Photo-oxidation of proteins†
David I.
Pattison
ab,
Aldwin Suryo
Rahmanto
a and
Michael J.
Davies
*ab
aThe Heart Research Institute, 7 Eliza Street, Newtown, Sydney, NSW 2042, Australia
bFaculty of Medicine, University of Sydney, Sydney, NSW 2006, Australia. E-mail: daviesm@hri.org.au
First published on 22nd August 2011
Abstract
Photo-induced damage to proteins occurs via multiple pathways. Direct damage induced by UVB (λ 280–320 nm) and UVA radiation (λ 320–400 nm) is limited to a small number of amino acid residues, principally tryptophan (Trp), tyrosine (Tyr), histidine (His) and disulfide (cystine) residues, with this occurring via both excited state species and radicals. Indirect protein damage can occur via singlet oxygen (1O21Δg), with this resulting in damage to Trp, Tyr, His, cystine, cysteine (Cys) and methionine (Met) residues. Although initial damage is limited to these residues multiple secondary processes, that occur both during and after radiation exposure, can result in damage to other intra- and inter-molecular sites. Secondary damage can arise via radicals (e.g. Trp, Tyr and Cys radicals), from reactive intermediates generated by 1O2 (e.g. Trp, Tyr and His peroxides) and via molecular reactions of photo-products (e.g. reactive carbonyls). These processes can result in protein fragmentation, aggregation, altered physical and chemical properties (e.g. hydrophobicity and charge) and modulated biological turnover. Accumulating evidence implicates these events in cellular and tissue dysfunction (e.g.apoptosis, necrosis and altered cell signaling), and multiple human pathologies.
Introduction
Most forms of life on Earth are exposed to light on a daily basis, and in most cases this exposure is essential to viability and health. As the Earth's upper atmosphere absorbs UV radiation with wavelengths below ca. 295 nm (i.e. all of the UVC band, λ 200–280 nm, and a considerable proportion of the UVB band, λ 280–320 nm), the majority of exposure at the Earth's surface consists of UVA radiation (λ 320–400 nm; 95% of all UV exposure) and visible light, as well as the infra-red wavelengths. Despite the positive effects of light exposure, UV radiation and visible light in the presence of suitable chromophores can induce protein damage. This review discusses the mechanisms of these processes and its consequences.Mechanisms of photo-induced damage to proteins
Photo-induced damage to proteins can occur via two major pathways.1–4 The first of these is mediated predominantly by UVB (and to a lesser extent UVA) radiation, where direct absorption by particular amino acid residues (e.g.tryptophan (Trp), tyrosine (Tyr), phenylalanine (Phe), histidine (His), methionine (Met), cysteine (Cys) and disulfide (cystine) bonds) results in the formation of electronically-excited states and photo-ionization reactions. In addition to direct absorption of light, photosensitized reactions also occur through the absorption of UV and/or visible light by a sensitizing compound. These sensitizers can be endogenous species (e.g.porphyrins,5,6 aromatic lens filter compounds,7,8vitamins such as riboflavin9) or exogenous species (e.g.drugs, polyaromatic compounds and dye molecules10–15). The sensitizers are typically excited to a short lived excited singlet state that rapidly undergoes intersystem crossing to give a longer lived triplet. The latter either undergoes decay processes to the ground state, or reacts with other species by one of two pathways (known as Type I and Type II mechanisms) that can result in damage to target molecules (e.g.proteins). Type I damage results in free radical formation arising from either one electron oxidation or hydrogen atom abstraction from the substrate, whereas the Type II mechanism involves energy transfer from the excited sensitizer to molecular oxygen, to yield the first excited state of singlet oxygen (1O21Δg; reviewed in ref. 2, 16, and 17) which readily oxidizes surrounding targets. Sensitizers rarely react solely by a Type I or Type II mechanism due to variations in energy transfer efficiency, thus photosensitized reactions typically proceed via a mixture of 1O2 and radical species (e.g.O2−˙ and substrate radicals).16 The protein damage profile is typically affected by the proportion of each reactive species generated through photosensitization, and therefore depends critically on the nature of the sensitizer, the excitation wavelength and the reaction conditions.16Damage to proteins induced by UV/visible irradiation
Proteins are major cellular targets for direct photo-oxidation due to their high abundance and the presence of endogenous chromophores within the protein structure (certain amino acid side-chains as well as bound chromophores, such as flavins and porphyrins). A major pathway for oxidation of amino acids, peptides and proteins by UV light is via the absorption of the incident radiation by the protein. The major intrinsic chromophoric species present in proteins are Trp, Tyr, Phe, His, Cys and cystine side chains, with all other amino acid side chains and peptide bonds only absorbing radiation with λ < 230 nm. Upon absorption of UV light the chromophoric residues are typically converted to their first excited singlet states that have only short lifetimes.2 These states readily lose energy by inter-system crossing to the triplet state, and energy transfer to other groups through collisional deactivation. The resulting triplet states typically have much longer lifetimes (on the order of microseconds) than the singlet states and are able to undergo electron transfer and hydrogen abstraction mechanisms, rather than solely energy transfer processes. The (photo)physical properties of these triplets are well characterised (reviewed in ref. 2 and 4) and will only be reviewed briefly below. The chemistry of the (hydrated) electron resulting from direct photo-ionisation is also well characterized,18–20 with addition to O2, to give O2−˙, being a major and fast reaction (k = ca. 1010 dm3 mol−1 s−1).18 Hydrated electrons can also add to free carboxyl groups (e.g. the C-terminus, or Asp/Glu side-chains) and amine groups (e.g. the N-terminus, or Lys side-chains) in processes that result in deamination and elimination of H˙ respectively. Addition can also occur at other sites, particularly with cystine (to give a radical-anion, RSSR−˙), and protonated His side-chains. In peptides, the hydrated electron can also add to the carbonyl groups of the peptide backbone to give a radical-anion that can subsequently give rise to backbone cleavage. These reactions have been reviewed.18–21Indirect photo-oxidation of proteins occurs through the formation and subsequent reactions of 1O2 with Trp, His, Tyr, Met and Cys side-chains. These reactions are facile as 1O2 reacts with these protein side-chains more rapidly than with most other cellular targets at pH 7.4.3,4,16,17,22 Furthermore, proteins are also present in most biological systems at much higher concentrations than other potential targets for 1O2 (reviewed in ref. 3, 4, and 17). Under alkaline conditions (typically at pH > 10) 1O2 also reacts with Arg and Lys in their neutral (unprotonated) forms.23 Whilst direct 1O2-mediated damage to proteins primarily occurs at these residues, consumption of other amino acids can also occur through reaction of 1O2-induced intermediates (such as hydroperoxides) with these residues.
The following sections discuss the nature and mechanism of UV damage to each of these susceptible amino acids in more detail.
Tryptophan side-chains
The indolic side-chain of Trp is the strongest UV chromophore in proteins (with the absorbance band exhibiting a high molar absorptivity which extends to greater wavelengths than any of the other residues), but the overall significance of Trp photo-oxidation is attenuated by its relatively low abundance in many proteins. Nevertheless, Trp exhibits a rich photochemistry. The first triplet state of Trp (3Trp) readily undergoes electron transfer reactions with suitable electron acceptors24 to give, for example, with disulfides the corresponding radical anion (RSSR−˙) and a Trp radical-cation (Trp+˙; reviewed in ref. 2). The Trp radical-cations formed via these mechanisms rapidly deprotonate to the neutral indolyl radical (Trp˙; Fig. 1).25 Subsequent reactions of the indolyl (Trp˙) radicals include reaction with O2 to give a ring C-3 peroxyl radical that can undergo further hydrogen atom abstraction reactions (Fig. 1).21,25–27 Evidence has been presented for the repair of both the indolyl and peroxyl radicals by antioxidants such as ascorbate, selenocysteine and GSH (e.g.28–32).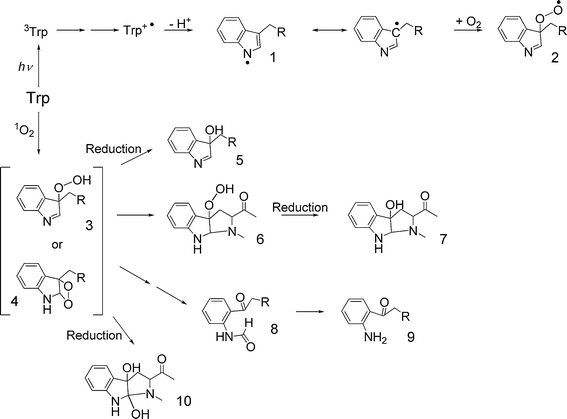 | ||
| Fig. 1 Mechanisms of oxidation of Trp by direct UV absorption and 1O2-mediated pathways. Key to structures (where the α-amino acid moiety is represented by R = CH(CO2H)NH2): 1, indolyl radical; 2, C-3 peroxyl radical; 3, C-3 hydroperoxide; 4, dioxetane intermediate; 5, C-3 alcohol; 6, 3α-hydroperoxypyrroloindole; 7, 3α-hydroxypyrroloindole; 8, N-formylkynurenine; 9, kynurenine; 10, 3α-dihydroxypyrroloindole. | ||
Trp residues can also undergo direct photo-ionization processes. There has been some debate over whether these are mono- or bi-photonic processes,24,33 but recent studies suggest that both mono- and bi-photonic mechanisms occur simultaneously,34,35 and extensive research continues on the multi-photon chemistry of Trp.
In addition to direct photo-oxidation, Trp residues are also highly susceptible to 1O2-mediated damage. Oxidation of Trp by 1O2 initially results in the formation of either a dioxetane intermediate across the C2–C3 double bond of Trp, or a hydroperoxide at the C3 position (Fig. 1).36,37 These intermediates can subsequently decompose to N-formylkynurenine (and hence kynurenineviahydrolysis) or can undergo ring closure reactions to form 3α-hydroperoxypyrroloindoles and 3α-dihydroxypyrroloindoles.38–41Decomposition of the peroxide intermediates may involve non-radical reactions to yield, for example, 3α-hydroxypyrroloindoles, or homolysis of the –O–O– bond to give radicals. Metal ions, heat and UV light have been shown to catalyze Trp peroxide decomposition to radicals,38 and the mechanism and products of such reactions has been reviewed.37 These processes have recently been reinvestigated in detail using isotopically 18O-labeled 1O2 in H2O to generate the various intermediates; these studies have allowed the origin of incorporated oxygen atoms to be accurately assessed.40,41 Similar reactions appear to occur with both the free amino acid and N-blocked Trp residues, and hence analogous materials are formed on Trp-containing peptides and proteins, though the yields of some of the intermediates is modulated.38,39
Two of the major products of Trp photo-oxidation, N-formylkynurenine and kynurenine, are more effective photosensitizing agents than the parent amino acid42 and can produce a variety of reactive species that further degrade these and other structures. Prolonged exposure (over several hours) of Trp to UV/visible light results in extensive Trp photo-degradation leading to products arising from dehydrogenation and cyclisation reactions39,43 as well as hydroxylated Trp and kynurenine derivatives that implicate hydroxyl radicals (or radical-cations, with subsequent rapid hydration to give the hydroxylated species) in the photo-oxidation process.43,44 A recent study has reported the detection of nitrotryptophan in such systems, suggesting that reactive nitrating species are formed as a result of UVA photolysis of Trp-containing peptides;43,44 the mechanism underlying this surprising and interesting observation, has not been elucidated. A number of oxidation/degradation products of free Trp have been shown to react with proteins, and particularly lens proteins, with this resulting in the formation of covalently-bound adducts at Cys, His and Lys residues.8,45–48 These materials have been shown to act as sensitizers of further oxidative damage.48,49 Thus, the formation of N-formylkynurenine, kynurenine and related materials on UV-exposed proteins may lead to an enhancement in the photo-oxidation of the target proteins due to further generation of reactive species.
Tyrosine side-chains
The first triplet state of Tyr undergoes similar electron transfer processes to those described above for Trp, leading to formation of Tyr+˙ and phenoxyl radicals following (rapid) deprotonation of the radical-cation (Fig. 2).2,25 As for Trp, the radical-cation of Tyr can also be generated via direct photo-ionization reactions.2,50 The Tyr phenoxyl radicals generated by these mechanisms can undergo a wide variety of reactions, including oxidation of susceptible substrates51–53 and dimerization (via C–O and C–C linkages) to yield di-tyrosine products and hydrogen atom abstraction reactions (see Fig. 2; reviewed in ref. 21). The Tyr phenoxyl radical can also be efficiently repaired by a range of low-molecular-mass antioxidants including ascorbate, nitric oxide, glutathione and selenocysteine.28–32,54,55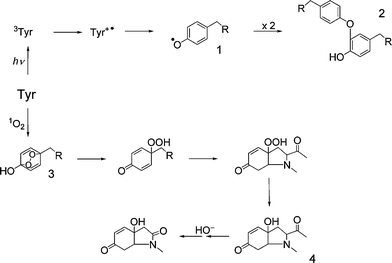 | ||
| Fig. 2 Oxidation pathways for Tyrvia direct UV absorption or 1O2 production. Key to structures (where the α-amino acid moiety is represented by R = CH(CO2H)NH2): 1, tyrosyl radical; 2, a C–O linked isomer of a dityrosine crosslink; 3, Tyr endoperoxide; 4, HOHICA (3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid). | ||
Photo-oxidation of free Tyr residues by 1O2 initially yields unstable endoperoxides (Fig. 2). These rapidly decompose via ring opening to give a C1 hydroperoxide, and cyclized products involving nucleophilic addition of the α-amino group.56–59 Thermal decay of these intermediates gives rise to a cyclized indolic product, 3a-hydroxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid (HOHICA; Fig. 2),56–59 which can be oxidized further to a decarboxylated keto compound under basic conditions.56,57 The decomposition of the initial peroxides and cyclized products can be accelerated by exposure to metal ions or UV light resulting in radical production, but the identities of the decomposition products generated via these radical pathways are unknown.58,60
In peptides and proteins, the nucleophilic ring closure reactions of the α-amino group are less favorable (due to its incorporation into a peptide bond), and consequently reaction with other nucleophiles can compete with this cyclization (A. Wright, C. L. Hawkins and M. J. Davies, unpublished data). Such addition reactions could play an important role in protein cross-linking if neighbouring Lys, Arg or Cys residues were to provide the nucleophilic centre. Previous studies have reported that di-tyrosine is not a major product of 1O2-mediated oxidation of Tyr on proteins,61,62 and it has been suggested that another Tyr-derived product, 3,4-dihydroxyphenylalanine (DOPA) is not generated by reaction of 1O2.61,62 However, more recent data have shown that photolysis of proteins in the absence or presence of a 1O2-generating photosensitizer (e.g. Rose Bengal) generates both of these materials in significant yields.43,44,63,64 Further oxidation and hydroxylation of DOPA residues to form dopaquinone and trihydroxyphenylalanine (TOPA) derivatives has also been demonstrated on prolonged exposure of Tyr solutions to UVA light.43,44
Phenylalanine side-chains
UV-mediated oxidation of Phe is relatively straightforward when compared to the other aromatic amino acids. Excitation of Phe to the first triplet state results in a simple photo-dissociation reaction that yields a benzyl radical (Fig. 3).65 Direct photo-ionization of Phe results in radical-cation formation, which undergoes rapid hydration to yield hydroxylated ring products (o-, m- and p-Tyr; Fig. 3; reviewed in ref. 50), though there is also evidence for deprotonation to yield benzyl radicals.25 These processes are however only significant with short wavelength UV radiation.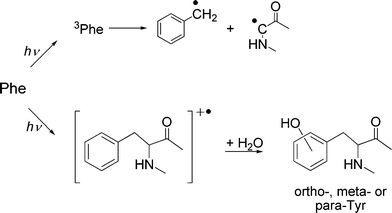 | ||
| Fig. 3 Direct UV absorption by Phe leads to formation of the triplet state and subsequent radical formation, or photo-ionization followed by hydration to form Tyr isomers. | ||
Histidine side-chains
Direct absorption of UV or visible light by His is not a major mechanism, and the majority of damage observed at such residues occurs via sensitized processes. Recent studies with model His compounds in the presence of flavins have demonstrated the formation of radicals on the imidazole function,66 and this has also been demonstrated with other sensitizers.67 The direct interaction (via a Type I mechanism) between the triplet state flavin and His (as determined by EPR spin trapping techniques) is markedly pH dependent,66 with this attributed to electronic effects arising from the differential protonation state of the imino group.66 The majority of His oxidation mediated by photo-excited flavins appears to occur via production of 1O2, consistent with studies with other sensitizers where evidence for 1O2 as the reactive intermediate has been obtained.67His oxidation, both free and in peptides, by 1O2 has been studied extensively, and occurs via the initial formation of one or more endoperoxides (Fig. 4).67,68 The structure of these endoperoxides has been determined by low temperature NMR in organic solvents,69,70 but at higher (ambient) temperatures they undergo rapid reactions that result in the formation of multiple decomposition products including aspartic acid, asparagine and urea (Fig. 4).68,71,72 These products arise from opening of the imidazole ring, and may occur via radical processes.67,73 In aqueous solutions, free His also yields bicyclic products generated by nucleophilic attack of the α-amino group onto the oxidized, but still intact, imidazole ring, as characterized by NMR.67 Many of these initial His-derived products can undergo further reactions, with this resulting in the formation of dimers from N-acetyl-His,67 and His-His and His-Lys cross-links following nucleophilic addition of a (non-oxidized) His or Lys side-chain, to a keto group on an oxidized His.74,75
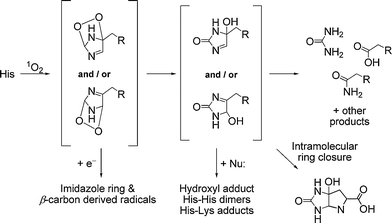 | ||
| Fig. 4 Reaction pathways for 1O2-mediated oxidation of His residues (where the α-amino acid moiety is represented by R = CH(CO2H)NH2). | ||
In addition to 1O2-mediated photo-oxidation of His residues, it has been shown that light exposure of proteins in the presence of stilbene-derived fluorescent whitening agents (FWA; commonly used in the textile and clothing industries) can lead to the formation of 2-oxo-His and nitrated His derivatives.76 These products suggest that radical species may be involved, and the formation of peroxynitrite (ONOO−/ΟNΟΟH) has been suggested,76 leading in turn to the formation of HO˙ and NO2. viaONOOH decomposition (or CO3−˙ and NO2. from the corresponding CO2 adduct of ONOO−). The mechanism of this photo-induced nitration of protein-bound His residues has not been definitively elucidated.
Cysteine and cystine groups
The direct absorption of UV radiation by disulfide bonds, and ensuing reactions, has primarily been studied with shorter UV wavelengths (λ < 260 nm) than those under consideration here. Under these conditions there is evidence that the initial singlet state undergoes homolytic cleavage of the disulfide bond to form two short-lived thiyl radicals that can recombine to regenerate the parent disulfide (reviewed in ref. 77). However, there is controversy over the identities of the radicals formed, with suggestions that the key intermediates are RSS˙, or even [RS-(SR)-SR]˙ formed via adduction of RSS˙ to a further cysteine moiety (see ref. 78). Short wavelength UV irradiation of cystine can also yield the radical-cation (RSSR+˙) and cystine S-monoxide (among other products), in the presence of O2.78 Evidence for homolytic cleavage of the C–S bond has also been presented following irradiation with UVC wavelengths.79 Recent studies using longer wavelength photolysis (λ ca. 300 nm) of a model peptide containing an intrachain disulfide bond, have demonstrated the formation of a thiyl radical pair that can rapidly recombine or undergo a series of complex rearrangement reactions leading to the formation of products such as cyclic thioethers and H2S.80A common mechanism of photo-oxidation of disulfide bonds is viaelectron transfer from 3Trp or 3Tyr species to cystine, with this resulting in the formation of disulfide radical anions (RSSR−˙).2,24,77,81–84 This species can readily dissociate, in a reversible reaction, to give the anion (RS−) and a radical (RS˙) or can undergo rapid electron transfer with O2 to give O2−˙.4,21Thiyl radicals formed via such dissociation have been shown to undergo multiple reactions including hydrogen abstraction from suitable C–H bonds (and hence propagation of damage and chain reactions, both within and between proteins and peptides, and with other biomolecules),85–89 recombination with the thiyl anion to regenerate the disulfide radical anion, or reaction with O2 to give a thiyl peroxyl radical (RSOO˙) (reviewed in ref. 21). The resulting thiyl peroxyl radicals can eliminate O2 thereby reverting to thiyl radicals, or can isomerize to a sulfonyl radical (RS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O˙) and hence give rise to sulfinic (RSO2H) and sulfonic (RSO3H) acids (reviewed in ref. 90,91).
O)O˙) and hence give rise to sulfinic (RSO2H) and sulfonic (RSO3H) acids (reviewed in ref. 90,91).
Production of 1O2 through photosensitization reactions is a major pathway to Cys oxidation, with this postulated to occur via formation of a zwitterion (RS+–OO−);92,93 subsequent decomposition of this short-lived intermediate yielding cystine, thiosulfinates and other species.92Cystine, is a major reaction product16,94 when further Cys residues are able to react with the initial intermediate. However, other products including cysteic acid (RSO3H) can also be formed, when such reactions are slow or unfavourable.16
Methionine side-chains
Photosensitized oxidation of Met residues has been studied extensively, and has been reviewed recently.95 Many of these studies have utilized aromatic ketones as sensitizers, excited with laser wavelengths in the 300–400 nm region. The triplet states that are formed undergo charge-transfer with Met to form the radical cation MetS+˙,96,97 which can undergo a range of reactions depending on the pH, including dimerization (to give the corresponding dimeric radical-cation with a two-centre, three-electron bond) and deprotonation reactions to form carbon-centred radicals from adjacent C–H bonds on the side-chain.98,99Met can also be oxidized by 1O2 arising from sensitizers.4,95,100 This leads to formation of a zwitterionic species (R2S+-OO−) which, depending on the pH, either oxidizes a second Met (to give two moles of Met sulfoxide; MetSO) or eliminates H2O2 to form a single mole of MetSO101 (reviewed in ref. 16). Stable cyclic intermediates involving the α-amino nitrogen and the sulfur atom of the thioether have also been reported (reviewed in ref. 16), with subsequent hydrolysis again yielding a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of MetSO and H2O2.101
:1 ratio of MetSO and H2O2.101
There is little evidence for direct UV/visible damage to Met residues, although a recent study has demonstrated, by X-ray absorption spectroscopy, that Met is converted to MetSO and MetS+˙ in the presence or absence of O2, respectively.102 As these studies have been carried out in the absence of a sensitizer, the formation of these products is attributed to UV absorption by weak bands at 262 and 285 nm that tail into the UVA region. However, these studies used a Xe arc lamp as the light source, and it is unclear as to what wavelengths are responsible for the observed oxidation products.
Damage transfer reactions within proteins
Transfer of damage between the initial sites of oxidation in peptides and proteins and other residues is well characterised and primarily involves aromatic and sulfur-containing residues. Examination of these processes has yielded important information about the transfer of oxidizing and reducing equivalents through proteins (reviewed in ref. 103–106). The reduction potentials of side-chain and backbone-derived radicals indicate that the ultimate sink for oxidising equivalents in proteins are Tyr and Cys residues (or Trp is the absence of these side-chains).104 Some of these reactions are equilibria and hence damage transfer can occur in both directions and the overall effects can be readily reversed. Thus Tyr phenoxyl radicals can both be generated by thiyl radicals and repaired by Cys reactions,104,107 with the balance of these processes determined, at least in part, by the concentration of thiol anions, as thiyl radicals can be removed via reaction with these species (to give the disulfide radical anion).Oxidation of Trp residues can yield indolyl radical, but it has been established that if such species are generated on proteins that also contain Tyr residues, rapid electron transfer can occur between these species to yield Tyr phenoxyl radicals;103,105 such transfer can occur over long distances within (and between) proteins, but the rate of these processes is critically dependent on the protein structure. Transfer can be slow even if the residues are close in distance, if they are present in unfavourable and rigid conformations.108 In contrast, transfer can occur rapidly over very long distances if the structure is flexible, or if there are suitable intervening residues that can act as a conduit for electron migration.104,108,109 In many proteins the exact transfer pathway is unclear due to the presence of multiple alternative pathways. These processes may result in a different pattern and rates of oxidation than might be initially expected on the basis of the reactivity of the initiating oxidant.104
The cystine disulfide bond is the major sink for photo-ejected electrons and reducing equivalents, with this competing with direct addition to O2 in some cases.103 Transfer to disulfide bonds can occur via both hydrogen bonding networks and via the backbone.106,110 The formation of disulfide radical-anions via such reactions can be of particular importance, as thiyl radicals formed from cleavage of the radical-anion can in turn oxidise Tyr residues, thereby providing a point of convergence of oxidation and reduction pathways.
Evidence has also been obtained for radical transfer from side-chain sites to the α-carbon, and for repair of α-carbon radicals by Cys residues. It has been shown that thiyl radicals generated from the Cys residue in GSH can abstract a hydrogen atom from backbone α-carbon sites,111–113 due to the stability of the α-carbon radical. Analogous thiyl to α-carbon transfer reactions have also been detected with the thiyl radical from homocysteine (when the amino group is deprotonated),114 and Cys itself, despite the apparent involvement of an (unfavourable) 1,4 rearrangement in the latter reaction.89,115,116
Structural and functional effects of photo-induced protein oxidation
The data reviewed above illustrate the diversity of damage that can be generated on proteins on exposure to UV/visible light in the presence or absence of a sensitizing molecule and O2. However, the consequences of photo-induced protein oxidation generally fall into a few discreet categories that are outlined below.Protein unfolding
Photo-oxidation of some proteins can result in an increased extent or susceptibility to unfolding and conformational changes, enhanced exposure of hydrophobic residues, and altered light scattering and optical rotation properties (e.g. ref. 16, 61, and 117–122). Studies on apo (i.e. non-heme containing) horseradish peroxidase have provided some insight into the mechanisms of unfolding, as this protein is highly resistant to photo-oxidation, with this attributed, at least in part, to the tertiary protein structure protecting susceptible residues from damage.123 Studies on milk proteins have demonstrated that random coil proteins (such as caseins) are more susceptible to individual amino acid damage than more densely-packed globular proteins (e.g. lactalbumin, lactoglobulin),120 consistent with this hypothesis. However in the study of milk proteins, the proteins displaying the greatest changes in secondary and/or tertiary structure were globular proteins that exhibited less measurable damage at the individual amino acid level.120 In contrast, photo-oxidation of lactoferrin resulted in a more compact protein structure, rather than protein unfolding.120 These data are consistent with low levels of damage to particular residues in globular proteins having major effects on protein structure, particularly if these are buried within a compact structure. In contrast, damage to exposed residues (e.g. in random coil structures) appears to have much less marked structural impact. These limited data indicate that there are multiple factors that contribute to photo-induced protein unfolding, with the contribution of each process likely to be dependent on the structure and conformation of individual proteins; the contribution of individual factors remain to be elucidated.Protein aggregation
The formation of high-molecular-mass aggregates (dimers and higher species) is a common and well-documented consequence of protein photo-oxidation.121,122,124–126 Aggregates may arise from radical–radical termination reactions, such as reaction of two Tyr-derived phenoxyl radicals to give di-tyrosine (alternatively notated as bi-tyrosine) (e.g. ref. 63, 120, and 127), but multiple other reactions have also been shown to lead to cross-link formation, with some of these occurring via “dark” reactions after the cessation of light exposure.128 Evidence has been presented for a role for oxidized His residues in cross-link formation,61,67,74,75,129,130 with this potentially involving His oxidation products that contain carbonyl functions which can undergo addition reactions with other nucleophiles.67,68 An increase in the yield of (unspecified) protein carbonyls has been reported on photo-oxidation of albumin by porphyrin sensitizers.6,131 It is likely that at least some of the reported cross-links arise from reaction of carbonyl compounds (on His and other residues) formed by photo-oxidation, with nucleophilic Lys, Arg and Cys side chains. The formation of such cross-links may explain the observed loss of Lys and Arg residues in some photo-oxidized proteins (reviewed in ref. 16). His residues have been suggested to be of particular importance in cross-link generation, as it has been reported that cross-links are not formed with proteins subject to photo-oxidation that lack His residues;61,62 the number of proteins that fall in to this category are however low, and hence other factors may also be important.Protein fragmentation
Photo-induced fragmentation of proteins appears to be of lesser importance than aggregation with many proteins, although there is evidence that it can occur under some circumstances.16,132,133Lysozyme has been shown to be fragmented by photolysis in the presence of a sensitizer due to backbone cleavage, with Trp oxidation and radical formation implicated in this process.134 Radicals formed at the α-carbon site on the peptide backbone are known to be key intermediates in chain cleavage in the presence of O2 (reviewed in ref. 4, 19, and 135), and peroxides formed on His and Tyr residues have been shown to be capable of generating such radicals at least in small peptides.58,60,67 Thus initial oxidation at side-chain sites may result in subsequent backbone cleavage via damage transfer within a protein (or possibly on other proteins, though this has not been examined in detail), with radicals as key intermediates. These transfer reactions may occur to nearby residues within a protein sequence, or to remote locations in the protein sequence, if the sites are close in space within the protein 3-D structure.Enzyme inactivation
Enzymes have long been known to be inactivated on exposure to UV light,136 and a considerable number of studies on such loss of activity have been published (reviewed in ref. 2, 16, and 17). Much of the photochemistry that occurs with free amino acids also appears to occur with proteins (except where cyclization reactions involving the free α-amino group are involved) and hence enzymes with active site (or otherwise critical) Cys, Met, Trp, His or Tyr residues are likely to be most susceptible to photo-inactivation. Direct photo-oxidation of such residues may only be one cause of enzyme inactivation, as a number of studies have shown that reactive protein peroxides, formed principally on His, Trp and Tyr residues, can give rise to secondary inactivation of other enzymes, at least in vitro (reviewed in ref. 3). Similar effects have recently been reported in cells exposed to visible light and sensitizers, with enzyme inactivation occurring both during, and after the cessation of light exposure.137–139 Photo-generated peroxides have been shown to be capable of oxidizing susceptible residues that lead to enzyme inactivation in crucial intracellular enzymes such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH),140 caspases,141proteintyrosine phosphatases (PTP),142 cathepsins and related proteases,143 as well as multi-enzyme complexes such as the 26S proteasome.144 Furthermore, protein peroxides also react rapidly with, and hence deplete, low-molecular-mass antioxidants such as GSH145 which may have important ramifications for the function of other antioxidant enzymes (e.g. the glutathione peroxidase family; GPxs) via depletion of crucial co-factors.138,145–147 Recent studies have shown that these protein- and peptide-derived peroxide reactions may be modulated by peroxiredoxins (Prx2/Prx3) via rapid competitive reaction of the peroxide with the Cys residues in the peroxiredoxins.148Consequences of protein photo-oxidation in biological systems
Modulation of protein structure and function, whether occurring via direct or indirect processes as outlined above, can affect a wide range of cellular and biochemical responses. The consequences of such modulation will be discussed in this section, in particular in the context of cellular signalling, redox homeostasis, proteolytic turnover of damaged molecules, and cell survival or death. This discussion is not exhaustive and only highlights some particular examples; further data in some of these effects can be found in other articles within this issue.Effects of photo-oxidation on cellular signalling
Multiple reactive intermediates (e.g.1O2, O2˙−, HO˙, H2O2, other peroxides) generated by light exposure can modulate various cellular signalling pathways including the mitogen-activated protein kinase family (MAPK), c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinases (ERK1/2) and nuclear factor-κB (NFκB).149–152Experimental studies with isolated proteins, cells, and tissues have documented that reactive species generated as a result of exposure to UV or visible light are capable of both activating or inactivating kinases, and inactivating phosphatases.153–155 This is likely to result in changes in the overall extent of protein phosporylation as a result of the complimentary action of these enzyme families in adding and removing phosphate groups respectively. The effect of exposure is likely to be system-specific due to the large number of kinases and phosphatases within cells/tissues. Furthermore the different members of these families are probably differentially susceptible to oxidative damage and changes in activity. It is therefore impossible to generalize as to what effect UV and/or visible light will have on phosphorylation status of any particular system. Phosphatase and tensin homolog (PTEN), protein tyrosine phosphatase 1B (PTP 1B), MAPK phosphatase, calcineurin, and low-molecular-weight (LMW) tyrosine phosphatase are a few examples of phosphatase enzymes whose activity is affected by UV and/or visible light exposure, with this reported to be due to oxidation of redox-sensitive Cys residues, particularly that present in the active sites of the phosphatases.156 This Cys is critical for enzyme activity, and is readily oxidised to multiple species (see above) some of which are reversible (e.g. formation of sulfenic acid or disulfide species), and some not (e.g. sulfinic and sulfonic acid formation). The former results in a temporary inhibition of activity, with this postulated to be a redox switch and potential mechanism to prevent irreversible oxidation that may occur under severe exposure.157 Such Cys residues have been shown to be susceptible to oxidation by both primary oxidant species (e.g.1O2)139,158–160 and secondary oxidation products such as aldehydes and hydroperoxides/H2O2.38,161 The latter reactions may account for secondary (dark phase) effects after the cessation of initial UV/visible light exposure.
Studies on the activity of a range of kinases have provided evidence of a more complex biphasic (i.e.activation–inhibition) regulatory mechanism,162 with the overall effect dependent on the nature and extent of exposure; this again is dependent on the particular system examined. This complex biphasic response has been proposed to arise from the presence of multiple redox-sensitive residues within these proteins.162 In the src family of non-receptor tyrosine kinases, and protein kinase C, these redox-sensitive residues appear to be present in regulatory and catalytic domains of the protein. With low levels of oxidant exposure, redox-sensitive residue(s) in the regulatory domain appear to be targeted with oxidation generally resulting in kinase activation and/or an enhancement in activity. In the case of protein kinase C, this activation/enhancement appears to be due to protein conformational changes, which lead to an “opening” of the kinase active site. On exposure to greater extents of oxidation, an inhibition or reduction in kinase activity has been detected with protein kinase C and creatine kinase.163,164 This inactivation may occur as a result of direct oxidation of the catalytic cysteine by oxidants, when present at high levels.
Zinc is a redox-inactive metal ion that is abundant in living organisms. It plays a critical role in the structure and function of many proteins and enzymes, including many transcription factors where it is often chelated by Cys or His residues, in Zn1Cys2His2 clusters; related clusters (e.g.Zn1Cys2His1) are also present in the active sites of some enzymes (e.g.alcohol dehydrogenases and matrix metalloproteinases, MMPs). Given the propensity of Cys and His to undergo photo-oxidative damage (see above) it is not surprising that UVA irradiation has been shown to increase the level of free Zn2+ in fibroblast cells,165 with the release of this metal ion linked with the oxidation of the Cys ligands. In mast cells, Zn2+ release was accompanied by a reduction in protein tyrosine phosphatase activity, and activation of stress-signalling ERK and JNK-MAPK pathways.166 It has been proposed that this Zn2+-mediated phosphatase inhibition is due to interaction of the active site Cys of the phosphatase with the released Zn2+,167 though the studies discussed above suggest that direct oxidant effects on the phosphatase enzymes may also occur.
In each of these cases it is clear that oxidative processes arising from UV irradiation, or visible light exposure in the presence of a sensitizer can result in modulation of cell signalling pathways as evidenced by changes in intracellular protein phosphorylation levels.168 In some cases antioxidants have been shown to attenuate these effects,150,169,170 but whether this arises from scavenging of radicals, and/or other oxidants (e.g.1O2, H2O2, other peroxides), modulation of oxidant formation (e.g. as a result of the absorption of the incident radiation), or changes in repair rates (e.g.reduction of sulfenic acids and disulfides by added thiols, either directly or enzymatically), is not always clear.
Effects of photo-oxidation on redox homeostasis
Alterations in redox status, and particularly changes in the ratio of reduced to oxidized thiols, is a well-documented consequence of exposure to UVA or visible light in the presence of a sensitizer. Whilst such alterations during the period of exposure are well characterized, it is clear from a number of studies that this phenomenon can occur after cessation of photo-oxidation and persist for a considerable period. Direct oxidation of thiols (both protein-bound and low molecular mass, e.g.GSH) is likely to account, at least in part, for the immediate changes in redox status. Thus a simultaneous increase in oxidizing species, detected using a redox-sensitive fluorescent probe, and depletion of intracellular reduced glutathione (GSH) have been detected immediately following UVA irradiation in multiple studies (e.g. ref. 146). Long-lived oxidants such as peroxides may contribute to subsequent changes and prolong the oxidative stress.138,139Changes in the levels of various intracellular low-molecular-mass antioxidants, and the expression and activity of antioxidant and repair enzyme systems, are also likely to play a key role in determining the overall effect of UVA/visible light plus sensitizer exposure. Effects of such changes on cell survival and mutagenicity have been studied in detail, but relatively little work has been carried out on understanding the mechanistic details of the effect of irradiation on intracellular antioxidant capacity. The data obtained to date, suggest that UVA irradiation, or visible light/sensitizer combinations, may impair intracellular antioxidant capacity by: (a) directly or indirectly activating or inactivating critical enzymes; (b) changing the expression of these species; (c) depleting the availability of intracellular reductants and co-factors; (d) inhibiting dehydrogenase enzymes responsible for the maintenance of NADPH/NADP+ and NADH/NAD+ ratios; (e) directly consuming antioxidant species; and (f) by giving rise to further reactive intermediates (e.g.peroxides).
It is well established that UVA irradiation can decrease the availability of cellular reductants, including GSH and NADPH, but whether this arises from direct oxidation of these species, or as a result of indirect depletion through enzymatic activity (e.g. via the GSH/GSH peroxidase/glutaredoxin or thioredoxin reductase/thioredoxin/peroxiredoxin systems) is unclear. Thus the intracellular availability of NADPH, a co-factor for many antioxidant enzymes, has been shown to be influenced by changes in the activity of dehydrogenase enzymes, with both glucose-6-phosphate dehydrogenase and isocitric dehydrogenase reported to be inhibited by 1O2in vitro.139,171,172 Direct inhibition of the protective activities of superoxide dismutase,173,174catalase,173,175glutathione reductase,147,176methionine sulfoxide reductase,177thioredoxin,139glutathione peroxidase,176,178 and thioredoxin reductase (A. Suryo Rahmanto and M. J. Davies, unpublished data) has been reported. In some cases, inhibition appears to involve oxidation of amino acids critical for conformational integrity,175 or the redox activity of the proteins,179,180 or both. For instance, Maresca et al. have reported that oxidation of Trp and Met residues (to 5-hydroxyTrp and MetSO respectively) in catalase on exposure to UVA radiation, is associated with changes in electrophoretic mobility and antioxidant activity.175 Recently, a correlation between the inhibition of glutathione peroxidase with protein fragmentation, and thioredoxin reductase with aggregation, has been detected following exposure of these enzymes to 1O2 (A. Suryo Rahmanto and M. J. Davies, unpublished data).
Recent studies have indicated that some of the secondary effects of photo-generated oxidants can be modulated by protective enzymes. Thus peroxiredoxins react with long-lived amino acid-, peptide- and protein hydroperoxides generated by 1O2, as well as their well-established substrate H2O2,181,182 with this resulting in thiol oxidation, formation of peroxiredoxin dimers, and ultimately loss of peroxidase activity.148 Similar evidence has emerged from studies with selenoproteins,183–185 with H2O2-mediated oxidation of the selenocysteine (Sec) residues in thioredoxin reductase and glutathione peroxidase resulting in the (reversible) formation of a selenenic acid, and thiodiselenide.184,186 Rhee et al. have also shown however that reaction at the active site Sec residue in glutathione peroxidase can result in irreversible loss of activity due to conversion of the Sec residue to dehydroalanine, with release of the selenium centre from the protein.185
UVA-induced oxidative stress can also result in the release of redox active iron from heme groups (either free on bound to proteins) and other sites of iron sequestration. The release of iron may contribute to secondary damage as a result of the activity of this metal ion in catalyzing oxidant formation,187,188 though upregulation of heme-degrading (e.g.heme oxygenase-1189) and iron-binding proteins (e.g. ferritin190) may minimize such effects with any increase in “free” iron (e.g. in human fibroblasts) being transient.190 These processes appear to involve, at least in part, the inactivation of iron regulatory protein-1 (IRP-1), which modulates the level of both the iron-binding protein transferrin, and the iron-sequestering protein ferritin.191 This inactivation of IRP-1 has been ascribed to oxidation of the Cys-437 residue in this protein.191 Release of redox-active iron from ferritin-iron complexes as a result of proteolytic degradation following oxidation has also been reported.190,192
A number of studies have also highlighted the importance of plasma membrane oxidoreductases, in particular NADPH oxidase-1 (Nox-1), in generating oxidants post-cessation of UVA irradiation. It has been reported that UVA irradiation increases NADPH oxidase activity, and that reduction of Nox1 activity by either pharmacological approaches (diphenyleneiodonium) or siRNA, reduces the level of oxidant formation in human keratinocytes significantly.193 Schauen et al. have also shown that inhibition of Nox-1 activity by diphenyleneiodonium improves the viability of human fibroblasts subject to UVA irradiation significantly.194
Amino acid-, peptide-, and protein hydroperoxides, common products generated by both radicals in the presence of O2, and by 1O2, can inhibit both the activity of various thiol-containing enzymes (e.g.glutathione reductase and peroxiredoxins145,148) and deplete reducing co-factors such as GSH.138
Effects of photo-oxidation on cellular proteolytic pathways
A number of studies have attempted to quantify protein damage in cells, and in skin, arising from photo-oxidation (e.g. ref. 195–198). This has been most commonly addressed by measurement of protein carbonyls, a generic marker of protein damage. It should however be noted that carbonyls are a relatively minor product of photo-oxidation, and that the resulting values reflect only the difference between the rates of generation and removal of this oxidation product; the total extent of formation may be markedly different, especially if protein catabolic systems are not significantly impaired. Two of the major systems that remove modified materials are the proteasomal system and the lysosomal pathway, both of which are intracellular systems (the latter can however remove damaged extracellular material after internalization). Turnover of extracellular protein damage is therefore less rapid and efficient. The proteasomal pathway has been reported to account for 70–80% of the turnover of oxidized proteins.199 A recent study has reported that UVA irradiation decreases the proteolytic activity of the proteasome in human fibroblasts and skin tissue, with 1O2 believed to be the mediator of damage and inactivation.200 However, studies with human keratinocytes and isolated proteasome preparations have indicated that the 20S core of the proteasome is resistant to oxidative inactivation.196,201 Studies with both the 20S and 26S forms using H2O2 suggest that redox modulation of proteasomal activity occurs via the 19S regulatory subunit present in the 26S (but not 20S) particle.196,201 Recent work has shown that oxidized proteins, and particularly those containing hydroperoxide groups generated by 1O2, can inhibit some of the proteolytic activities of the 26S proteasome,144 with this associated with the loss of proteasomal thiols.144 Thus both initial reactive intermediates, such as 1O2, and photo-products such as hydroperoxides, appear to be capable of modulating the catabolism of both oxidized materials (via the 20S proteasome) and normal proteins (via the 26S proteasome). The latter inhibition may contribute to alterations in cell signalling as a result of the key role of the 26S system in regulating cellular signalling systems.Oxidative damage may also impair proteasome activity by other mechanisms, including via the formation of large protein aggregates that are poorly digested for multiple reasons, including access into the catalytic core of the 20S proteasome that requires complete unfolding of the protein structure. Large protein aggregates, or aggresomes, which may result from extensive protein oxidation with consequent conformational changes, intra- and/or inter-molecular cross-links and enhanced hydrophobicity, appear to be particularly resistant to degradation. Lipofuscin (sometimes also known as age pigment) which contains modified proteins202 has been shown to inhibit proteasomal activity.195,200,203 This material may also act as a photo-sensitizer of further damage in some cell types possibly as a result of the presence of chromophoric structures generated by oxidation.204UVA radiation may also affect the proteasomal pathway by targeting the ubiquitinylation system, which is critical for the signalling and trafficking of modified proteins for turnover. A number of key ubiquitin-conjugating enzymes are Cys-dependent, which is likely to make these species susceptible to photo-oxidation by both initial oxidants and downstream reactive intermediates (e.g.hydroperoxides). Direct evidence for modulation of this pathway by UVA radiation, however, has yet to be reported.
The activity of the second major intracellular pathway for turnover of modified proteins, the lysosomal pathway is also susceptible to oxidative damage, though this has been less well studied than the proteasomal system. Photo-oxidation of human fibroblast using acridine orange, a sensitizer that accumulates in lysosomes, results in disruption of lysosomal membranes.205 Wan et al. reported that this loss of membrane integrity increased the lysosomal proton permeability and its sensitivity to osmotic damage.206,207 Disruption of lysosomal membranes has been associated with both membrane protein thiol oxidation and protein cross-linking.206 In addition to these structural changes, it has been shown that peptide and protein hydroperoxides generated by 1O2, can inhibit some of the major protease enzymes present in lysosomes; the activity of the Cys-dependent cathepsins B, L and S are particularly affected (both in the isolated form, and when present in cell lysates and in intact macrophage cells138,139,143). The activity of the Asp-dependent protease cathepsin D was less markedly inhibited.138,139,143 Such inhibition of the cellular protein catabolic systems may result in an accumulation of damaged materials within cells and enhanced cellular dysfunction.143
UVA radiation can also damage extracellular materials both directly and indirectly. It has been reported that extracellular matrix proteins including collagen and elastin can both potentiate the formation of H2O2 (possibly as a result of the presence of chromophoric materials on these proteins arising from oxidation and/or glycation), and be targets of damage.208 While the species formed on these proteins are yet to be characterized, subsequent incubation of human fibroblast cells with these oxidized proteins induced intracellular oxidant generation, inhibition of cell proliferation, and damage to DNA.208
Matrix metalloproteinases (MMP) are key extracellular proteases that can be up-regulated by UV irradiation; these enzymes play a key role in both the degradation of extracellular matrix proteins and matrix remodelling. In vitro and ex vivo studies have demonstrated that MMP up-regulation (both at transcriptional and translational levels) can occur via a redox-controlled process of activation/inhibition of phosphatases and kinases in the Ras-JNK phosphorylation signalling pathway.209 Koch et al. reported a redox-dependent negative regulation of MMP-1 activity by GSH and various low molecular mass thiols (Cys, homocysteine, and N-acetyl Cys), in addition to the well-established regulation by the endogenous tissue inhibitors of matrix metalloproteinases (TIMPs);210 this inhibition has been postulated to occur as a result of the interaction of the thiols with the active site Zn2+ of the MMPs. Photo-oxidation may however also directly inhibit these enzymes as a result of oxidation of the ligands at the Zn2+ active site (see above), though this has not been explored in detail.
Photo-oxidation and cell death
Oxidative stress caused by UVA irradiation initiates a series of cellular responses that can result in cell death either viaapoptosis or necrosis. Apoptosis appears to be the predominant mechanism with modest UV doses, with this process detected for both keratinocytes and fibroblastsin vivo (e.g. in sunburnt skin211).Extensive studies have examined the mechanism(s) of apoptosis following UVA irradiation. This has been detected as early as 30 min post-exposure in vitro, and within 8 h in skin tissue; long-wave UVA (320–400 nm) has been reported to be more effective than UVB and UVC in inducing early apoptosis.212 The pathways believed to be involved in such apoptosis have been reviewed elsewhere.213 Zhuang and Kochevar reported the activation of death receptor Fas, independent of Fas ligand, in human leukaemia HL-60 cells by UVA irradiation;214 this ligand-independent activation may occur as a result of 1O2-mediated protein oxidation and cross-linking, and thus may be responsible for the observed clustering of Fas receptors.214 Such oxidation and receptor cross-linking have also been demonstrated in ligand-activated TNF receptor; binding of CD95 and TRAIL to their respective TNF receptors stimulated the formation of inter-protein disulfide bonds and stabilisation of the active receptor trimer.215
Initiation of apoptosis commonly involves the activation of caspases, which are Cys-Asp proteases. Modulation of caspase activity may therefore affect the fate of exposed cells. Caspases in UVA-irradiated cells may be favoured targets for oxidants generated by photo-oxidation as a result of their active site Cys residues, and a number of studies have provided evidence for 1O2, H2O2, and/or amino acid and peptide hydroperoxides as potent caspase inhibitorsin vitro and in intact cells.141,216,217
A number of studies have suggested that UVA irradiation has only minimal direct effects on mitochondria with regard to the triggering of cell apoptosis.194,218 Thus loss of mitochondria membrane potential and release of the mitochondrially-derived apoptosis inducing factor cytochrome c, after UV irradiation, may be mediated by cellular responses rather than direct effects.
Protein photo-oxidation in human disease: deleterious effects
Protein photo-oxidation occurs primarily in tissues routinely exposed to sunlight, with skin and eyes being major target organs. Long-term photo-exposure of skin is known to result in decreased elasticity, an increased leathery texture and changes in pigmentation (reviewed in ref. 152, 197, and 219–221). These macroscopic effects are believed to be consequences of changes at the molecular level, with at least some of these due to photo-damage to structural proteins such as collagen and elastin, and/or repetitive induction of the inflammatory response.152,197,219–222 These processes may also be integral in the development of skin cancers upon prolonged exposure to UVA radiation (e.g. ref. 223–225). DNA damage is clearly a major potential driving force, but epigenetic factors and UV-induced damage to critical proteins (e.g. inactivation of DNA repair enzymes, altered cell signalling, inhibition of caspases and hence inhibition of apoptosis in damaged cells) may also play a major role (reviewed in ref. 50).Photosensitization reactions have been postulated to contribute to the development of lens cataracts and macular degeneration in the eye (reviewed in ref. 4 and 226–229). Many of the UV filters in lenses, which appear to act by absorbing incident radiation with subsequent physical quenching of the excited states without significant formation of reactive species, are derivatives of Trp (e.g.kynurenine, 3-hydroxykynurenine, 3-hydroxykynurenine-O-β-D-glucoside and 4-(2-amino-3-hydroxyphenyl)-4-oxobutanoic acid O-β-D-glucoside7,8). The first step in the production of these filter compounds is oxidation of free Trp mediated by the enzyme indoleamine 2,3-dioxygenase.230 Despite the efficiency of physical quenching processes, some chemical oxidation of the filter compounds occurs through continued exposure to UV/visible light, and this can result in the formation of oxidation products, including deaminated unsaturated aldehydes. These can become covalently attached to lens proteins, such as crystallins, viaMichael addition reactions and other processes (e.g. ref. 8, 45, and 231–233). The modified filters, both free and protein-bound, can act as photosensitizers of further protein damage, with formation of 1O2, peroxides and radicals implicated in these mechanisms.48,64,234–236
Protein photo-oxidation in human disease: beneficial effects
In addition to being implicated as a causative feature in numerous pathologies, photo-oxidation of proteins has been utilized beneficially in a variety of medical treatments. The technique of photodynamic therapy (PDT) has been available for several decades, with much research and development continuing into this technique (reviewed in ref. 10–15). The basic premise of PDT is to administer a photosensitizing drug (or a precursor that is metabolized into a photosensitizer), before illuminating with a light source such as a lamp, laser or light emitting diode (LED) in the presence of O2. The excitation of the drug leads to 1O2 and radical formation (as described above) that damages neighbouring biomolecules. This controlled production of oxidants has applications, for example, in treating a wide range of cancers, in bacterial inactivation (e.g. in dentistry), in photoangioplasty and treatment of a variety of skin disorders, including psoriasis and acne.10–15,237 An exciting related development is that of “photochemical tissue bonding” in which a sensitizer and light can be used to suture together pieces of tissue with little scarring of tissue; recent success for this technique has been demonstrated in the repair of peripheral nerves and other tissue damage.238–240 This process appears to occur without significant photo-toxicity.239Protein photo-oxidation in food, agriculture and commercial materials
The photo-oxidation of proteins occurs in a wide variety of non-medical or health-related scenarios and can have beneficial or adverse effects. In all cases gaining further insight into the photo-induced mechanisms involved, is essential to promote or prevent these reactions as required, and some of these research areas are highlighted briefly below.Over recent years food scientists have taken increasing interest in the effect of photo-induced protein oxidation on food quality and preservation (reviewed in ref. 241). Dairy products such as milk and cheese have received much attention as riboflavin, which is present at significant levels in many of these products, sensitizes the production of reactive oxygen species in the presence of light. The resulting photo-oxidation that can occur during processing and/or storage can result in the production of “off-flavours” or discolouration and can also inhibit enzymes involved in clotting (e.g. discussed in ref. 9, 120, 242, and 243). The effects of protein oxidation have also been investigated in muscle tissues such as red meat (reviewed in ref. 244), with photo-oxidation leading to changes in tenderness, palatability and colour.
Photo-oxidation of proteins also has implications in the textile industry, with the fluorescence whitening agents (FWA) that are frequently used in textile processing, and in washing powders, acting as photosensitizers. Recent research has shown that photo-oxidation of wool mediated by these compounds results in undesirable yellowing through oxidation of Trp and His residues on keratin and other proteins.78,245–247
Conclusions
The majority of protein damage initiated by UVA and visible light is mediated by photo-sensitization reactions and the production of 1O2 and other reactive oxygen species. 1O2 reacts preferentially with the aromatic and sulfur-containing side-chains of Trp, His, Tyr, Met, Cys and cystine residues, with endoperoxides and zwitterionic species the main primary products. However, these species are typically moderately reactive intermediates that undergo further reactions, including via radical mechanisms and ring opening reactions that result in disruption of protein structure. Cross-linking and aggregation of proteins predominates over fragmentation processes for 1O2-mediated oxidation. In addition to photosensitized reactions, direct photo-oxidation reactions can occur with some protein residues (e.g. Trp), resulting in the formation of excited states and radical species that can also play a role in photo-induced protein degradation and its biological sequelae.Knowledge of the mechanisms involved in photo-degradation of proteins is increasingly being utilized to our benefit in the areas of food science. Whilst the chemical modifications and resulting structural ramifications of UV/visible radiation on proteins are increasingly well characterized, much remains to be done in unraveling the biological consequences of these changes.
Acknowledgements
This work was supported by grants from the Australian Research Council under the ARC Centres of Excellence (CE0561607) and Discovery (DP0988311) programs.References
- R. V. Bensasson, E. J. Land and T. G. Truscott, Pulse radiolysis and flash photolysis: Contributions to the chemistry of biology and medicine, Pergamon Press, Oxford, 1983 Search PubMed.
- R. V. Bensasson, E. J. Land and T. G. Truscott, Excited states and free radicals in biology and medicine, Oxford University Press, Oxford, 1993 Search PubMed.
- M. J. Davies, Reactive species formed on proteins exposed to singlet oxygen, Photochem. Photobiol. Sci., 2004, 3, 17–25 CAS.
- M. J. Davies and R. J. W. Truscott, Photo-oxidation of protein and its role in cataractogenesis, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS.
- S. G. Afonso, R. E. de Salamanca and A. M. D. Batlle, The photodynamic and non-photodynamic actions of porphyrins, Braz. J. Med. Biol. Res., 1999, 32, 255–266 CrossRef CAS.
- J. A. Silvester, G. S. Timmins and M. J. Davies, Photodynamically-generated bovine serum albumin radicals: Evidence for damage transfer and oxidation at cysteine and tryptophan residues, Free Radical Biol. Med., 1998, 24, 754–766 CrossRef CAS.
- L. Bova, A. M. Wood, J. F. Jamie and R. J. W. Truscott, UV filter compounds in human lenses: the origin of AHBG, Invest. Ophthalmol. Vis. Sci., 1999, 40, 3237–3244 CAS.
- A. Korlimbinis and R. J. Truscott, Identification of 3-hydroxykynurenine bound to proteins in the human lens. A possible role in age-related nuclear cataract, Biochemistry, 2006, 45, 1950–1960 CrossRef CAS.
- M. R. Clausen, K. Huvaere, L. H. Skibsted and J. Stagsted, Characterization of peroxides formed by riboflavin and light exposure of milk. Detection of urate hydroperoxide as a novel oxidation product, J. Agric. Food Chem., 2010, 58, 481–487 CrossRef CAS.
- D. Phillips, Light relief: Photochemistry and medicine, Photochem. Photobiol. Sci., 2010, 9, 1589–1596 CAS.
- E. S. Nyman and P. H. Hynninen, Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy, J. Photochem. Photobiol., B, 2004, 73, 1–28 CrossRef CAS.
- T. Maisch, J. Baier, B. Franz, M. Maier, M. Landthaler, R. M. Szeimies and W. Baumler, The role of singlet oxygen and oxygen concentration in photodynamic inactivation of bacteria, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7223–7228 CrossRef CAS.
- S. Pervaiz and M. Olivo, Art and science of photodynamic therapy, Clin. Exp. Pharmacol. Physiol., 2006, 33, 551–556 CrossRef CAS.
- K. Konopka and T. Goslinski, Photodynamic therapy in dentistry, J. Dent. Res., 2007, 86, 694–707 CrossRef CAS.
- S. Choudhary, K. Nouri and M. L. Elsaie, Photodynamic therapy in dermatology: A review, Lasers Med. Sci., 2009, 24, 971–980 CrossRef.
- R. C. Straight and J. D. Spikes, Photosensitized oxidation of biomolecules, in Singlet O2, ed. A. A. Frimer, CRC Press, Boca Raton, 1985, pp. 91–143 Search PubMed.
- M. J. Davies, Singlet oxygen-mediated damage to proteins and its consequences, Biochem. Biophys. Res. Commun., 2003, 305, 761–770 CrossRef CAS.
- E. J. Hart, M. Anbar, The hydrated electron, Wiley-Interscience, New York, 1970 Search PubMed.
- W. M. Garrison, Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- C. von Sonntag, The chemical basis of radiation biology, Taylor and Francis, London, 1987 Search PubMed.
- M. J. Davies and R. T. Dean, Radical-mediated protein oxidation: From chemistry to medicine, Oxford University Press, Oxford, 1997 Search PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, Rate constants for the decay and reactions of the lowest electronically excited state of molecular oxygen in solution. An expanded and revised compilation, J. Phys. Chem. Ref. Data, 1995, 24, 663–1021 CrossRef CAS.
- G. Papeschi, M. Monici and S. Pinzauti, Ph effect on dye sensitized photooxidation of aminoacids and albumins, Med. Biol. Environ., 1982, 10, 245–250 CAS.
- D. Creed, The photophysics and photochemistry of the near-UV absorbing amino acids. I. Tryptophan and its simple derivatives, Photochem. Photobiol., 2008, 39, 537–562 CrossRef.
- M. J. Davies and B. C. Gilbert, Free radical reactions. Fragmentation and rearrangements in aqueous solution, Adv. Detailed React. Mech., 1991, 1, 35–81 CAS.
- L. P. Candeias, P. Wardman and R. P. Mason, The reaction of oxygen with radicals from oxidation of tryptophan and indole-3-acetic acid, Biophys. J., 1997, 67, 229–237 CrossRef CAS.
- D. J. Kelman, J. A. DeGray and R. P. Mason, Reaction of myoglobin with hydrogen peroxide forms a peroxyl radical which oxidizes substrates, J. Biol. Chem., 1994, 269, 7458–7463 CAS.
- T. Nauser, W. H. Koppenol and J. M. Gebicki, The kinetics of oxidation of GSH by protein radicals, Biochem. J., 2005, 392, 693–701 CrossRef CAS.
- D. Steinmann, T. Nauser, J. Beld, M. Tanner, D. Gunther, P. L. Bounds and W. H. Koppenol, Kinetics of tyrosyl radical reduction by selenocysteine, Biochemistry, 2008, 47, 9602–9607 CrossRef CAS.
- A. S. Domazou, W. H. Koppenol and J. M. Gebicki, Efficient repair of protein radicals by ascorbate, Free Radical Biol. Med., 2009, 46, 1049–1057 CrossRef CAS.
- J. M. Gebicki, T. Nauser, A. Domazou, D. Steinmann, P. L. Bounds and W. H. Koppenol, Reduction of protein radicals by GSH and ascorbate: Potential biological significance, Amino Acids, 2010, 39, 1131–1137 CrossRef CAS.
- B. M. Hoey and J. Butler, The repair of oxidized amino acids by antioxidants, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1984, 791, 212–218 CrossRef CAS.
- D. N. Nikogosyan and H. Gorner, Photolysis of aromatic amino acids in aqueous solution by nanosecond 248 and 193 nm laser light, J. Photochem. Photobiol., B, 1992, 13, 219–234 CrossRef CAS.
- K. L. Stevenson, G. A. Papadantonakis and P. R. LeBreton, Nanosecond UV laser photoionization of aqueous tryptophan: Temperature dependence of quantum yield, mechanism, and kinetics of hydrated electron decay, J. Photochem. Photobiol., A, 2000, 133, 159–167 CrossRef CAS.
- Y. P. Tsentalovich, O. A. Snytnikova and R. Z. Sagdeev, Properties of excited states of aqueous tryptophan, J. Photochem. Photobiol., A, 2004, 162, 371–379 CrossRef CAS.
- M. Nakagawa, H. Watanabe, S. Kodato, H. Okajima, T. Hino, J. L. Flippen and B. Witkop, A valid model for the mechanism of oxidation of tryptophan to formylkynurenine -25 years later, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4730–4733 CrossRef CAS.
- I. Saito, T. Matsuura, M. Nakagawa and T. Hino, Peroxidic intermediates in photosensitized oxygenation of tryptophan derivatives, Acc. Chem. Res., 1977, 10, 346–352 CrossRef CAS.
- M. Gracanin, C. L. Hawkins, D. I. Pattison and M. J. Davies, Singlet oxygen-mediated amino acid and protein oxidation: Formation of tryptophan peroxides and decomposition products, Free Radical Biol. Med., 2009, 47, 92–102 CrossRef CAS.
- A. Posadaz, A. Biasutti, C. Casale, J. Sanz, F. Amat-Guerri and N. A. Garcia, Rose bengal-sensitized photooxidation of the dipeptides l-tryptophyl-l-phenylalanine, l-tryptophyl-l-tyrosine and l-tryptophyl-l-tryptophan: Kinetics, mechanism and photoproducts, Photochem. Photobiol., 2004, 80, 132–138 CrossRef CAS.
- G. E. Ronsein, M. C. de Oliveira, M. H. de Medeiros and P. Di Mascio, Characterization of O2(1Δg)-derived oxidation products of tryptophan: A combination of tandem mass spectrometry analyses and isotopic labeling studies, J. Am. Soc. Mass Spectrom., 2009, 20, 188–197 CrossRef CAS.
- G. E. Ronsein, M. C. Oliveira, S. Miyamoto, M. H. Medeiros and P. Di Mascio, Tryptophan oxidation by singlet molecular oxygen [O2(1Δg)]: Mechanistic studies using 18O-labeled hydroperoxides, mass spectrometry, and light emission measurements, Chem. Res. Toxicol., 2008, 21, 1271–1283 CrossRef CAS.
- R. W. Redmond and J. N. Gamlin, A compilation of singlet oxygen yields from biologically relevant molecules, Photochem. Photobiol., 1999, 70, 391–475 CAS.
- A. J. Grosvenor, J. D. Morton and J. M. Dyer, Profiling of residue-level photo-oxidative damage in peptides, Amino Acids, 2009, 39, 285–296 CrossRef.
- A. J. Grosvenor, J. D. Morton and J. M. Dyer, Isobaric labeling approach to the tracking and relative quantitation of peptide damage at the primary structural level, J. Agric. Food Chem., 2010, 58, 12672–12677 CrossRef CAS.
- B. D. Hood, B. Garner and R. J. Truscott, Human lens coloration and aging. Evidence for crystallin modification by the major ultraviolet filter, 3-hydroxy-kynurenine O-beta-d-glucoside, J. Biol. Chem., 1999, 274, 32547–32550 CrossRef CAS.
- S. Vazquez, J. A. Aquilina, J. F. Jamie, M. M. Sheil and R. J. Truscott, Novel protein modification by kynurenine in human lenses, J. Biol. Chem., 2001, 277, 4867–4873 CrossRef.
- N. R. Parker, A. Korlimbinis, J. F. Jamie, M. J. Davies and R. J. Truscott, Reversible binding of kynurenine to lens proteins: Potential protection by glutathione in young lenses, Invest. Ophthalmol. Visual Sci., 2007, 48, 3705–3713 Search PubMed.
- J. Mizdrak, P. G. Hains, R. J. Truscott, J. F. Jamie and M. J. Davies, Tryptophan-derived ultraviolet filter compounds covalently bound to lens proteins are photosensitizers of oxidative damage, Free Radical Biol. Med., 2008, 44, 1108–1119 CrossRef CAS.
- N. R. Parker, J. F. Jamie, M. J. Davies and R. J. Truscott, Protein-bound kynurenine is a photosensitizer of oxidative damage, Free Radical Biol. Med., 2004, 37, 1479–1489 CrossRef CAS.
- D. I. Pattison and M. J. Davies, Actions of ultraviolet light on cellular structures, Exs, 2006, 96, 131–157 CAS.
- J. A. Irwin, H. Ostdal and M. J. Davies, Myoglobin-induced oxidative damage: Evidence for radical transfer from oxidized myoglobin to other proteins and antioxidants, Arch. Biochem. Biophys., 1999, 362, 94–104 CrossRef CAS.
- H. Ostdal, M. J. Davies and H. J. Andersen, Reaction between protein radicals and other biomolecules, Free Radical Biol. Med., 2002, 33, 201–209 CrossRef CAS.
- H. Ostdal, L. H. Skibsted and H. J. Andersen, Formation of long-lived protein radicals in the reaction between H2O2-activated metmyoglobin and other proteins, Free Radical Biol. Med., 1997, 23, 754–761 CrossRef CAS.
- L. K. Folkes, M. Trujillo, S. Bartesaghi, R. Radi and P. Wardman, Kinetics of reduction of tyrosine phenoxyl radicals by glutathione, Arch. Biochem. Biophys., 2011, 506, 242–249 CrossRef CAS.
- J. P. Eiserich, J. Butler, A. von der Vliet, C. E. Cross and B. Halliwell, Nitric oxide rapidly scavenges tyrosine and tryptophan radicals, Biochem. J., 1995, 310, 745–749 CAS.
- S. Criado, A. T. Soltermann, J. M. Marioli and N. A. Garcia, Sensitized photooxidation of di- and tripeptides of tyrosine, Photochem. Photobiol., 1998, 68, 453–458 CrossRef CAS.
- E. Katsuya, K. Seya and H. Hikino, Photo-oxidation of L-tyrosine, an efficient, 1,4-chirality transfer reaction, J. Chem. Soc., Chem. Commun., 1988, 934–935 Search PubMed.
- A. Wright, W. A. Bubb, C. L. Hawkins and M. J. Davies, Singlet oxygen-mediated protein oxidation: Evidence for the formation of reactive side-chain peroxides on tyrosine residues, Photochem. Photobiol., 2002, 76, 35–46 CrossRef CAS.
- F. M. Jin, J. Leitich and C. von Sonntag, The photolysis (λ = 254 nm) of tyrosine in aqueous solutions in the absence and presence of oxygen - the reaction of tyrosine with singlet oxygen, J. Photochem. Photobiol., A, 1995, 92, 147–153 CrossRef CAS.
- A. Wright, C. L. Hawkins and M. J. Davies, Singlet oxygen-mediated protein oxidation: Evidence for the formation of reactive peroxides, Redox Rep., 2000, 5, 159–161 CrossRef CAS.
- D. Balasubramanian, X. Du and J. S. J. Zigler, The reaction of singlet oxygen with proteins, with special reference to crystallins, Photochem. Photobiol., 1990, 52, 761–768 CrossRef CAS.
- D. Balasubramanian and R. Kanwar, Molecular pathology of dityrosine cross-links in proteins: Structural and functional analysis of four proteins, Mol. Cell. Biochem., 2002, 234/235, 27–38 CrossRef CAS.
- M. A. Lam, D. I. Pattison, S. E. Bottle, D. J. Keddie and M. J. Davies, Nitric oxide and nitroxides can act as efficient scavengers of protein-derived free radicals, Chem. Res. Toxicol., 2008, 21, 2111–2119 CrossRef CAS.
- N. R. Parker, J. F. Jamie, M. J. Davies and R. J. W. Truscott, Protein-bound kynurenine is a photosensitizer of oxidative damage, Free Radical Biol. Med., 2004, 37, 1479–1489 CrossRef CAS.
- D. V. Bent and E. Hayon, Excited state chemistry of aromatic amino acids and related peptides. II. Phenylalanine, J. Phys. Chem., 1975, 97, 2606–2612 CAS.
- K. Huvaere and L. H. Skibsted, Light-induced oxidation of tryptophan and histidine. Reactivity of aromatic N-heterocycles toward triplet-excited flavins, J. Am. Chem. Soc., 2009, 131, 8049–8060 CrossRef CAS.
- V. V. Agon, W. A. Bubb, A. Wright, C. L. Hawkins and M. J. Davies, Sensitizer-mediated photooxidation of histidine residues: Evidence for the formation of reactive side-chain peroxides, Free Radical Biol. Med., 2006, 40, 698–710 CrossRef CAS.
- M. Tomita, M. Irie and T. Ukita, Sensitized photooxidation of histidine and its derivatives. Products and mechanism of the reaction, Biochemistry, 1969, 8, 5149–5160 CrossRef CAS.
- P. Kang and C. S. Foote, Synthesis of a C-13, N-15 labeled imidazole and characterization of the 2,5-endoperoxide and its decomposition, Tetrahedron Lett., 2000, 41, 9623–9626 CrossRef CAS.
- P. Kang and C. S. Foote, Photosensitized oxidation of C-13,N-15-labeled imidazole derivatives, J. Am. Chem. Soc., 2002, 124, 9629–9638 CrossRef CAS.
- S. Kai and M. Suzuki, Dye-sensitized photooxidation of 2,4-disubstituted imidazoles - the formation of isomeric imidazolinones, Heterocycles, 1996, 43, 1185–1188 CrossRef CAS.
- M. Tomita, M. Irie and T. Ukita, Sensitized photooxidation of N-benzoyl histidine, Tetrahedron Lett., 1968, 9, 4933–4936 CrossRef.
- S. H. Chang, G. M. Teshima, T. Milby, B. Gillece-Castro and E. Canova-Davis, Metal-catalyzed photooxidation of histidine in human growth hormone, Anal. Biochem., 1997, 244, 221–227 CrossRef CAS.
- H. R. Shen, J. D. Spikes, P. Kopecekova and J. Kopecek, Photodynamic crosslinking of proteins. I. Model studies using histidine- and lysine-containing N-(2-hydroxypropyl)methacrylamide copolymers, J. Photochem. Photobiol., B, 1996, 34, 203–210 CrossRef CAS.
- H.-R. Shen, J. D. Spikes, C. J. Smith and J. Kopecek, Photodynamic cross-linking of proteins IV. Nature, of the His-His bond(s) formed in the Rose Bengal-photosensitized cross-linking of N-benzoyl-l-histidine, J. Photochem. Photobiol., A, 2000, 130, 1–6 CrossRef CAS.
- J. M. Dyer, S. Clerens, C. D. Cornellison, C. J. Murphy, G. Maurdev and K. R. Millington, Photoproducts formed in the photoyellowing of collagen in the presence of a fluorescent whitening agent, Photochem. Photobiol., 2009, 85, 1314–1321 CrossRef CAS.
- D. Creed, The photophysics and photochemistry of the near-UV absorbing amino acids. III. Cystine and its simple derivatives, Photochem. Photobiol., 1984, 39, 577–583 CrossRef CAS.
- K. R. Millington and J. S. Church, The photodegradation of wool keratin. 2. Proposed mechanisms involving cystine, J. Photochem. Photobiol., B, 1997, 39, 204–212 CrossRef CAS.
- C. Kolano, J. Helbing, G. Bucher, W. Sander and P. Hamm, Intramolecular disulfide bridges as a phototrigger to monitor the dynamics of small cyclic peptides, J. Phys. Chem. B, 2007, 111, 11297–11302 CrossRef CAS.
- O. Mozziconacci, B. A. Kerwin and C. Schöneich, Photolysis of an intrachain peptide disulfide bond: Primary and secondary processes, formation of H2S, and hydrogen transfer reactions, J. Phys. Chem. B, 2010, 114, 3668–3688 CrossRef CAS.
- D. V. Bent and E. Hayon, Excited state chemistry of aromatic amino acids and related peptides. I. Tyrosine, J. Phys. Chem., 1975, 97, 2599–2606 CAS.
- D. V. Bent and E. Hayon, Excited state chemistry of aromatic amino acids and related peptides. III. Tryptophan, J. Phys. Chem., 1975, 97, 2612–2619 CAS.
- D. Creed, The photophysics and photochemistry of the near-UV absorbing amino acids. II. Tyrosine and its simple derivatives, Photochem. Photobiol., 1984, 39, 363–375 Search PubMed.
- A. Vanhooren, K. De Vriendt, B. Devreese, A. Chedad, A. Sterling, H. Van Dael, J. Van Beeumen and I. Hanssens, Selectivity of tryptophan residues in mediating photolysis of disulfide bridges in goat alpha-lactalbumin, Biochemistry, 2006, 45, 2085–2093 CrossRef CAS.
- C. Schöneich and K.-D. Asmus, Determination of absolute rate constants for the reversible hydrogen-atom transfer between thiyl radicals and alcohols or ethers, J. Chem. Soc., Faraday Trans., 1995, 91, 1923–1930 RSC.
- C. Schöneich and K. D. Asmus, Reaction of thiyl radicals with alcohols, ethers and polyunsaturated fatty acids: A possible role of thiyl free radicals in thiol mutagenesis?, Radiat. Environ. Biophys., 1990, 29, 263–271 CrossRef.
- C. Schöneich, Mechanisms of protein damage induced by cysteine thiyl radical formation, Chem. Res. Toxicol., 2008, 21, 1175–1179 CrossRef.
- O. Mozziconacci, V. Sharov, T. D. Williams, B. A. Kerwin and C. Schöneich, Peptide cysteine thiyl radicals abstract hydrogen atoms from surrounding amino acids: The photolysis of a cystine containing model peptide, J. Phys. Chem. B, 2008, 112, 9250–9257 CrossRef CAS.
- T. Nauser, G. Casi, W. H. Koppenol and C. Schöneich, Reversible intramolecular hydrogen transfer between cysteine thiyl radicals and glycine and alanine in model peptides: Absolute rate constants derived from pulse radiolysis and laser flash photolysis, J. Phys. Chem. B, 2008, 112, 15034–15044 CrossRef CAS.
- P. Wardman and C. von Sonntag, Kinetic factors that control the fate of thiyl radicals in cells, Methods Enzymol., 1995, 251, 31–45 CAS.
- C. Schöneich, Kinetics of thiol reactions, Methods Enzymol., 1995, 251, 45–55 Search PubMed.
- R. W. Murray and S. L. Jindal, Photosensitized oxidation of disulfides related to cystine, Photochem. Photobiol., 1972, 16, 147–151 CrossRef CAS.
- C. S. Foote and J. W. Peters, Chemistry of singlet oxygen. XIV. A reactive intermediate in sulfide photooxidation, J. Am. Chem. Soc., 1971, 93, 3795–3796 CrossRef CAS.
- M. Rougee, R. V. Bensasson, E. J. Land and R. Pariente, Deactivation of singlet molecular oxygen by thiols and related compounds, possible protectors against skin photosensitivity, Photochem. Photobiol., 1988, 47, 485–489 CrossRef CAS.
- C. Schöneich, Methionine oxidation by reactive oxygen species: Reaction mechanisms and relevance to alzheimer's disease, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1703, 111–119 CrossRef.
- G. L. Hug, K. Bobrowski, H. Kozubek and B. Marciniak, Photooxidation of methionine derivatives by the 4-carboxybenzophenone triplet state in aqueous solution. Intracomplex proton transfer involving the amino group, Photochem. Photobiol., 1998, 68, 785–796 CrossRef CAS.
- G. L. Hug, K. Bobrowski, H. Kozubek and B. Marciniak, Photo-oxidation of methionine-containing peptides by the 4-carboxybenzophenone triplet state in aqueous solution. Competition between intramolecular two-centered three-electron bonded (S … S)(+) and (S … N)(+) formation, Photochem. Photobiol., 2000, 72, 1–9 CrossRef CAS.
- T. Pedzinski, A. Markiewicz and B. Marciniak, Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies, Res. Chem. Intermed., 2009, 35, 497–506 CrossRef CAS.
- H. Yashiro, R. C. White, A. V. Yurkovskaya and M. D. E. Forbes, Methionine radical cation: Structural studies as a function of pH using X- and Q-band time-resolved electron paramagnetic resonance spectroscopy, J. Phys. Chem. A, 2005, 109, 5855–5864 CrossRef CAS.
- G. E. Ronsein, S. Miyamoto, E. Bechara, P. Di Mascio and G. R. Martinez, Singlet oxygen-mediated protein oxidation: Damage mechanisms, detection techniques and biological implications, Quim. Nova, 2006, 29, 563–568 CrossRef CAS.
- P. K. Sysak, C. S. Foote and T.-Y. Ching, Chemistry of singlet oxygen - XXV. Photooxygenation, of methionine, Photochem. Photobiol., 1977, 26, 19–27 CrossRef CAS.
- A. Karunakaran-Datt and P. Kennepohl, Redox photochemistry of methionine by sulfur K-edge X-ray absorption spectroscopy: Potential implications for cataract formation, J. Am. Chem. Soc., 2009, 131, 3577–3582 CrossRef CAS.
- M. H. Klapper and M. Faraggi, Application of pulse radiolysis to protein chemistry, Q. Rev. Biophys., 1979, 12, 465–519 CrossRef CAS.
- W. A. Prutz, Free radical transfer involving sulphur peptide functions, in Sulfur-centered reactive intermediates in chemistry and biology, ed. C. Chatgilialoglu and K.-D. Asmus, Plenum Press, New York, 1990, pp. 389–399 Search PubMed.
- M. R. DeFelippis, M. Faraggi and M. H. Klapper, Evidence for through-bond long-range electron transfer in peptides, J. Am. Chem. Soc., 1990, 112, 5640–5462 CrossRef CAS.
- H. B. Gray and J. R. Winkler, Electron transfer in proteins, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS.
- W. A. Prutz, J. Butler and E. J. Land, Phenol coupling initiated by one-electron oxidation of tyrosine units in peptides and histone, Int. J. Radiat. Biol., 1983, 44, 183–196 CrossRef CAS.
- W. A. Prutz, F. Siebert, J. Butler, E. J. Land, A. Menez and T. Montenay-Garestier, Charge transfer in peptides. Intramolecular radical transformations involving methionine, tryptophan and tyrosine, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1982, 705, 139–149 CrossRef.
- M. Faraggi, M. R. DeFelippis and M. H. Klapper, Long-range electron transfer between tyrosine and tryptophan in peptides, J. Am. Chem. Soc., 1989, 111, 5141–5145 CrossRef CAS.
- M. Faraggi, J. P. Steiner and M. H. Klapper, Intramolecular electron and proton transfer in proteins: CO2− reduction of riboflavin binding protein and ribonuclease A, Biochemistry, 1985, 24, 3273–3279 CrossRef CAS.
- L. Grierson, K. Hildenbrand and E. Bothe, Intramolecular transformation reaction of the glutathione thiyl radical into a non-sulphur-centred radical: A pulse-radiolysis and EPR study, Int. J. Radiat. Biol., 1992, 62, 265–277 CrossRef CAS.
- R. Zhao, J. Lind, G. Merenyi and T. E. Eriksen, Significance of the intramolecular transformation of glutathione thiyl radicals to α-aminoalkyl radicals. Thermochemical and biological implications, J. Chem. Soc., Perkin Trans. 2, 1997, 569–574 RSC.
- A. Rauk, D. Yu and D. A. Armstrong, Oxidative damage to and by cysteine in proteins: An ab initio study of the radical structures, C–H, S–H, and C–C bond dissociation energies, and transition structures for H abstraction by thiyl radicals, J. Am. Chem. Soc., 1998, 120, 8848–8855 CrossRef CAS.
- R. Zhao, J. Lind, G. Merényi and T. E. Eriksen, Kinetics of one-electron oxidation of thiols and hydrogen abstraction by thiyl radicals from α-amino C–H bonds, J. Am. Chem. Soc., 1994, 116, 12010–12015 CrossRef CAS.
- O. Mozziconacci, B. A. Kerwin and C. Schöneich, Reversible hydrogen transfer between cysteine thiyl radical and glycine and alanine in model peptides: Covalent H/D exchange, radical–radical reactions, and L- to D-Ala conversion, J. Phys. Chem. B, 2010, 114, 6751–6762 CrossRef CAS.
- O. Mozziconacci, T. D. Williams, B. A. Kerwin and C. Schöneich, Reversible intramolecular hydrogen transfer between protein cysteine thiyl radicals and alpha C–H bonds in insulin: Control of selectivity by secondary structure, J. Phys. Chem. B, 2008, 112, 15921–15932 CrossRef CAS.
- E. Silva, C. De Landea, A. M. Edwards and E. Lissi, Lysozyme photo-oxidation by singlet oxygen: Properties of the partially inactivated enzyme, J. Photochem. Photobiol., B, 2000, 55, 196–200 CrossRef CAS.
- C. Prinsze, T. M. Dubbelman and J. Van Steveninck, Protein damage, induced by small amounts of photodynamically generated singlet oxygen or hydroxyl radicals, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1038, 152–157 CrossRef CAS.
- C. Prinsze, T. M. Dubbelman and J. Van Steveninck, Potentiation of thermal inactivation of glyceraldehyde-3-phosphate dehydrogenase by photodynamic treatment. A possible model for the synergistic interaction between photodynamic therapy and hyperthermia, Biochem. J., 1991, 276, 357–362 CAS.
- T. K. Dalsgaard, D. Otzen, J. H. Nielsen and L. B. Larsen, Changes in structures of milk proteins upon photo-oxidation, J. Agric. Food Chem., 2007, 55, 10968–10976 CrossRef CAS.
- L. Redecke, S. Binder, M. I. Y. Elmallah, R. Broadbent, C. Tilkorn, B. Schulz, P. May, A. Goos, A. Eich, M. Rubhausen and C. Betzel, UV-light-induced conversion and aggregation of prion proteins, Free Radical Biol. Med., 2009, 46, 1353–1361 CrossRef CAS.
- S. Roy, B. D. Mason, C. S. Schöneich, J. F. Carpenter, T. C. Boone and B. A. Kerwin, Light-induced aggregation of type I soluble tumor necrosis factor receptor, J. Pharm. Sci., 2009, 98, 3182–3199 CrossRef CAS.
- E. Silva and M. Barrera, The riboflavin-sensitized photooxidation of horseradish apoperoxidase, Radiat. Environ. Biophys., 1985, 24, 57–61 CrossRef CAS.
- J. D. Goosey, J. S. Zigler, Jr. and J. H. Kinoshita, Cross-linking of lens crystallins in a photodynamic system: A process mediated by singlet oxygen, Science, 1980, 208, 1278–1280 CAS.
- M. Francis Simpanya, R. R. Ansari, V. Leverenz and F. J. Giblin, Measurement of lens protein aggregation in vivo using dynamic light scattering in a guinea pig/UVA model for nuclear cataract, Photochem. Photobiol., 2008, 84, 1589–1595 CrossRef.
- W. Wang, S. Nema and D. Teagarden, Protein aggregation-pathways and influencing factors, Int. J. Pharm., 2010, 390, 89–99 CrossRef CAS.
- H. Shen, J. D. Spikes, C. J. Smith and J. Kopecek, Photodynamic cross-linking of proteins V. Nature, of the tyrosine-tyrosine bonds formed in the fmn-sensitized intermolecular cross-linking of N-acetyl-L-tyrosine, J. Photochem. Photobiol., A, 2000, 133, 115–122 CrossRef CAS.
- T. M. Dubbelman, C. Haasnoot and J. van Steveninck, Temperature dependence of photodynamic red cell membrane damage, Biochim. Biophys. Acta, Biomembr., 1980, 601, 220–227 CrossRef CAS.
- H. Verweij and J. van Steveninck, Model studies on photodynamic crosslinking, Photochem. Photobiol., 1982, 35, 265–267 CrossRef CAS.
- J. Dillon, R. Chiesa, R. H. Wang and M. McDermott, Molecular changes during the photooxidation of alpha-crystallin in the presence of uroporphyrin, Photochem. Photobiol., 1993, 57, 526–530 CrossRef CAS.
- J. A. Silvester, G. S. Timmins and M. J. Davies, Protein hydroperoxides and carbonyl groups generated by porphyrin-induced photo-oxidation of bovine serum albumin, Arch. Biochem. Biophys., 1998, 350, 249–258 CrossRef CAS.
- A. Michaeli and J. Feitelson, Reactivity of singlet oxygen toward amino acids and peptides, Photochem. Photobiol., 1994, 59, 284–289 CrossRef CAS.
- S. Rinalducci, N. Campostrini, P. Antonioli, P. G. Righetti, P. Roepstorff and L. Zolla, Formation of truncated proteins and high-molecular-mass aggregates upon soft illumination of photosynthetic proteins, J. Proteome Res., 2005, 4, 2327–2337 CrossRef CAS.
- T. Gomyo and M. Fujimaki, Studies on changes in protein by dye sensitized photooxidation. Part 3. On the photodecomposition products of lysozyme, Agric. Biol. Chem., 1970, 34, 302–309 CrossRef CAS.
- C. L. Hawkins and M. J. Davies, Generation and propagation of radical reactions on proteins, Biochim. Biophys. Acta, Bioenerg., 2001, 1504, 196–219 CrossRef CAS.
- H. von Tappeiner, Uber die wirkung fluoreszierender substanzen auf fermente und toxine, Ber. Dtsch. Chem. Ges., 1903, 36, 3035–3038 CrossRef CAS.
- A. Wright, C. L. Hawkins and M. J. Davies, Photo-oxidation of cells generates long-lived intracellular protein peroxides, Free Radical Biol. Med., 2003, 34, 637–647 CrossRef CAS.
- A. Suryo Rahmanto, P. E. Morgan, C. L. Hawkins and M. J. Davies, Cellular effects of peptide and protein hydroperoxides, Free Radical Biol. Med., 2010, 48, 1071–1078 CrossRef.
- A. Suryo Rahmanto, P. E. Morgan, C. L. Hawkins and M. J. Davies, Cellular effects of photo-generated oxidants and long-lived, reactive, hydroperoxide photo-products, Free Radical Biol. Med., 2010, 49, 1505–1515 CrossRef.
- P. E. Morgan, R. T. Dean and M. J. Davies, Inhibition of glyceraldehyde-3-phosphate dehydrogenase by peptide and protein peroxides generated by singlet oxygen attack, Eur. J. Biochem., 2003, 269, 1916–1925 CrossRef.
- M. B. Hampton, P. E. Morgan and M. J. Davies, Inactivation of cellular caspases by peptide-derived tryptophan and tyrosine peroxides, FEBS Lett., 2002, 527, 289–292 CrossRef CAS.
- M. Gracanin and M. J. Davies, Inhibition of protein tyrosine phosphatases by amino acid, peptide and protein hydroperoxides: Potential modulation of cell signaling by protein oxidation products, Free Radical Biol. Med., 2007, 42, 1543–1551 CrossRef CAS.
- H. A. Headlam, M. Gracanin, K. J. Rodgers and M. J. Davies, Inhibition of cathepsins and related proteases by amino acid, peptide, and protein hydroperoxides, Free Radical Biol. Med., 2006, 40, 1539–1548 CrossRef CAS.
- M. Gracanin, M. A. Lam, P. E. Morgan, K. J. Rodgers, C. L. Hawkins and M. J. Davies, Amino acid, peptide, and protein hydroperoxides and their decomposition products modify the activity of the 26 s proteasome, Free Radical Biol. Med., 2011, 50, 389–399 CrossRef CAS.
- P. E. Morgan, R. T. Dean and M. J. Davies, Protective mechanisms against peptide and protein peroxides generated by singlet oxygen, Free Radical Biol. Med., 2004, 36, 484–496 CrossRef CAS.
- M. Tanito, A. Nishiyama, T. Tanaka, H. Masutani, H. Nakamura, J. Yodoi and A. Ohira, Change of redox status and modulation by thiol replenishment in retinal photooxidative damage, Invest. Ophthalmol. Vis. Sci., 2002, 43, 2392–2400 Search PubMed.
- M. Linetsky, J. M. Hill, V. G. Chemoganskiy, F. Hu and B. J. Ortwerth, Studies on the mechanism of the UVA light-dependent loss of glutathione reductase activity in human lenses, Invest. Ophthalmol. Visual Sci., 2003, 44, 3920–3926 Search PubMed.
- A. V. Peskin, A. G. Cox, P. Nagy, P. E. Morgan, M. B. Hampton, M. J. Davies and C. C. Winterbourn, Removal of amino acid, peptide and protein hydroperoxides by reaction with peroxiredoxins 2 and 3, Biochem. J., 2010, 432, 313–321 CrossRef CAS.
- V. Muthusamy and T. J. Piva, The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways, Arch. Dermatol. Res., 2009, 302, 5–17 CrossRef.
- A. J. Ridley, J. R. Whiteside, T. J. McMillan and S. L. Allinson, Cellular and sub-cellular responses to UVA in relation to carcinogenesis, Int. J. Radiat. Biol., 2009, 85, 177–195 CrossRef CAS.
- C. J. Bertling, F. Lin and A. W. Girotti, Role of hydrogen peroxide in the cytotoxic effects of UVA/B radiation on mammalian cells, Photochem. Photobiol., 1996, 64, 137–142 CrossRef CAS.
- G. T. Wondrak, M. K. Jacobson and E. L. Jacobson, Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection, Photochem. Photobiol. Sci., 2006, 5, 215–237 CAS.
- S. Gross, A. Knebel, T. Tenev, A. Neininger, M. Gaestel, P. Herrlich and F. D. Bohmer, Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction, J. Biol. Chem., 1999, 274, 26378–26386 CrossRef CAS.
- Y. Xu, Y. Shao, J. J. Voorhees and G. J. Fisher, Oxidative inhibition of receptor-type protein-tyrosine phosphatase kappa by ultraviolet irradiation activates epidermal growth factor receptor in human keratinocytes, J. Biol. Chem., 2006, 281, 27389–27397 CrossRef CAS.
- P. Gulati, B. Markova, M. Gottlicher, F. D. Bohmer and P. A. Herrlich, UVA inactivates protein tyrosine phosphatases by calpain-mediated degradation, EMBO Rep., 2004, 5, 812–817 CrossRef CAS.
- P. Chiarugi, M. L. Taddei and G. Ramponi, Oxidation and tyrosine phosphorylation: Synergistic or antagonistic cues in protein tyrosine phosphatase, Cell. Mol. Life Sci., 2005, 62, 931–936 CrossRef CAS.
- R. L. van Montfort, M. Congreve, D. Tisi, R. Carr and H. Jhoti, Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1b, Nature, 2003, 423, 773–777 CrossRef CAS.
- J. M. Denu and K. G. Tanner, Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation, Biochemistry, 1998, 37, 5633–5642 CrossRef CAS.
- C. von Montfort, V. S. Sharov, S. Metzger, C. Schöneich, H. Sies and L. O. Klotz, Singlet oxygen inactivates protein tyrosine phosphatase-1b by oxidation of the active site cysteine, Biol. Chem., 2006, 387, 1399–1404 CrossRef CAS.
- A. Caselli, R. Marzocchini, G. Camici, G. Manao, G. Moneti, G. Pieraccini and G. Ramponi, The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2, J. Biol. Chem., 1998, 273, 32554–32560 CrossRef CAS.
- A. Hernandez-Hernandez, M. N. Garabatos, M. C. Rodriguez, M. L. Vidal, A. Lopez-Revuelta, J. I. Sanchez-Gallego, M. Llanillo and J. Sanchez-Yague, Structural characteristics of a lipid peroxidation product, trans-2-nonenal, that favour inhibition of membrane-associated phosphotyrosine phosphatase activity, Biochim. Biophys. Acta, Gen. Subj., 2005, 1726, 317–325 CrossRef CAS.
- G. A. Knock and J. P. Ward, Redox regulation of protein kinases as a modulator of vascular function, Antioxid. Redox Signal., 2011 DOI:10.1089/ars.2010.3614.
- R. Gopalakrishna and S. Jaken, Protein kinase c signaling and oxidative stress, Free Radical Biol. Med., 2000, 28, 1349–1361 CrossRef CAS.
- H. Lenz, M. Schmidt, V. Welge, U. Schlattner, T. Wallimann, H. P. Elsasser, K. P. Wittern, H. Wenck, F. Stab and T. Blatt, The creatine kinase system in human skin: Protective effects of creatine against oxidative and UV damage in vitro and in vivo, J. Invest. Dermatol., 2005, 124, 443–452 CrossRef CAS.
- E. Pirev, C. Calles, P. Schroeder, H. Sies and K. D. Kroncke, Ultraviolet-A irradiation but not ultraviolet-B or infrared-A irradiation leads to a disturbed zinc homeostasis in cells, Free Radical Biol. Med., 2008, 45, 86–91 CrossRef CAS.
- S. Yamasaki, K. Sakata-Sogawa, A. Hasegawa, T. Suzuki, K. Kabu, E. Sato, T. Kurosaki, S. Yamashita, M. Tokunaga, K. Nishida and T. Hirano, Zinc is a novel intracellular second messenger, J. Cell Biol., 2007, 177, 637–645 CrossRef CAS.
- K. D. Kroncke and L. O. Klotz, Zinc fingers as biologic redox switches?, Antioxid. Redox Signaling, 2009, 11, 1015–1027 CrossRef.
- A. Knebel, H. J. Rahmsdorf, A. Ullrich and P. Herrlich, Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents, EMBO J., 1996, 15, 5314–5325 CAS.
- P. Larsson, K. Ollinger and I. Rosdahl, Ultraviolet (UV)A- and UVB-induced redox alterations and activation of nuclear factor-kappaB in human melanocytes-protective effects of alpha-tocopherol, Br. J. Dermatol., 2006, 155, 292–300 CrossRef CAS.
- S. Gonzalez, M. Fernandez-Lorente and Y. Gilaberte-Calzada, The latest on skin photoprotection, Clin. Dermatol., 2008, 26, 614–626 CrossRef.
- S. Y. Kim, J. K. Tak and J. W. Park, Inactivation of NADP+-dependent isocitrate dehydrogenase by singlet oxygen derived from photoactivated Rose Bengal, Biochimie, 2004, 86, 501–507 CrossRef CAS.
- S. M. Lee, T. L. Huh and J. W. Park, Inactivation of NADP+-dependent isocitrate dehydrogenase by reactive oxygen species, Biochimie, 2001, 83, 1057–1065 CrossRef CAS.
- J. Luo, L. Li, Y. P. Zhang, D. R. Spitz, G. R. Buettner, L. W. Oberley and F. E. Domann, Inactivation of primary antioxidant enzymes in mouse keratinocytes by photodynamically generated singlet oxygen, Antioxid. Redox Signaling, 2006, 8, 1307–1314 CrossRef CAS.
- K. Punnonen, C. T. Jansen, A. Puntala and M. Ahotupa, Effects of in vitro UVA irradiation and PUVA treatment on membrane fatty acids and activities of antioxidant enzymes in human keratinocytes, J. Invest. Dermatol., 1991, 96, 255–259 CAS.
- V. Maresca, E. Flori, S. Briganti, E. Camera, M. Cario-Andre, A. Taieb and M. Picardo, UVA-induced modification of catalase charge properties in the epidermis is correlated with the skin phototype, J. Invest. Dermatol., 2006, 126, 182–190 CrossRef CAS.
- S. R. Johar, U. M. Rawal, N. K. Jain and A. R. Vasavada, Sequential effects of ultraviolet radiation on the histomorphology, cell density and antioxidative status of the lens epithelium–an in vivo study, Photochem. Photobiol., 2003, 78, 306–311 CrossRef CAS.
- C. R. Picot, M. Moreau, M. Juan, E. Noblesse, C. Nizard, I. Petropoulos and B. Friguet, Impairment of methionine sulfoxide reductase during UV irradiation and photoaging, Exp. Gerontol., 2007, 42, 859–863 CrossRef CAS.
- M. T. Leccia, M. Yaar, N. Allen, M. Gleason and B. A. Gilchrest, Solar simulated irradiation modulates gene expression and activity of antioxidant enzymes in cultured human dermal fibroblasts, Exp. Dermatol., 2001, 10, 272–279 CrossRef CAS.
- S. Ma, R. M. Caprioli, K. E. Hill and R. F. Burk, Loss of selenium from selenoproteins: Conversion of selenocysteine to dehydroalanine in vitro, J. Am. Soc. Mass Spectrom., 2003, 14, 593–600 CrossRef CAS.
- H. T. Le, A. F. Chaffotte, E. Demey-Thomas, J. Vinh, B. Friguet and J. Mary, Impact of hydrogen peroxide on the activity, structure, and conformational stability of the oxidized protein repair enzyme methionine sulfoxide reductase A, J. Mol. Biol., 2009, 393, 58–66 CrossRef CAS.
- F. M. Low, M. B. Hampton, A. V. Peskin and C. C. Winterbourn, Peroxiredoxin 2 functions as a noncatalytic scavenger of low-level hydrogen peroxide in the erythrocyte, Blood, 2007, 109, 2611–2617 CrossRef CAS.
- A. V. Peskin, F. M. Low, L. N. Paton, G. J. Maghzal, M. B. Hampton and C. C. Winterbourn, The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents, J. Biol. Chem., 2006, 282, 11885–11892 CrossRef.
- G. Snider, L. Grout, E. L. Ruggles and R. J. Hondal, Methaneseleninic acid is a substrate for truncated mammalian thioredoxin reductase: Implications for the catalytic mechanism and redox signaling, Biochemistry, 2010, 49, 10329–10338 CrossRef CAS.
- S. R. Lee, S. Bar-Noy, J. Kwon, R. L. Levine, T. C. Stadtman and S. G. Rhee, Mammalian thioredoxin reductase: Oxidation of the C-terminal cysteine/selenocysteine active site forms a thioselenide, and replacement of selenium with sulfur markedly reduces catalytic activity, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2521–2526 CrossRef CAS.
- S. G. Rhee and C. S. Cho, Blot-based detection of dehydroalanine-containing glutathione peroxidase with the use of biotin-conjugated cysteamine, Methods Enzymol., 2010, 474, 23–34 CAS.
- A. Bindoli, J. M. Fukuto and H. J. Forman, Thiol chemistry in peroxidase catalysis and redox signaling, Antioxid. Redox Signaling, 2008, 10, 1549–1564 CrossRef CAS.
- G. F. Vile and R. M. Tyrrell, UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen, Free Radical Biol. Med., 1995, 18, 721–730 CrossRef CAS.
- E. Kvam, V. Hejmadi, S. Ryter, C. Pourzand and R. M. Tyrrell, Heme oxygenase activity causes transient hypersensitivity to oxidative ultraviolet A radiation that depends on release of iron from heme, Free Radical Biol. Med., 2000, 28, 1191–1196 CrossRef CAS.
- S. M. Keyse and R. M. Tyrrell, Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 99–103 CrossRef CAS.
- C. Pourzand, R. D. Watkin, J. E. Brown and R. M. Tyrrell, Ultraviolet A radiation induces immediate release of iron in human primary skin fibroblasts: The role of ferritin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6751–6756 CrossRef CAS.
- G. Cairo, E. Castrusini, G. Minotti and A. Bernelli-Zazzera, Superoxide and hydrogen peroxide-dependent inhibition of iron regulatory protein activity: A protective stratagem against oxidative injury, FASEB J., 1996, 10, 1326–1335 CAS.
- O. Reelfs, I. M. Eggleston and C. Pourzand, Skin protection against UVA-induced iron damage by multiantioxidants and iron chelating drugs/prodrugs, Curr. Drug Metab., 2010, 11, 242–249 CrossRef CAS.
- A. Valencia and I. E. Kochevar, Nox1-based NADPH oxidase is the major source of UVA-induced reactive oxygen species in human keratinocytes, J. Invest. Dermatol., 2007, 128, 214–222 CrossRef.
- M. Schauen, H. T. Hornig-Do, S. Schomberg, G. Herrmann and R. J. Wiesner, Mitochondrial electron transport chain activity is not involved in ultraviolet A (UVA)-induced cell death, Free Radical Biol. Med., 2007, 42, 499–509 CrossRef CAS.
- B. Catalgol, I. Ziaja, N. Breusing, L. O. Klotz, J. Krutmann and T. Grune, Protein oxidation and proteasome inhibition during UVA irradiation, Free Radical Res., 2008, 42, S33 Search PubMed.
- A. L. Bulteau, M. Moreau, C. Nizard and B. Friguet, Impairment of proteasome function upon UVA- and UVB-irradiation of human keratinocytes, Free Radical Biol. Med., 2002, 32, 1157–1170 CrossRef CAS.
- C. S. Sander, H. Chang, S. Salzmann, C. S. Muller, S. Ekanayake-Mudiyanselage, P. Elsner and J. J. Thiele, Photoaging is associated with protein oxidation in human skin in vivo, J. Invest. Dermatol., 2002, 118, 618–625 CrossRef CAS.
- R. Haywood, C. Andrady, N. Kassouf and N. Sheppard, Intensity-dependent direct solar radiation- and UVA-induced radical damage to human skin and DNA, lipids and proteins, Photochem. Photobiol., 2011, 87, 117–130 CrossRef CAS.
- F. Kriegenburg, E. G. Poulsen, A. Koch, E. Kruger and R. Hartmann-Petersen, Redox control of the ubiquitin-proteasome system: From molecular mechanisms to functional significance, Antioxid. Redox Signal., 2011 DOI:10.1089/ars.2010.3590.
- B. Catalgol, I. Ziaja, N. Breusing, T. Jung, A. Hohn, B. Alpertunga, P. Schroeder, N. Chondrogianni, E. S. Gonos, I. Petropoulos, B. Friguet, L. O. Klotz, J. Krutmann and T. Grune, The proteasome is an integral part of solar ultraviolet A radiation-induced gene expression, J. Biol. Chem., 2009, 284, 30076–30086 CrossRef CAS.
- T. Reinheckel, N. Sitte, O. Ullrich, U. Kuckelkorn, K. J. Davies and T. Grune, Comparative resistance of the 20S and 26S proteasome to oxidative stress, Biochem. J., 1998, 335, 637–642 CAS.
- U. T. Brunk and A. Terman, Lipofuscin: Mechanisms of age-related accumulation and influence on cell function, Free Radical Biol. Med., 2002, 33, 611–619 CrossRef CAS.
- T. Grune, T. Jung, K. Merker and K. J. Davies, Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofuscin, ceroid, and ‘aggresomes’ during oxidative stress, aging, and disease, Int. J. Biochem. Cell Biol., 2004, 36, 2519–2530 CrossRef CAS.
- S. Davies, M. H. Elliott, E. Floor, T. G. Truscott, M. Zareba, T. Sarna, F. A. Shamsi and M. E. Boulton, Photocytotoxicity of lipofuscin in human retinal pigment epithelial cells, Free Radical Biol. Med., 2001, 31, 256–265 CrossRef CAS.
- U. T. Brunk, H. Dalen, K. Roberg and H. B. Hellquist, Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts, Free Radical Biol. Med., 1997, 23, 616–626 CrossRef CAS.
- F. Y. Wan, L. Yang, Y. G. Zhong, W. Zhu, Y. N. Wang and G. J. Zhang, Enhancement of lysosomal osmotic sensitivity induced by the photooxidation of membrane thiol groups, Photochem. Photobiol., 2002, 75, 134–139 CrossRef CAS.
- F. Y. Wan and G. J. Zhang, Enhancement of lysosomal proton permeability induced by photooxidation of membrane thiol groups, Arch. Biochem. Biophys., 2002, 402, 268–274 CrossRef CAS.
- G. T. Wondrak, M. J. Roberts, D. Cervantes-Laurean, M. K. Jacobson and E. L. Jacobson, Proteins of the extracellular matrix are sensitizers of photo-oxidative stress in human skin cells, J. Invest. Dermatol., 2003, 121, 578–586 CrossRef CAS.
- S. Kar, S. Subbaram, P. M. Carrico and J. A. Melendez, Redox-control of matrix metalloproteinase-1: A critical link between free radicals, matrix remodeling and degenerative disease, Respir. Physiol. Neurobiol., 2010, 174, 299–306 CrossRef CAS.
- S. Koch, C. M. Volkmar, V. Kolb-Bachofen, H. G. Korth, M. Kirsch, A. H. Horn, H. Sticht, N. Pallua and C. V. Suschek, A new redox-dependent mechanism of MMP-1 activity control comprising reduced low-molecular-weight thiols and oxidizing radicals, J. Mol. Med., 2008, 87, 261–272 CrossRef.
- A. Van Laethem, S. Claerhout, M. Garmyn and P. Agostinis, The sunburn cell: Regulation of death and survival of the keratinocyte, Int. J. Biochem. Cell Biol., 2005, 37, 1547–1553 CrossRef CAS.
- J. M. Sheehan and A. R. Young, The sunburn cell revisited: An update on mechanistic aspects, Photochem. Photobiol. Sci., 2002, 1, 365–377 CAS.
- S. W. Ryter, H. P. Kim, A. Hoetzel, J. W. Park, K. Nakahira, X. Wang and A. M. Choi, Mechanisms of cell death in oxidative stress, Antioxid. Redox Signaling, 2007, 9, 49–89 CrossRef CAS.
- S. Zhuang and I. E. Kochevar, Ultraviolet A radiation induces rapid apoptosis of human leukemia cells by Fas ligand-independent activation of the Fas death pathways, Photochem. Photobiol., 2003, 78, 61–67 CAS.
- D. Berg, M. Lehne, N. Muller, D. Siegmund, S. Munkel, W. Sebald, K. Pfizenmaier and H. Wajant, Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95l, Cell Death Differ., 2007, 14, 2021–2034 CrossRef CAS.
- M. B. Hampton, I. Stamenkovic and C. C. Winterbourn, Interaction with substrate sensitises caspase-3 to inactivation by hydrogen peroxide, FEBS Lett., 2002, 517, 229–232 CAS.
- D. Suto, Y. Iuchi, Y. Ikeda, K. Sato, Y. Ohba and J. Fujii, Inactivation of cysteine and serine proteases by singlet oxygen, Arch. Biochem. Biophys., 2007, 461, 151–158 CrossRef CAS.
- R. Gniadecki, T. Thorn, J. Vicanova, A. Petersen and H. C. Wulf, Role of mitochondria in ultraviolet-induced oxidative stress, J. Cell. Biochem., 2001, 80, 216–222 CrossRef.
- J. H. Rabe, A. J. Mamelak, P. J. S. McElgunn, W. L. Morison and D. N. Sauder, Photoaging: Mechanisms and repair, J. Am. Acad. Dermatol., 2006, 55, 1–19 CrossRef.
- M. Wlaschek, I. Tantcheva-Poor, L. Naderi, W. Ma, L. A. Schneider, Z. Razi-Wolf, J. Schuller and K. Scharffetter-Kochanek, Solar UV irradiation and dermal photoaging, J. Photochem. Photobiol., B, 2001, 63, 41–51 CrossRef CAS.
- G. T. Wondrak, Let the sun shine in: Mechanisms and potential for therapeutics in skin photodamage, Curr. Opin. Invest. Drugs, 2007, 8, 390–400 CAS.
- G. J. Smith, Photodegradation of keratin and other structural proteins, J. Photochem. Photobiol., B, 1995, 27, 187–198 CrossRef CAS.
- H. S. Black, F. R. deGruijl, P. D. Forbes, J. E. Cleaver, H. N. Ananthaswamy, E. C. deFabo, S. E. Ullrich and R. M. Tyrrell, Photocarcinogenesis: An overview, J. Photochem. Photobiol., B, 1997, 40, 29–47 CrossRef CAS.
- E. Kvam and R. M. Tyrrell, Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation, Carcinogenesis, 1997, 18, 2379–2384 CrossRef CAS.
- D. L. Narayanan, R. N. Saladi and J. L. Fox, Ultraviolet radiation and skin cancer, Int. J. Dermatol., 2010, 49, 978–986 CrossRef.
- J. E. Roberts, Hazards of sunlight exposure to the eye, in Sun protection in man, ed. P. U. Giacomoni, Elsevier, Amsterdam, 2001, pp. 155–174 Search PubMed.
- J. E. Roberts, Ocular phototoxicity, J. Photochem. Photobiol., B, 2001, 64, 136–143 CrossRef CAS.
- D. N. Hu, J. D. Simon and T. Sarna, Role of ocular melanin in ophthalmic physiology and pathology, Photochem. Photobiol., 2008, 84, 639–644 CrossRef CAS.
- B. Rozanowski, J. Cuenco, S. Davies, F. A. Shamsi, A. Zadlo, P. Dayhaw-Barker, M. Rozanowska, T. Sarna and M. E. Boulton, The phototoxicity of aged human retinal melanosomes, Photochem. Photobiol., 2008, 84, 650–657 CrossRef CAS.
- O. Takikawa, T. Littlejohn and R. J. W. Truscott, Indoleamine 2.3-dioxygenase in the human lens, the first enzyme in the synthesis of UV filters, Exp. Eye Res., 2001, 72, 271–277 CrossRef CAS.
- J. A. Aquilina and R. J. Truscott, Cysteine is the initial site of modification of α-crystallin by kynurenine, Biochem. Biophys. Res. Commun., 2000, 276, 216–223 CrossRef CAS.
- B. Garner, D. C. Shaw, R. A. Lindner, J. A. Carver and R. J. Truscott, Non-oxidative modification of lens crystallins by kynurenine: A novel post-translational protein modification with possible relevance to ageing and cataract, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1476, 265–278 CrossRef CAS.
- J. A. Aquilina and R. J. W. Truscott, Identifying sites of attachment of UV filters to proteins in older human lenses, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2002, 1596, 6–15 CrossRef CAS.
- D. Balasubramanian, Photodynamics of cataract: An update on endogenous chromophores and antioxidants, Photochem. Photobiol., 2005, 81, 498–501 CrossRef CAS.
- A. Korlimbinis, P. G. Hains, R. J. Truscott and J. A. Aquilina, 3-hydroxykynurenine oxidizes alpha-crystallin: Potential role in cataractogenesis, Biochemistry, 2006, 45, 1852–1860 CrossRef CAS.
- B. J. Ortwerth, J. Bhattacharyya and E. Shipova, Tryptophan metabolites from young human lenses and the photooxidation of ascorbic acid by UVA light, Invest. Ophthalmol. Visual Sci., 2009, 50, 3311–3319 Search PubMed.
- T. D. Mody and J. L. Sessler, Texaphyrins: A new approach to drug development, J. Porphyrins Phthalocyanines, 2001, 5, 134–142 CrossRef CAS.
- T. S. Johnson, A. C. O'Neill, P. M. Motarjem, C. Amann, T. Nguyen, M. A. Randolph, J. M. Winograd, I. E. Kochevar and R. W. Redmond, Photochemical tissue bonding: A promising technique for peripheral nerve repair, J. Surg. Res., 2007, 143, 224–229 CrossRef CAS.
- M. Yao, A. Yaroslavsky, F. P. Henry, R. W. Redmond and I. E. Kochevar, Phototoxicity is not associated with photochemical tissue bonding of skin, Lasers Surg. Med., 2010, 42, 123–131 CrossRef.
- R. A. Franco, J. R. Dowdall, K. Bujold, C. Amann, W. Faquin, R. W. Redmond and I. E. Kochevar, Photochemical repair of vocal fold microflap defects, Laryngoscope, 2011, 121, 1244–1251 CrossRef.
- E. Choe and D. B. Min, Chemistry and reactions of reactive oxygen species in foods, Crit. Rev. Food Sci. Nutr., 2006, 46, 1–22 CrossRef CAS.
- B. H. Ostdal, M. R. Weisbjerg, L. H. Skibsted and J. H. Nielsen, Protection against photooxidation of milk by high urate content, Milchwissenschaft-Milk Sci. Internat., 2008, 63, 119–122 Search PubMed.
- D. Scheidegger, R. P. Pecora, P. M. Radici and S. C. Kivatinitz, Protein oxidative changes in whole and skim milk after ultraviolet or fluorescent light exposure, J. Dairy Sci., 2010, 93, 5101–5109 CrossRef CAS.
- M. N. Lund, M. Heinonen, C. P. Baron and M. Estevez, Protein oxidation in muscle foods: A review, Mol. Nutr. Food Res., 2011, 55, 83–95 CAS.
- J. M. Dyer, S. D. Bringans and W. G. Bryson, Determination of photo-oxidation products within photoyellowed bleached wool proteins, Photochem. Photobiol., 2006, 82, 551–557 CrossRef CAS.
- J. M. Dyer, J. E. Plowman, G. L. Krsinic, S. Deb-Choudhury, H. Koehn, K. R. Millington and S. Clerens, Proteomic evaluation and location of UVB-induced photo-oxidation in wool, J. Photochem. Photobiol., B, 2010, 98, 118–127 CrossRef CAS.
- K. R. Millington, Photoyellowing of wool. Part 1: Factors affecting photoyellowing and experimental techniques, Color. Technol., 2006, 122, 169–186 CAS.
Footnote |
| † Contribution to the themed issue on the biology of UVA. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |