klessen and G. Rassner, Activity, expression, and transcription rate of the cathepsins B, D, H, and L in cutaneous malignant melanoma, Cancer, 2001, 91, 972–982 CrossRef CAS.
- M. Bylaite, H. Moussali, I. Marciukaitiene, T. Ruzicka and M. Walz, Expression of cathepsin L and its inhibitor hurpin in inflammatory and neoplastic skin diseases, Exp. Dermatol., 2006, 15, 110–118 CrossRef CAS.
- M. J. Quintanilla-Dieck, K. Codriansky, M. Keady, J. Bhawan and T. M. Runger, Cathepsin K in melanoma invasion, J. Invest. Dermatol., 2008, 128, 2281–2288 CrossRef CAS.
- A. Schonefuss, W. Wendt, B. Schattling, R. Schulten, K. Hoffmann, M. Stuecker, C. Tigges, H. Lubbert and C. Stichel, Upregulation of cathepsin S in psoriatic keratinocytes, Exp. Dermatol., 2009, 19, e80–88 CrossRef.
- K. A. Codriansky, M. J. Quintanilla-Dieck, S. Gan, M. Keady, J. Bhawan and T. M. Runger, Intracellular degradation of elastin by cathepsin K in skin fibroblasts-a possible role in photoaging, Photochem. Photobiol., 2009, 85, 1356–1363 CrossRef CAS.
- E. Cavarra, M. Fimiani, G. Lungarella, L. Andreassi, M. de Santi, C. Mazzatenta and L. Ciccoli, UVA light stimulates the production of cathepsin G and elastase-like enzymes by dermal fibroblasts: a possible contribution to the remodeling of elastotic areas in sun-damaged skin, Biol. Chem., 2002, 383, 199–206 CrossRef CAS.
- A. Klose, A. Wilbrand-Hennes, J. Brinckmann and N. Hunzelmann, Alternate trafficking of cathepsin L in dermal fibroblasts induced by UVA radiation, Exp. Dermatol., 2009, 19, e117–123 CrossRef.
- S. D. Lamore, S. Qiao, D. Horn and G. T. Wondrak, Proteomic identification of cathepsin B and nucleophosmin as novel UVA-targets in human skin fibroblasts, Photochem. Photobiol., 2010, 86, 1307–1317 CrossRef CAS.
- W. Lai, Y. Zheng, Z. Z. Ye, X. Y. Su, M. J. Wan, Z. J. Gong, X. Y. Xie and W. Liu, Changes of cathepsin B in human photoaging skin both in vivo and in vitro, Chin. Med. J. (Engl), 2010, 123, 527–531 CAS.
- Y. Zheng, W. Lai, M. Wan and H. I. Maibach, Expression of Cathepsins in Human Skin Photoaging, Skin Pharmacol. Physiol., 2011, 24, 10–21 CrossRef CAS.
- S. D. Lamore, S. Azimian, D. Horn, B. L. Anglin, K. Uchida, C. M. Cabello and G. T. Wondrak, The malondialdehyde-derived fluorophore DHP-lysine is a potent sensitizer of UVA-induced photooxidative stress in human skin cells, J. Photochem. Photobiol., B, 2010, 101, 251–264 CrossRef CAS.
- S. D. Lamore, C. M. Cabello and G. T. Wondrak, The topical antimicrobial zinc pyrithione is a heat shock response inducer that causes DNA damage and PARP-dependent energy crisis in human skin cells, Cell Stress Chaperones, 2009, 15, 309–322 CrossRef.
- E. L. Eskelinen, C. K. Schmidt, S. Neu, M. Willenborg, G. Fuertes, N. Salvador, Y. Tanaka, R. Lullmann-Rauch, D. Hartmann, J. Heeren, K. von Figura, E. Knecht and P. Saftig, Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts, Mol. Biol. Cell, 2004, 15, 3132–3145 CrossRef CAS.
- Y. Ichimura, E. Kominami, K. Tanaka and M. Komatsu, Selective turnover of p62/A170/SQSTM1 by autophagy, Autophagy, 2008, 4, 1063–1066 CAS.
- T. Johansen and T. Lamark, Selective autophagy mediated by autophagic adapter proteins, Autophagy, 2011, 7 Search PubMed.
- T. Kabuta, A. Furuta, S. Aoki, K. Furuta and K. Wada, Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy, J. Biol. Chem., 2008, 283, 23731–23738 CrossRef CAS.
- D. Sevlever, P. Jiang and S. H. Yen, Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species, Biochemistry, 2008, 47, 9678–9687 CrossRef CAS.
- P. G. Mastroberardino and M. Piacentini, Type 2 transglutaminase in Huntington's disease: a double-edged sword with clinical potential, J. Intern. Med., 2010, 268, 419–431 CrossRef CAS.
- M. D'Eletto, M. G. Farrace, L. Falasca, V. Reali, S. Oliverio, G. Melino, M. Griffin, G. M. Fimia and M. Piacentini, Transglutaminase 2 is involved in autophagosome maturation, Autophagy, 2009, 5, 1145–1154 CrossRef CAS.
- J. Wang, Beclin 1 bridges autophagy, apoptosis and differentiation, Autophagy, 2008, 4, 947–948 CAS.
- J. L. Zhong, A. Yiakouvaki, P. Holley, R. M. Tyrrell and C. Pourzand, Susceptibility of skin cells to UVA-induced necrotic cell death reflects the intracellular level of labile iron, J. Invest. Dermatol., 2004, 123, 771–780 CrossRef CAS.
- C. A. Bivik, P. K. Larsson, K. M. Kagedal, I. K. Rosdahl and K. M. Ollinger, UVA/B-induced apoptosis in human melanocytes involves translocation of cathepsins and Bcl-2 family members, J. Invest. Dermatol., 2006, 126, 1119–1127 CrossRef CAS.
- S. D. Ha, B. Ham, J. Mogridge, P. Saftig, S. Lin and S. O. Kim, Cathepsin B-mediated autophagy flux facilitates the anthrax toxin receptor 2-mediated delivery of anthrax lethal factor into the cytoplasm, J. Biol. Chem., 2009, 285, 2120–2129 CrossRef.
- J. Moscat and M. T. Diaz-Meco, p62 at the crossroads of autophagy, apoptosis, and cancer, Cell, 2009, 137, 1001–1004 CrossRef CAS.
- A. Duran, M. Serrano, M. Leitges, J. M. Flores, S. Picard, J. P. Brown, J. Moscat and M. T. Diaz-Meco, The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis, Dev. Cell, 2004, 6, 303–309 CrossRef CAS.
- S. Pankiv, T. H. Clausen, T. Lamark, A. Brech, J. A. Bruun, H. Outzen, A. Overvatn, G. Bjorkoy and T. Johansen, p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy, J. Biol. Chem., 2007, 282, 24131–24145 CrossRef CAS.
- M. Komatsu, S. Waguri, M. Koike, Y. S. Sou, T. Ueno, T. Hara, N. Mizushima, J. Iwata, J. Ezaki, S. Murata, J. Hamazaki, Y. Nishito, S. Iemura, T. Natsume, T. Yanagawa, J. Uwayama, E. Warabi, H. Yoshida, T. Ishii, A. Kobayashi, M. Yamamoto, Z. Yue, Y. Uchiyama, E. Kominami and K. Tanaka, Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice, Cell, 2007, 131, 1149–1163 CrossRef CAS.
- J. L. Webb, B. Ravikumar, J. Atkins, J. N. Skepper and D. C. Rubinsztein, Alpha-Synuclein is degraded by both autophagy and the proteasome, J. Biol. Chem., 2003, 278, 25009–25013 CrossRef CAS.
- K. S. Hoek, N. C. Schlegel, O. M. Eichhoff, D. S. Widmer, C. Praetorius, S. O. Einarsson, S. Valgeirsdottir, K. Bergsteinsdottir, A. Schepsky, R. Dummer and E. Steingrimsson, Novel MITF targets identified using a two-step DNA microarray strategy, Pigm. Cell Melanoma Res., 2008, 21, 665–676 CrossRef CAS.
- M. Koike, M. Shibata, S. Waguri, K. Yoshimura, I. Tanida, E. Kominami, T. Gotow, C. Peters, K. von Figura, N. Mizushima, P. Saftig and Y. Uchiyama, Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease), Am. J. Pathol., 2005, 167, 1713–1728 CrossRef CAS.
- A. Jain, T. Lamark, E. Sjottem, K. B. Larsen, J. A. Awuh, A. Overvatn, M. McMahon, J. D. Hayes and T. Johansen, p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription, J. Biol. Chem., 2010, 285, 22576–22591 CrossRef CAS.
- M. Komatsu, H. Kurokawa, S. Waguri, K. Taguchi, A. Kobayashi, Y. Ichimura, Y. S. Sou, I. Ueno, A. Sakamoto, K. I. Tong, M. Kim, Y. Nishito, S. Iemura, T. Natsume, T. Ueno, E. Kominami, H. Motohashi, K. Tanaka and M. Yamamoto, The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1, Nat. Cell Biol., 2010, 12, 213–223 CAS.
- C. Settembre, A. Fraldi, L. Jahreiss, C. Spampanato, C. Venturi, D. Medina, R. de Pablo, C. Tacchetti, D. C. Rubinsztein and A. Ballabio, A block of autophagy in lysosomal storage disorders, Hum. Mol. Genet., 2007, 17, 119–129 CrossRef.
- T. Otomo, K. Higaki, E. Nanba, K. Ozono and N. Sakai, Inhibition of autophagosome formation restores mitochondrial function in mucolipidosis II and III skin fibroblasts, Mol. Genet. Metab., 2009, 98, 393–399 CrossRef CAS.
- A. L. Bulteau, M. Moreau, C. Nizard and B. Friguet, Impairment of proteasome function upon UVA- and UVB-irradiation of human keratinocytes, Free Radical Biol. Med., 2002, 32, 1157–1170 CrossRef CAS.
- B. Catalgol, I. Ziaja, N. Breusing, T. Jung, A. Hohn, B. Alpertunga, P. Schroeder, N. Chondrogianni, E. S. Gonos, I. Petropoulos, B. Friguet, L. O. Klotz, J. Krutmann and T. Grune, The proteasome is an integral part of solar ultraviolet a radiation-induced gene expression, J. Biol. Chem., 2009, 284, 30076–30086 CrossRef CAS.
- A. Hohn, T. Jung, S. Grimm, B. Catalgol, D. Weber and T. Grune, Lipofuscin inhibits the proteasome by binding to surface motifs, Free Radical Biol. Med., 2011, 50, 585–591 CrossRef.
- K. Moin, L. Demchik, J. Mai, J. Duessing, C. Peters and B. F. Sloane, Observing proteases in living cells, Adv. Exp. Med. Biol., 2000, 477, 391–401 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |