Activation of p38 MAP kinase and JNK pathways by UVA irradiation†
Jack
Zhang
a and
G. Tim
Bowden
*bc
aDepartment of Medical Pharmacology, University of Arizona, Tucson, Arizona, USA
bDepartment of Cell Biology and Anatomy, University of Arizona, Tucson, Arizona, USA
cArizona Cancer Center, University of Arizona, Tucson, Arizona, USA
First published on 22nd August 2011
Abstract
There are more than two million new cases of non-melanoma skin cancers (NMSCs) diagnosed each year in the United States of America. The clear etiological factor is chronic exposure to solar radiation from the sun. The wavelengths of solar light that reach the earth's surface include UVB (280–320 nm), which accounts for 1–10%, and UVA (320–400 nm), which accounts for 90–99% of the radiation. While most published research has focused on the effects of UVB, little is known concerning UVA-mediated signal transduction pathways, and their role in skin tumor promotion and progression, giving rise to squamous cell carcinomas (SCCs). Here, we focus on UVA-mediated activation of p38 MAP kinase and c-Jun N-terminal kinase (JNK), and their roles in activator protein-1 (AP-1) mediated transcription, cyclooxygenase-2 (COX-2) and Bcl-XL expression. Since p38 MAP kinase and JNK play major roles in the expression of UVA-induced AP-1, COX-2 and Bcl-XL, pharmacological inhibitors of these kinases may be useful in the chemoprevention of SCC skin cancer.
1. Introduction
According to the American Cancer Society, skin cancer is the most common cancer, accounting for about half of all cancers in the United States of America. Skin cancer is classified as melanoma (4% of total cases) and non-melanoma skin cancers (NMSCs, 96% of total cases). NMSCs is further divided into basal-cell carcinomas (BCCs) and squamous-cell carcinomas (SCCs), accounting for 80% and 16% of skin cancers respectively.1,2 BCC and SCC are both derived from the basal layer of the epidermis of the skin, but whereas BCCs are slow growing and rarely metastasize, SCCs can be highly invasive and can metastasize.1It is estimated that there are more than two million NMSC cases in the US each year.3,4 Although typically not lethal, NMSCs are responsible for tissue deformity and substantial morbidity, particularly in high-risk populations, such as people with light skin or with organ transplantation. The direct cost for treatment of NMSCs in the United States of America was estimated to be $1.5 billion in 2004.5 Besides, actinic keratosis (AK) is also considered as an early stage of invasive SCC and carcinoma in situ. If AK is included, the direct cost would increase to more than $2.3 billion.5 The incidence of NMSCs continues to rise in recent years. This trend is in sharp contrast to most other malignancies which in general are declining.3,4,6 A plausible explanation for this trend is that exposure to ultraviolet irradiation (UVR) from sunlight has increased, due to the gradual depletion of the ozone layer in the past decades. Thus, it is critical to better understand the mechanisms and development of NMSCs for better prevention and treatment.
The UV irradiation of sunlight is subdivided based on wavelength: UVA (320–400 nm), UVB (280–320 nm) and UVC (200–280 nm). As UVC is shielded by the ozone layer, only UVA (90-99%) and UVB (1–10%) wavelengths can reach the earth's surface. Although UVA has less energy than UVB, it constitutes the vast majority of UV irradiation from sunlight and penetrates deeper in human skin than UVB.7
Mounting evidence has clearly shown that UVA irradiation causes DNA lesions through both direct and indirect mechanisms. It is believed that the primary genotoxic effect of UVA results from generation of reactive oxygen species (ROS), including superoxide radical, hydrogen peroxide and hydroxyl radical, with singlet oxygen as the major ROS species. The ROS in turn, causes oxidative damage of macromolecules including DNA and cellular structures, e.g. gap junctional intercellular communication (GJIC) in normal human keratinocytes.8 However, a study published by Mouret et al. challenged the above notion.9 They found that in UVA exposed human skin, the predominant DNA lesion is cyclobutane pyrimidine dimers (CPDs), which are products of direct photosensitization. Overall, UVA is able to cause genomic instability, i.e. mutations of oncogenes and tumor suppressors. For example, p53 is the most extensively studied example for UVA induced gene mutation. Huang et al. demonstrated that the predominant mutation induced by UVA is A:T>C:G transversions in p53 gene in EHS (engineered human skin).10 p53 loss-of-function/mutations have been associated with 60% of cancers. Likewise, another tumor suppressor, PTEN (phosphatase and tensin homologue deleted on chromosome 10) was also found to be downregulated in HaCaT cells treated with chronic UVA exposure 11 and in heterozygous PTEN knockout mice.12 The carcinogenicity of UVA has been demonstrated in cultured cells which acquired resistance to apoptosis and became transformed upon chronic exposure to low dose of UVA.11 Importantly, various mouse models have clearly demonstrated that UVA alone can induce papillomas and SCC in vivo.12–14 These results highlighted the notion that UVA is also a complete carcinogen as is UVB.
2. Signaling pathways activated by UVA in human kertinocytes
2.1 Activation of p38 MAP kinase
p38 MAP kinase is activated by the upstream dual specific MKKs, primarily MKK3 and MKK6, which phosphorylate a conserved Thr–X–Tyr motif in the activation loop (T180 and Y182 in p38α).18,19 Upon activation, p38 migrates into the nucleus and phosphorylates its downstream targets including, MAPKAP2 (MK2), MNK1 and 2, ATF1/2/6, Elk-1, GADD153, p53, and MSK1/2, which are involved in stress response and cell survival.19
There are four p38 isoforms of p38 identified up to date: p38α (MAPK14), p38β (MAPK11), p38γ (MAPK13) and p38δ (MAPK12), with p38α is the most dominant isoform (commonly referred to as p38). Although these isoforms are homologous, they differ in several aspects: tissue specific expression, substrates, sensitivity to pyridinyl imidazole inhibitors (e.g. SB203580) and functions. For example, the expression of p38α and p38β isoforms is ubiquitous while p38δ isoform is also expressed in many tissues. p38γ, however, is mainly expressed in skeletal muscles.19,20
In skin, only the p38α, p38β and 38δ isoforms are detected while p38γ is absent.21 It has been concluded that p38α and p38β are important for stress response and inflammation in skin and p38δ plays a critical role in the differentiation of keratinocytes.22
2.1.2.1 Activation of p38 by ASK1 kinase. Considerable published data demonstrated that ROS production leads to activation of p38 MAP kinase.8,23–26 There is evidence indicating that ROS induces p38 MAP kinase activation through ASK1 (apoptosis signal regulated kinase-1), a MAPK kinase kinase (MKKK).26 This protein kinase is activated by oligomerization and phosphorylation. In untreated cells, thiol-disulphide oxidoreductase thioredoxin-1 (Trx1) covalently binds to ASK1 and inhibits its activity. However, upon H2O2 treatment, ASK1 is rapidly activated through dissociation from Trx1 and multimerization by forming interchain disulfide bonds.27
2.1.2.2 Activation of p38 through inhibition of phosphatases. Activity of MAP kinases depends upon the balance between upstream kinases and phosphatases.20 For example, MEF cells lack DUSP1/MKP1 showed sustained activation of p38 and JNK upon 30 J m−2 UVC irradiation.28 There are ten MKPs (mitogen-activated protein kinase phosphatases) that have been shown to counteract MAPK activities by dephosphorylating their phosphothreonine and phosphotyrosine residues within the Thr-X-Tyr activation motif at the same time.29 For this reason, these MKPs are classified as DUSPs (dual-specificity phosphatases) that belong to the PTP (protein tyrosine phosphatase) family.29,30 Each of these MKPs has relative specificity for its substrates. For instance, MKP1 (DUSP1) has a substrate preference as follows: JNK > p38 ≫ ERK1/2.29 MKPs also have different subcellular distribution, e.g. MKP1 (DUSP1) and MKP2 (DUSP4) are primarily nuclear while MKP3 (DUSP6) is cytoplamsmic.30
Compelling evidence has shown that PTPs are subject to inactivation by ROS.28,31–33 PTP1B is well characterized for this mechanism. It turned out that the cysteine at the catalytic center is nucleophilic and easily oxidized to form cysteine sulfenic acid intermediates (Cys-SOH) and sulphenyl-amide, leading to significant change of conformation and loss of substrate binding capacity.31,33 The activity can be restored by reducing agents or scavenging ROS by Trx-dependent reductases.32
Since the PTP family members are all cysteine-based,31 ROS may inactivate the whole family through a similar mechanism. Indeed, inhibition of MKP1, MKP3, MKP5 and MKP7 was reported in mouse fibroblast cells treated with H2O2.32 Gross et al. showed that UVC (2 kJ m−2) caused a 7-fold decrease in PTP activity in TRMP cells.34 Similarly, Xu et al. found that 50 mJ cm−2 of UVC causes significant oxidization of RPTP-κ (receptor-type protein tyrosine phosphatase κ), leading to a 60% reduction of its activity in primary human keratinocytes. Moreover, this inhibition can be reversed by the reducing agent, DTT.35
2.2 MK2 is a critical player downstream of p38 in skin
Recently, MK2 has been clearly demonstrated to play a critical role in skin carcinogenesis in mice treated with DMBA/TPA.40 The onset of papillomas was 8 weeks after the start of TPA treatment in the wild-type C57BL/6 mice, whereas papillomas were not found in the MK2 K/O mice until 12 weeks. At week 18, 93% of the wild type mice developed papillomas with an average of 5.4 per mouse. In contrast, only 7% of the K/O mice developed one papilloma, indicating that MK2 is required for the development of skin tumor.40We and others have demonstrated that UVA irradiation activates p38 MAP kinase in multiple cell types treated with different UVA dose levels. It has been found that p38 MAP kinase is activated in JB6 cells (mouse epidermal) with UVA at 160 kJ m−2.41 Similarly, activation of p38 reaches maximum within 15–30 min in human skin fibroblasts irradiated with 300 kJ m−2 UVA.42 In immortalized human keratinocytes (HaCaT), our laboratory also observed that p38 is activated within 5 min with the peak between 30–45 min in response to 250 kJ m−2 UVA.43,44 In synergizing with arsenite, dose levels as low as 10 kJ m−2 UVA can boost p38 activation dramatically in HaCaT cells.45
2.3 Anti-apoptotic role of p38 activation
Activated p38 MAP kinase has a broad range of substrates that are implicated in multiple biological processes including protein and mRNA degradation, cytoskeleton organization, endocytosis, cell migration.19,20 It is also implicated in apoptosis with either pro- or anti-apoptotic effect. For example, UVB promotes apoptosis in human keratinocytes by inducing Bax translocation to the mitochondria and cytochrome c release. However, this apoptotic response can be blocked by inhibition of p38 MAPK with either dominant negative p38 or SB203580 in HaCaT cells.46 Furthermore, p38 is also critical to induce Noxa, a BH-3 only pro-apoptotic molecule which is indispensible for UVB induced apoptosis in NHK cells.47 Consistently, inhibition of p38 with SB202190 significantly reduces sunburn cells and subcorneal pustules in the mouse skin with acute UVB irradiation.48 These data indicate that p38 plays a pro-apoptotic role in keratinocytes.Our laboratory demonstrated that p38 plays an anti-apoptotic function in HaCaT cells, because inhibition of p38 with 2 μM of SB202190 leads to extensive apoptosis in UVA irradiated cells.49 These cells showed typical apoptotic features, e.g. condensed chromatin, nuclear fragmentation and apoptotic body formation, release of cytochrome c, and cleavages of caspases.49 Possible explanations for these controversial data may include differential responses to UVA and UVB, and dependence upon p53 since HaCaT cells bear a non-functional version of p53.7,16
We further explored the mechanism by which UVA upregulates Bcl-XL gene expression through p38 MAP kinase. It turns out that p38, upon activation, stabilizes Bcl-XL mRNA.49,52 Bcl-XL mRNA contains a relatively long 3′-UTR, which possesses three canonical ARE (AU-rich) elements. The ARE elements can recruit RNA binding proteins that will either stabilize or destabilize mRNA molecules depending upon cellular context.53 We also demonstrated that UVA irradiation increases the reporter activity in cells transiently transfected with the pRL-TK harboring Bcl-XL 3′-UTR downstream of Renilla luciferase.49 The induction of the reporter activity was abolished when the cells were cotransfected with a dominant-negative p38 MAP kinase. Our findings strongly support the notion that UVA stabilization of Bcl-XL mRNA depends upon the 3′-UTR and is regulated by p38 MAP kinase.49
Published evidence has shown that mRNA 3′-UTRs confer stability through their interactions with different RNA binding proteins and microRNAs.54,55 Therefore, we tried to identify possible RNA binding proteins that may stabilize Bcl-XL mRNA in UVA irradiated HaCaT cells with a proteomic approach. One such protein identified was nucleolin, a ubiquitously expressed protein participating in numerous biological functions, e.g. ribosomal biogenesis, cell cycle regulation and apoptosis.56 Nucleolin has been implicated in stabilizing multiple mRNAs including p53, IL-2, GADD45 and Bcl-2.57–59 We demonstrated that nucleolin specifically binds to the Bcl-XL 3′-UTR and stabilizes Bcl-XL mRNA.52 Moreover, nucleolin interacts with PABP (poly(A) binding protein) through its RGG (Arginine-Glycine-Glycine) motif. Although the mechanism of how nucleolin stabilizes Bcl-XL mRNA is not clear, it is possible that p38 directly phosphorylates nucleolin,60 changing its subcellular localization or binding properties. Consequently, nucleolin may stabilizing Bcl-XL mRNA through its interaction with PABP to protect the poly(A) tail and to facilitate the mRNA molecule to form a closed-loop structure.52
It is possible that mRNA stabilization is dependent upon activation of MAPKAP kinase-2 (MK2),62 which confers p38 biological functions and plays a critical role in regulating stability of numerous mRNAs.63 Indeed, phosphorylation of MK2 was observed in UVA irradiated HaCaT cells in our previous studies.43,44 Inhibition of p38 by either dominant negative p38 or SB202190 completely blocked MK2 phosphorylation in UVA treated HaCaT cells.43,44 Alternatively, COX-2 mRNA may be stabilized in UVA irradiated cells by binding of HuR, a conserved protein that stabilizes a number of mRNAs.64 We and others found that COX-2 mRNA was stabilized through HuR binding to the 3′-UTR using UVB irradiated HaCaT cells.65,66 HuR is a nucleo-cytoplasm shuttling protein primarily localized in the nucleus. Upon phosphorylation by p38 at T118, it translocates into the cytoplasm and stabilizes its mRNA targets.67,68 Since UVA irradiation activates p38, it is likely that HuR cytoplasmic localization is also enhanced in UVA irradiated HaCaT cells.
2.4 p38 MAP kinase and skin carcinogenesis
The implication of p38 MAP kinase in skin carcinogenesis has been supported by multiple animal and clinical studies. For example, since p38α knockout mice are non-viable due to placenta defects and abnormal vascular development.69,70 Our laboratory constructed skin-specific transgenic mice in SKH1 background expressing dominant negative p38α to overcome this problem.71 We found that tumorigenesis was significantly inhibited in these mice.71 Importantly, aberrant activation of p38α has been regarded as a biomarker for skin carcinogenesis.72,73In contrast to p38α, knockout mice of p38β,74 p38γ 75 and p38δ 76 isoforms are viable and normal. Currently, there is no report regarding skin carcinogenesis in p38β KO mice. While displaying normal skin phenotype, the p38δ knockout mice in C57BL/6 background had lower tumor incidence, multiplicity, and size in comparison with the wild type in a typical TPA/DMBA two-stage carcinogenesis assay,76 indicating that p38δ is indispensible for skin carcinogenesis. Consistently, p38δ activity is increased in human primary cutaneous SCCs 77 and HNSCC tumors.78
2.5 Activation of JNK
JNK is also activated in response to various stress signals including ultraviolet irradiation.79 The JNK family is comprised of three members JNK1-3, encoded by MAPK8-10 genes, respectively.20 JNK1 and 2 are ubiquitously expressed whereas JNK3 is primarily expressed in brain.79 The upstream kinases for JNKs are mainly MKK4 and MKK7.80 The JNK family plays a critical role in proliferation, survival and differentiation.79 However, opposite results were observed dependent on the nature of stimuli, cellular context and duration of extracellular signals.20,79In a previous study, we found that 250 kJ m−2 UVA irradiation induces JNK phosphorylation in 5 min and reaches maximum in 30 min in HaCaT cells, a similar pattern as p38 activation.81 Inhibition of JNK with SP600125 leads to a decrease of c-Jun phosphorylation in a dose-dependent manner in UVA irradiated HaCaT cells.81 At 125 nM, c-Jun phosphorylation was significantly reduced. Moreover, phosphorylation of c-Jun induced by UVA was only inhibited by JNK specific inhibitor SP600125, but not p38 inhibitor SB202190,81 indicating that activation of JNK is not dependent on p38.81 The importance of JNK in skin carcinogenesis was later verified by Dong′s group who showed that JNK2 is required for UVA-mediated skin carcinogenesis since knockout of JNK2 in SKH-1 hairless mice developed significantly fewer papillomas than the wild type control after DMBA treatment and 18 weeks of UVA exposure.82 Interesting, knockout of JNK1 enhances formation of papillomas,82 indicating that JNK1 and JNK2 function distinctly in the cell as illustrated in an early study.83
2.6 Activation of AP-1
The JNK family has been shown to phosphorylate and upregulate the transcription factor AP-1 (activator protein 1),84 which functions as a dimer and recognizes TPA-responsive elements (TREs consensus sequence 5′-TGAG/CTCA-3′).85 AP-1 is made up of structurally and functionally related protein super families, i.e. Jun protein family (c-Jun, JunB and JunD), Fos protein family (c-Fos, FosB, Fra-1 and Fra-2), ATF (ATFa, ATF-2 and ATF-3), MAF (musculoaponeurotic fibrosarcoma) and JDP (JDP-1 and JDP-2) families.84,86 Depending upon the cellular context, these family members can interact with each other through their Leucine-zipper domains and form various homodimers or heterodimers to regulate their target genes, promoting cell survival, proliferation and therefore malignant transformation.1,86As a primary target of JNK, c-Jun is upregulated in both its abundance and phosphorylation in UVA irradiated HaCaT cells.81,87 Specifically, c-Jun abundance and phosphorylation starts to rise at 15 min and reaches maximum at 4 h after treatment of 250 kJ m−2 UVA. Similarly, we found that expression of other AP-1 components is also elevated in UVA irradiated HaCaT cells, e.g. c-fos, Fra-1 and Fra-2. Subsequently, EMSA experiments showed that binding of the AP-1 probe with the lysates extracted from UVA irradiated HaCaT cells is also enhanced.87 To examine if increase of c-Jun abundance and phosphorylation leads to AP-1 activation, we used the HCL14 cell line in which stably expresses luciferase reporter driven by AP-1 binding site.88 We found that UVA increases AP-1 activity in HCL14 cells in both dose and time-dependent manner, with a peak activity at 250 kJ m−2 and at 4 h, the same pattern as c-Jun protein abundance.87 We further demonstrated that JNK activation is required for cell survival in UVA irradiated HaCaT cells, since inhibition of UVA-induced JNK activity with 125 nM SP600125 in both the immortalized keratinocyte cell line HaCaT and primary human keratinocytes resulted in the cleavage of caspase 8, 9, 3 and PARP. As a result, these cells showed massive apoptotic responses as determined by morphology and flow cytometry.43 Our data showed that p38 MAP kinase was also required for AP-1 activity in UVA response,81 since inhibition of p38 by SB202190 decreased UVA induced AP-1 activity dramatically, suggesting that both pathways are required for activation of AP-1.81
Carcinogenesis is divided into three independent stages: initiation, promotion, and progression with tumor promotion/progress as the rate-limiting steps.1 It has also been well established that AP-1 plays a causative role in skin carcinogenesis, specifically in tumor promotion and/or progression steps both in vitro and in vivo.89–92 As a result, inhibition of AP-1 by TAM67 (dominant negative version of c-Jun with a deletion of its transactivation domain) blocked skin carcinogenesis induced by the complete carcinogen, UVB light.93 However, at this time we do not have similar direct evidence that AP-1 plays a functional role in UVA induced skin carcinogenesis.
3. Summary
UVA is the predominant irradiation from sunlight and causes DNA lesions and photodamage of other macromolecules. It is able to penetrate skin deeper into the basal layer of the skin where actively dividing keratinocytes reside. As a result, UVA induced DNA lesions predominately accumulate in these dividing cells which are potentially initiated cells for skin tumors.10 The causative effect of UVA in skin carcinogenesis has been well established in numerous studies. For example, chronic exposure of low dose of UVA causes malignant transformation of HaCaT cells in culture 11 and induces skin carcinogenesis in different mouse models as well as human skin.13,14,94,95To summarize, our laboratory has delineated p38 MAP kinase and JNK pathways in the UVA response in human keratinocytes (Fig. 1). UVA irradiation activates these pathways leading to resistance to apoptosis. Using dominant negative p38α transgenic mice, we showed that UVB induced skin carcinogenesis was significantly reduced in comparison to the wild type SKH-1 hairless mice.71 This result suggests the potential of p38 as a chemoprevention target in UVB and perhaps also UVA induced skin carcinogenesis. Although no data are currently available, we anticipate that pharmacologic inhibition of p38 and/or JNK would be able to inhibit UVA induced non-melanoma skin tumors (i.e. SCCs). Consistently, knockout of MK2, downstream target of p38, also significantly promotes resistance to skin carcinogenesis induced by DMBA/TPA in mice.40 These data provided the rationale for targeting p38MAP kinase pathway for chemoprevention of NMSC.
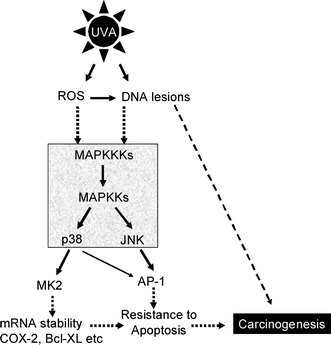 | ||
| Fig. 1 Scheme of UVA signaling in human keratinocytes UVA from sunlight can produce DNA damage through direct photodamage or generation of reactive oxygen species (ROS). Both DNA lesions and ROS subsequent contribute to activation of p38 MAP kinase or JNK through the MAP kinase module (boxed and shaded). Activated p38 MAP kinase phosphorylates its substrates, including MK2, which is critical for stabilization of numerous mRNA molecules, e.g. COX-2 and Bcl-XL. Meanwhile, activation of JNK promotes AP-1 activity. As a result, both p38 and JNK activation render cells more resistant to apoptosis. This critical step may lead to accumulation DNA damage and consequently, carcinogenesis. Solid arrows indicate direct interaction; Dashed arrows denote multiple steps. | ||
Studies on UVA induced biological effects are important for at risk population(s), i.e. people with light skin and frequent exposure to sunlight, or those frequently using tanning booths (primarily UVA). In recent years, these studies have become increasingly important for organ transplantation patients who take thiopurine derived drugs, such as azathiopurine. The incidence of skin cancer, primarily SCC, has increased (50–200 fold) in these patients.96 It turned out that the product of thiopurine, 6-thioguanine (6-TG) nucleotides, incorporates into DNA and absorbs UVA primarily at 340 nm, yielding singlet oxygen (1O2) in cultured human cells.97 Recently, it has been shown that UVA covalently crosslinks 6-TG in DNA and nuclear proteins, e.g. DNA repair and replication factors, blocking DNA damage replication repair mechanisms.98
UVA can also be an important environmental hazard for some regions with arsenite contamination of drinking water, since UVA and arsenite are synergistic in inducing skin cancers in SKH1 hairless mice.99,100 These regions include several US southwestern states (California, Nevada and Utah) and some developing countries, such as India, Bangladesh, Mexico, Chile and China.101 Given the fact that approximately 40–100 million people living in these regions are still consuming arsenite contaminated water,101 further study is warranted to better understand UVA responses and its interaction with arsenite.
References
- G. T. Bowden, Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling, Nat. Rev. Cancer, 2004, 4, 23–35 CrossRef CAS.
- A. I. Rubin, E. H. Chen and D. Ratner, Basal-cell carcinoma, N. Engl. J. Med., 2005, 353, 2262–2269 CrossRef CAS.
- R. V. Patel, A. Frankel and G. Goldenberg, An update on nonmelanoma skin cancer, J. Clin. Aesthet. Dermatol., 2011, 4, 20–27 Search PubMed.
- J. A. Cafardi, R. Shafi, M. Athar and C. A. Elmets, Prospects for skin cancer treatment and prevention: the potential contribution of an engineered virus, J. Invest. Dermatol., 2011, 131, 559–561 CrossRef CAS.
- D. R. Bickers, H. W. Lim, D. Margolis, M. A. Weinstock, C. Goodman, E. Faulkner, C. Gould, E. Gemmen and T. Dall, The burden of skin diseases: 2004 a joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology, J. Am. Acad. Dermatol., 2006, 55, 490–500 CrossRef.
- R. Marks, An overview of skin cancers. Incidence and causation, Cancer, 1995, 75, 607–612 CrossRef CAS.
- F. R. de Gruijl, Photocarcinogenesis: UVA vs. UVB, Methods Enzymol., 2000, 319, 359–366 CAS.
- B. Bellei, A. Mastrofrancesco, S. Briganti, N. Aspite, N. Ale-Agha, H. Sies and M. Picardo, Ultraviolet A induced modulation of gap junctional intercellular communication by P38 MAPK activation in human keratinocytes, Exp. Dermatol., 2008, 17, 115–124 CrossRef CAS.
- S. Mouret, C. Baudouin, M. Charveron, A. Favier, J. Cadet and T. Douki, Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13765–13770 CrossRef CAS.
- X. X. Huang, F. Bernerd and G. M. Halliday, Ultraviolet A within sunlight induces mutations in the epidermal basal layer of engineered human skin, Am. J. Pathol., 2009, 174, 1534–1543 CrossRef CAS.
- Y. Y. He, J. Pi, J. L. Huang, B. A. Diwan, M. P. Waalkes and C. F. Chignell, Chronic UVA irradiation of human HaCaT keratinocytes induces malignant transformation associated with acquired apoptotic resistance, Oncogene, 2006, 25, 3680–3688 CrossRef CAS.
- M. Ming, C. R. Shea, L. Feng, K. Soltani and Y. Y. He, UVA Induces Lesions Resembling Seborrheic Keratoses in Mice with Keratinocyte-Specific PTEN Downregulation, J. Invest. Dermatol., 2011, 131, 1583–1586 CrossRef CAS.
- A. de Laat, J. C. van der Leun and F. R. de Gruijl, Carcinogenesis induced by UVA (365-nm) radiation: the dose-time dependence of tumor formation in hairless mice, Carcinogenesis, 1997, 18, 1013–1020 CrossRef CAS.
- H. J. van Kranen, A. de Laat, J. van de Ven, P. W. Wester, A. de Vries, R. J. Berg, C. F. van Kreijl and F. R. de Gruijl, Low incidence of p53 mutations in UVA (365-nm)-induced skin tumors in hairless mice, Cancer Res., 1997, 57, 1238–1240 CAS.
- D. Hanahan and R. A. Weinberg, The hallmarks of cancer, Cell, 2000, 100, 57–70 CrossRef CAS.
- F. R. de Gruijl, p53 mutations as a marker of skin cancer risk: comparison of UVA and UVB effects, Exp. Dermatol., 2002, 11(s1), 37–39 CrossRef CAS.
- H. J. van Kranen and F. R. de Gruijl, Mutations in cancer genes of UV-induced skin tumors of hairless mice, J. Epidemiol., 1999, 9, S58–65 CrossRef CAS.
- A. Cuenda and S. Rousseau, p38 MAP-kinases pathway regulation, function and role in human diseases, Biochim. Biophys. Acta, Mol. Cell Res., 2007, 1773, 1358–1375 CrossRef CAS.
- A. Cuadrado and A. R. Nebreda, Mechanisms and functions of p38 MAPK signalling, Biochem. J., 2010, 429, 403–417 CrossRef CAS.
- E. F. Wagner and A. R. Nebreda, Signal integration by JNK and p38 MAPK pathways in cancer development, Nat. Rev. Cancer, 2009, 9, 537–549 CrossRef CAS.
- C. Johansen, K. Kragballe, M. Westergaard, J. Henningsen, K. Kristiansen and L. Iversen, The mitogen-activated protein kinases p38 and ERK1/2 are increased in lesional psoriatic skin, Br. J. Dermatol., 2005, 152, 37–42 CrossRef CAS.
- R. L. Eckert, T. Efimova, S. Balasubramanian, J. F. Crish, F. Bone and S. Dashti, p38 Mitogen-activated protein kinases on the body surface–a function for p38 delta, J. Invest. Dermatol., 2003, 120, 823–828 CrossRef CAS.
- T. Finkel, Oxygen radicals and signaling, Curr. Opin. Cell Biol., 1998, 10, 248–253 CrossRef CAS.
- A. Kulisz, N. Chen, N. S. Chandel, Z. Shao and P. T. Schumacker, Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes, Am. J. Physiol. Lung Cell Mol. Physiol., 2002, 282, L1324–L1329 CAS.
- P. Heusch, M. Canton, S. Aker, A. van de Sand, I. Konietzka, T. Rassaf, S. Menazza, O. E. Brodde, F. Di Lisa, G. Heusch and R. Schulz, The contribution of reactive oxygen species and p38 mitogen-activated protein kinase to myofilament oxidation and progression of heart failure in rabbits, Br. J. Pharmacol., 2010, 160, 1408–1416 CAS.
- I. Dolado, A. Swat, N. Ajenjo, G. De Vita, A. Cuadrado and A. R. Nebreda, p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis, Cancer Cell, 2007, 11, 191–205 CrossRef CAS.
- P. J. Nadeau, S. J. Charette, M. B. Toledano and J. Landry, Disulfide Bond-mediated multimerization of Ask1 and its reduction by thioredoxin-1 regulate H(2)O(2)-induced c-Jun NH(2)-terminal kinase activation and apoptosis, Mol. Biol. Cell, 2007, 18, 3903–3913 CrossRef CAS.
- C. J. Staples, D. M. Owens, J. V. Maier, A. C. Cato and S. M. Keyse, Cross-talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation, J. Biol. Chem., 2010, 285, 25928–25940 CrossRef CAS.
- D. M. Owens and S. M. Keyse, Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases, Oncogene, 2007, 26, 3203–3213 CrossRef CAS.
- K. I. Patterson, T. Brummer, P. M. O'Brien and R. J. Daly, Dual-specificity phosphatases: critical regulators with diverse cellular targets, Biochem. J., 2009, 418, 475–489 CAS.
- J. M. Denu and K. G. Tanner, Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: evidence for a sulfenic acid intermediate and implications for redox regulation, Biochemistry, 1998, 37, 5633–5642 CrossRef CAS.
- H. Kamata, S. Honda, S. Maeda, L. Chang, H. Hirata and M. Karin, Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases, Cell, 2005, 120, 649–661 CrossRef CAS.
- A. Salmeen, J. N. Andersen, M. P. Myers, T. C. Meng, J. A. Hinks, N. K. Tonks and D. Barford, Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate, Nature, 2003, 423, 769–773 CrossRef CAS.
- S. Gross, A. Knebel, T. Tenev, A. Neininger, M. Gaestel, P. Herrlich and F. D. Bohmer, Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction, J. Biol. Chem., 1999, 274, 26378–26386 CrossRef CAS.
- Y. Xu, Y. Shao, J. J. Voorhees and G. J. Fisher, Oxidative inhibition of receptor-type protein-tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes, J. Biol. Chem., 2006, 281, 27389–27397 CrossRef CAS.
- I. A. Manke, A. Nguyen, D. Lim, M. Q. Stewart, A. E. Elia and M. B. Yaffe, MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation, Mol. Cell, 2005, 17, 37–48 CrossRef CAS.
- H. C. Reinhardt, A. S. Aslanian, J. A. Lees and M. B. Yaffe, p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage, Cancer Cell, 2007, 11, 175–189 CrossRef CAS.
- H. C. Reinhardt and M. B. Yaffe, Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2, Curr. Opin. Cell Biol., 2009, 21, 245–255 CrossRef CAS.
- B. B. Zhou and S. J. Elledge, The DNA damage response: putting checkpoints in perspective, Nature, 2000, 408, 433–439 CrossRef CAS.
- C. Johansen, C. Vestergaard, K. Kragballe, G. Kollias, M. Gaestel and L. Iversen, MK2 regulates the early stages of skin tumor promotion, Carcinogenesis, 2009, 30, 2100–2108 CrossRef CAS.
- Y. Zhang, S. Zhong, Z. Dong, N. Chen, A. M. Bode and W. Ma, UVA induces Ser381 phosphorylation of p90RSK/MAPKAP-K1 via ERK and JNK pathways, J. Biol. Chem., 2001, 276, 14572–14580 CrossRef CAS.
- L. O. Klotz, C. Pellieux, K. Briviba, C. Pierlot, J. M. Aubry and H. Sies, Mitogen-activated protein kinase (p38-, JNK-, ERK-) activation pattern induced by extracellular and intracellular singlet oxygen and UVA, Eur. J. Biochem., 1999, 260, 917–922 CrossRef CAS.
- A. L. Silvers, J. S. Finch and G. T. Bowden, Inhibition of UVA-induced c-Jun N-terminal kinase activity results in caspase-dependent apoptosis in human keratinocytes, Photochem. Photobiol., 2006, 82, 423–431 CrossRef CAS.
- M. A. Bachelor, A. L. Silvers and G. T. Bowden, The role of p38 in UVA-induced cyclooxygenase-2 expression in the human keratinocyte cell line, HaCaT, Oncogene, 2002, 21, 7092–7099 CrossRef CAS.
- K. L. Cooper, K. J. Liu and L. G. Hudson, Enhanced ROS production and redox signaling with combined arsenite and UVA exposure: contribution of NADPH oxidase, Free Radical Biol. Med., 2009, 47, 381–388 CrossRef CAS.
- A. Van Laethem, S. Van Kelst, S. Lippens, W. Declercq, P. Vandenabeele, S. Janssens, J. R. Vandenheede, M. Garmyn and P. Agostinis, Activation of p38 MAPK is required for Bax translocation to mitochondria, cytochrome c release and apoptosis induced by UVB irradiation in human keratinocytes, FASEB J., 2004, 18, 1946–1948 CAS.
- K. Nys, A. Van Laethem, C. Michiels, N. Rubio, J. G. Piette, M. Garmyn and P. Agostinis, A p38(MAPK)/HIF-1 pathway initiated by UVB irradiation is required to induce Noxa and apoptosis of human keratinocytes, J. Invest. Dermatol., 2010, 130, 2269–2276 CrossRef CAS.
- J. Hildesheim, R. T. Awwad and A. J. Fornace Jr., p38 Mitogen-activated protein kinase inhibitor protects the epidermis against the acute damaging effects of ultraviolet irradiation by blocking apoptosis and inflammatory responses, J. Invest. Dermatol., 2004, 122, 497–502 CrossRef CAS.
- M. A. Bachelor and G. T. Bowden, Ultraviolet A-induced modulation of Bcl-XL by p38 MAPK in human keratinocytes: post-transcriptional regulation through the 3′-untranslated region, J. Biol. Chem., 2004, 279, 42658–42668 CrossRef CAS.
- A. Mahns, R. Wolber, F. Stab, L. O. Klotz and H. Sies, Contribution of UVB and UVA to UV-dependent stimulation of cyclooxygenase-2 expression in artificial epidermis, Photochem. Photobiol. Sci., 2004, 3, 257–262 CAS.
- S. Manandhar, A. You, E. S. Lee, J. A. Kim and M. K. Kwak, Activation of the Nrf2-antioxidant system by a novel cyclooxygenase-2 inhibitor furan-2-yl-3-pyridin-2-yl-propenone: implication in anti-inflammatory function by Nrf2 activator, J. Pharm. Pharmacol., 2008, 60, 879–887 CrossRef CAS.
- J. Zhang, G. Tsaprailis and G. T. Bowden, Nucleolin stabilizes Bcl-X L messenger RNA in response to UVA irradiation, Cancer Res., 2008, 68, 1046–1054 CrossRef CAS.
- E. Espel, The role of the AU-rich elements of mRNAs in controlling translation, Semin. Cell Dev. Biol., 2005, 16, 59–67 CrossRef CAS.
- M. R. Fabian, N. Sonenberg and W. Filipowicz, Regulation of mRNA translation and stability by microRNAs, Annu. Rev. Biochem., 2010, 79, 351–379 CrossRef CAS.
- J. Houseley and D. Tollervey, The many pathways of RNA degradation, Cell, 2009, 136, 763–776 CrossRef CAS.
- M. Srivastava and H. B. Pollard, Molecular dissection of nucleolin's role in growth and cell proliferation: new insights, FASEB J., 1999, 13, 1911–1922 CAS.
- C. Y. Chen, R. Gherzi, J. S. Andersen, G. Gaietta, K. Jurchott, H. D. Royer, M. Mann and M. Karin, Nucleolin and YB-1 are required for JNK-mediated interleukin-2 mRNA stabilization during T-cell activation, Genes Dev., 2000, 14, 1236–1248 CAS.
- T. K. Sengupta, S. Bandyopadhyay, D. J. Fernandes and E. K. Spicer, Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization, J. Biol. Chem., 2004, 279, 10855–10863 CrossRef CAS.
- Y. Zhang, D. Bhatia, H. Xia, V. Castranova, X. Shi and F. Chen, Nucleolin links to arsenic-induced stabilization of GADD45alpha mRNA, Nucleic Acids Res., 2006, 34, 485–495 CrossRef CAS.
- C. Yang, D. A. Maiguel and F. Carrier, Identification of nucleolin and nucleophosmin as genotoxic stress-responsive RNA-binding proteins, Nucleic Acids Res., 2002, 30, 2251–2260 CrossRef CAS.
- J. L. Dean, G. Sully, A. R. Clark and J. Saklatvala, The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilisation, Cell. Signalling, 2004, 16, 1113–1121 CrossRef CAS.
- M. Lasa, K. R. Mahtani, A. Finch, G. Brewer, J. Saklatvala and A. R. Clark, Regulation of cyclooxygenase 2 mRNA stability by the mitogen-activated protein kinase p38 signaling cascade, Mol. Cell. Biol., 2000, 20, 4265–4274 CrossRef CAS.
- M. Gaestel, MAPKAP kinases - MKs - two's company, three's a crowd, Nat. Rev. Mol. Cell Biol., 2006, 7, 120–130 CrossRef CAS.
- K. Abdelmohsen, A. Lal, H. H. Kim and M. Gorospe, Posttranscriptional orchestration of an anti-apoptotic program by HuR, Cell Cycle, 2007, 6, 1288–1292 CrossRef CAS.
- J. Zhang and G. T. Bowden, UVB irradiation regulates Cox-2 mRNA stability through AMPK and HuR in human keratinocytes, Mol. Carcinog., 2008, 47, 974–983 CrossRef CAS.
- N. S. Fernau, D. Fugmann, M. Leyendecker, K. Reimann, S. Grether-Beck, S. Galban, N. Ale-Agha, J. Krutmann and L. O. Klotz, Role of HuR and p38MAPK in ultraviolet B-induced post-transcriptional regulation of COX-2 expression in the human keratinocyte cell line HaCaT, J. Biol. Chem., 2010, 285, 3896–3904 CrossRef CAS.
- F. Farooq, S. Balabanian, X. Liu, M. Holcik and A. MacKenzie, p38 Mitogen-activated protein kinase stabilizes SMN mRNA through RNA binding protein HuR, Hum. Mol. Genet., 2009, 18, 4035–4045 CrossRef CAS.
- V. Lafarga, A. Cuadrado, I. Lopez de Silanes, R. Bengoechea, O. Fernandez-Capetillo and A. R. Nebreda, p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint, Mol. Cell. Biol., 2009, 29, 4341–4351 CrossRef CAS.
- J. S. Mudgett, J. Ding, L. Guh-Siesel, N. A. Chartrain, L. Yang, S. Gopal and M. M. Shen, Essential role for p38alpha mitogen-activated protein kinase in placental angiogenesis, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10454–10459 CrossRef CAS.
- R. H. Adams, A. Porras, G. Alonso, M. Jones, K. Vintersten, S. Panelli, A. Valladares, L. Perez, R. Klein and A. R. Nebreda, Essential role of p38alpha MAP kinase in placental but not embryonic cardiovascular development, Mol. Cell, 2000, 6, 109–116 CAS.
- S. E. Dickinson, E. R. Olson, J. Zhang, S. J. Cooper, T. Melton, P. J. Criswell, A. Casanova, Z. Dong, C. Hu, K. Saboda, E. T. Jacobs, D. S. Alberts and G. T. Bowden, p38 MAP kinase plays a functional role in UVB-induced mouse skin carcinogenesis, Mol. Carcinog., 2011, 50, 469–478 CrossRef CAS.
- J. G. Einspahr, G. T. Bowden, D. S. Alberts, N. McKenzie, K. Saboda, J. Warneke, S. Salasche, J. Ranger-Moore, C. Curiel-Lewandrowski, R. B. Nagle, B. J. Nickoloff, C. Brooks, Z. Dong and S. P. Stratton, Cross-validation of murine UV signal transduction pathways in human skin, Photochem. Photobiol., 2008, 84, 463–476 CrossRef CAS.
- J. G. Einspahr, S. P. Stratton, G. T. Bowden and D. S. Alberts, Chemoprevention of human skin cancer, Crit. Rev. Oncol. Hematol., 2002, 41, 269–285 CrossRef.
- V. A. Beardmore, H. J. Hinton, C. Eftychi, M. Apostolaki, M. Armaka, J. Darragh, J. McIlrath, J. M. Carr, L. J. Armit, C. Clacher, L. Malone, G. Kollias and J. S. Arthur, Generation and characterization of p38beta (MAPK11) gene-targeted mice, Mol. Cell. Biol., 2005, 25, 10454–10464 CrossRef CAS.
- G. Sabio, J. S. Arthur, Y. Kuma, M. Peggie, J. Carr, V. Murray-Tait, F. Centeno, M. Goedert, N. A. Morrice and A. Cuenda, p38gamma regulates the localisation of SAP97 in the cytoskeleton by modulating its interaction with GKAP, EMBO J., 2005, 24, 1134–1145 CrossRef CAS.
- E. M. Schindler, A. Hindes, E. L. Gribben, C. J. Burns, Y. Yin, M. H. Lin, R. J. Owen, G. D. Longmore, G. E. Kissling, J. S. Arthur and T. Efimova, p38delta Mitogen-activated protein kinase is essential for skin tumor development in mice, Cancer Res., 2009, 69, 4648–4655 CrossRef CAS.
- A. S. Haider, S. B. Peters, H. Kaporis, I. Cardinale, J. Fei, J. Ott, M. Blumenberg, A. M. Bowcock, J. G. Krueger and J. A. Carucci, Genomic analysis defines a cancer-specific gene expression signature for human squamous cell carcinoma and distinguishes malignant hyperproliferation from benign hyperplasia, J. Invest. Dermatol., 2006, 126, 869–881 CrossRef CAS.
- M. R. Junttila, R. Ala-Aho, T. Jokilehto, J. Peltonen, M. Kallajoki, R. Grenman, P. Jaakkola, J. Westermarck and V. M. Kahari, p38alpha and p38delta mitogen-activated protein kinase isoforms regulate invasion and growth of head and neck squamous carcinoma cells, Oncogene, 2007, 26, 5267–5279 CrossRef CAS.
- A. M. Bode and Z. Dong, The functional contrariety of JNK, Mol. Carcinog., 2007, 46, 591–598 CrossRef CAS.
- S. Lawler, Y. Fleming, M. Goedert and P. Cohen, Synergistic activation of SAPK1/JNK1 by two MAP kinase kinases in vitro, Curr. Biol., 1998, 8, 1387–1390 CrossRef CAS.
- A. L. Silvers, M. A. Bachelor and G. T. Bowden, The role of JNK and p38 MAPK activities in UVA-induced signaling pathways leading to AP-1 activation and c-Fos expression, Neoplasia, 2003, 5, 319–329 CAS.
- H. S. Choi, A. M. Bode, J. H. Shim, S. Y. Lee and Z. Dong, c-Jun N-terminal kinase 1 phosphorylates Myt1 to prevent UVA-induced skin cancer, Mol. Cell. Biol., 2009, 29, 2168–2180 CrossRef CAS.
- K. Sabapathy, K. Hochedlinger, S. Y. Nam, A. Bauer, M. Karin and E. F. Wagner, Distinct roles for JNK1 and JNK2 in regulating JNK activity and c-Jun-dependent cell proliferation, Mol. Cell, 2004, 15, 713–725 CrossRef CAS.
- E. F. Wagner, AP-1–Introductory remarks, Oncogene, 2001, 20, 2334–2335 CrossRef CAS.
- P. Angel and M. Karin, The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation, Biochim. Biophys. Acta, 1991, 1072, 129–157 CAS.
- R. Eferl and E. F. Wagner, AP-1: a double-edged sword in tumorigenesis, Nat. Rev. Cancer, 2003, 3, 859–868 CrossRef CAS.
- A. L. Silvers and G. T. Bowden, UVA irradiation-induced activation of activator protein-1 is correlated with induced expression of AP-1 family members in the human keratinocyte cell line HaCaT, Photochem. Photobiol., 2002, 75, 302–310 CrossRef CAS.
- W. Chen, A. H. Borchers, Z. Dong, M. B. Powell and G. T. Bowden, UVB irradiation-induced activator protein-1 activation correlates with increased c-fos gene expression in a human keratinocyte cell line, J. Biol. Chem., 1998, 273, 32176–32181 CrossRef CAS.
- F. E. Domann, J. P. Levy, M. J. Birrer and G. T. Bowden, Stable expression of a c-JUN deletion mutant in two malignant mouse epidermal cell lines blocks tumor formation in nude mice, Cell Growth Differ., 1994, 5, 9–16 CAS.
- F. E. Domann, Jr., J. P. Levy, J. S. Finch and G. T. Bowden, Constitutive AP-1 DNA binding and transactivating ability of malignant but not benign mouse epidermal cells, Mol. Carcinog., 1994, 9, 61–66 CrossRef.
- Z. Dong, M. J. Birrer, R. G. Watts, L. M. Matrisian and N. H. Colburn, Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 609–613 CrossRef CAS.
- M. R. Young, J. J. Li, M. Rincon, R. A. Flavell, B. K. Sathyanarayana, R. Hunziker and N. Colburn, Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9827–9832 CrossRef CAS.
- S. J. Cooper, J. MacGowan, J. Ranger-Moore, M. R. Young, N. H. Colburn and G. T. Bowden, Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model, Mol. Cancer Res., 2003, 1, 848–854 CAS.
- R. M. Tyrrell and S. M. Keyse, New trends in photobiology. The interaction of UVA radiation with cultured cells, J. Photochem. Photobiol., B, 1990, 4, 349–361 CrossRef CAS.
- J. G. Einspahr, D. S. Alberts, J. A. Warneke, P. Bozzo, J. Basye, T. M. Grogan, M. A. Nelson and G. T. Bowden, Relationship of p53 mutations to epidermal cell proliferation and apoptosis in human UV-induced skin carcinogenesis, Neoplasia, 1999, 1, 468–475 CAS.
- S. Euvrard, J. Kanitakis and A. Claudy, Skin cancers after organ transplantation, N. Engl. J. Med., 2003, 348, 1681–1691 CrossRef.
- X. Zhang, G. Jeffs, X. Ren, P. O'Donovan, B. Montaner, C. M. Perrett, P. Karran and Y. Z. Xu, Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light, DNA Repair, 2007, 6, 344–354 CrossRef CAS.
- Q. Gueranger, A. Kia, D. Frith and P. Karran, Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA, Nucleic Acids Res., 2011, 39, 5057–5066 CrossRef CAS.
- T. G. Rossman, A. N. Uddin and F. J. Burns, Evidence that arsenite acts as a cocarcinogen in skin cancer, Toxicol. Appl. Pharmacol., 2004, 198, 394–404 CrossRef CAS.
- T. G. Rossman, A. N. Uddin, F. J. Burns and M. C. Bosland, Arsenite is a cocarcinogen with solar ultraviolet radiation for mouse skin: an animal model for arsenic carcinogenesis, Toxicol. Appl. Pharmacol., 2001, 176, 64–71 CrossRef CAS.
- C. H. Tseng, Arsenic methylation, urinary arsenic metabolites and human diseases: current perspective, J. Environ. Sci. Health C: Environ. Carcinog. Ecotoxicol. Rev., 2007, 25, 1–22 CrossRef CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |