UVA photosensitization of thiopurines and skin cancer in organ transplant recipients†
Natalie R.
Attard‡
and
Peter
Karran
Cancer Research UK London Research Institute, Clare Hall Laboratories, South Mimms, Hertfordshire EN6 3LD, UK
First published on 23rd August 2011
Abstract
The thiopurines azathioprine, 6-mercaptopurine and 6-thioguanine (6-TG) are important medications for cancer and inflammatory disorders. They are also widely prescribed as immunosuppressants in organ transplant patients. Their metabolism results in the incorporation of 6-TG into patients' DNA, and this increases skin sensitivity to incident UVA. Unlike the canonical DNA bases, which do not absorb UVA to a significant degree, DNA 6-TG is a strong UVA chromophore. It acts as a Type II UVA photosensitizer, and the combination of 6-TG and UVA treatment induces a synergistic toxicity in cultured human cells. Here, we review some of the damage that this interaction causes. Photochemical activation of DNA 6-TG triggers DNA and protein oxidation; it induces DNA breakage, DNA crosslinking, oxidation of DNA bases and the covalent attachment of proteins to DNA. Many of these photochemical DNA lesions are difficult for cells to deal with, and we review the evidence linking thiopurine immunosuppression with genome instability and the high incidence of skin cancer in organ transplant recipients.
Introduction
The thiopurines 6-mercaptopurine (6-MP), its prodrug azathioprine and 6-thioguanine (6-TG) are effective anticancer drugs and immunosuppressants. 6-Mercaptopurine was one of the first effective treatments for childhood leukaemia and dramatically improved the previously dismal prognosis of this disease.1 The impact on organ transplant patient survival with azathioprine immunosuppression has been equally impressive since its introduction, first as monotherapy and later combined with corticosteroids, in pioneering transplant trials in the 1960s. Following the development of ciclosporin A a decade later, combinations of corticosteroids with azathioprine and/or ciclosporin were used for many years until the relatively recent introduction of a new generation of unrelated immunosuppressants such as mycophenolate mofetil, tacrolimus and sirolimus. Despite the trend towards the first-line use of these newer agents, there remain many thousands of patients who continue on azathioprine-based therapy. In addition to their use in transplant patients, azathioprine and 6-MP are widely employed as immune modulators in the treatment of a range of inflammatory conditions.2The cost of this successful immunosuppression is an increased incidence of malignancy, which is significantly higher for organ transplant recipients than for the general population.3 This observation was first made as long ago as 19694 in kidney transplant patients, and more recently5 cancer was reported to be the cause of death in one quarter of kidney transplant recipients 10 years after transplantation. Immunosuppression per se is undoubtedly a factor in this high cancer incidence but it is not the full story. This is illustrated by the differing patterns of malignancy in individuals infected with HIV compared with pharmacologically immunosuppressed transplant patients.6 Whilst both groups are highly susceptible to cancers with a known or suspected viral aetiology, organ transplantation carries a proportionately greater increased risk of skin cancer. In this article, we consider how azathioprine treatment may contribute directly to cancer risk. We review the important photochemical properties of azathioprine metabolites, describe some of the molecular damage that can arise when they interact with UVA, and suggest mechanisms by which these interactions may contribute to mutagenesis and cancer.
Epidemiology of skin cancer in organ transplant patients
As increasing numbers of organ transplant patients have achieved long-term survival with a functioning allograft, the incidence of skin cancer in this patient group has increased. Skin cancers are by far the most frequent malignancies, and over 90% are of the non-melanoma sub-type (NMSC). The frequency of squamous cell carcinoma (SCC)—an example of which is shown in Fig. 1—is more than 100 times that of the general population.7 Skin cancer represents a considerable source of morbidity for transplant patients. Their tumours occur at a younger age than in the general population, are often multiple and behave more aggressively.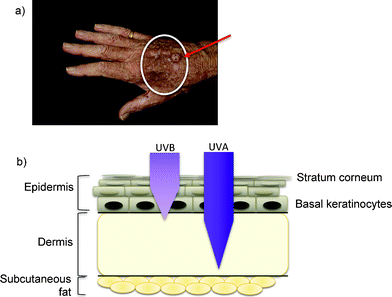 | ||
| Fig. 1 Skin cancer and ultraviolet radiation: (a) An example of SCC (arrowed) on a sun-exposed site in a transplant patient treated with azathioprine. In addition to the overt SCC, within the white circle there are many hyperkeratotic premaligant lesions and viral warts (HPV). (b) Ultraviolet penetration of skin. Both UVB and UVA radiation from sunlight reach replicating keratinocytes in the basal layer of the epidermis (a stem cell compartment). Longer wavelength UVA is able to penetrate deeper into the dermis. | ||
Epidemiological studies implicate both exposure to solar UVR and duration/intensity of immunosuppression in this increased risk. The majority of cancers occur on sun-exposed sites such as the head and neck, backs of hands and forearms. Their geographical distribution also reflects the importance of sunlight. The cumulative risk of NMSC at 10 years post-transplant is 50% in Queensland, Australia8 compared to 10% in the more temperate Netherlands.9 After 20 years, the risk increases to 40% in the Netherlands and to 80% in Queensland. It has been suggested that more intense immunosuppression regimens—such as those in cardiac transplantation, which involve higher immunosuppressant doses—are also associated with a greater NMSC risk,10 but this may instead reflect differences in age at transplantation.
Pathogenesis of OTR-associated skin cancer
The role of human papilloma virus (HPV) infection in the development of NMSC in transplant patients is much debated and is beyond the scope of this review (for a review see ref. 12). Any direct causal role for HPV remains to be established, but it is interesting that, akin to SCC development, the prevalence of HPV infection increases steadily with time after transplant.13 In addition, HPV and SCC frequently co-exist in the same sun-exposed sites in transplant patients (Fig. 1). The mechanism by which HPV could drive SCC development is not clear. HPV viral load is typically low in transplant-associated NMSCs, consistent with a role in tumour initiation,14 possibly as a co-factor with UVR.15 They may, however, simply be innocent bystanders.
Recently, increasing attention has been paid to UVA (wavelengths 320–400 nm) as a potential carcinogen.23UVA also induces potentially mutagenic cyclobutane pyrimidine dimers24 (in this case, predominantly T<>T dimers). It does so by a different mechanism to UVB and with several orders of magnitude lower efficiency. This is because, unlike UVB, UVA is absorbed poorly by DNA. Despite this property, T<>T dimers can arise by direct UVA absorption, which is greater in native than in single-stranded DNA.25 The biological effects of UVA are mediated partly through cellular chromophores, such as porphyrins or haem-containing proteins, which act as photosensitizers. These photosensitized reactions generate reactive oxygen species (ROS) that are extremely damaging to DNA. One important DNA lesion, 8-oxoguanine—the major oxidized DNA base,—is highly mutagenic and its formation probably underlies the increase in G→T transversion mutations in skin cancers.26
It follows from this that DNA damage caused directly by UVB or indirectly via photosensitized reactions involving UVA can both contribute to skin cancer development. It is also apparent that exogenous UVA chromophores that act as UVA photosensitizers and promote the formation of ROS are likely to increase the risk of mutation and skin cancer. In the next section, we describe how through their metabolism, the thiopurine class of drugs introduce a UVA chromophore into DNA and review the potential adverse biological consequences of its photoactivation.
Thiopurine metabolism
Thiopurines are prodrugs that require complex bioactivation to exert their pharmacological effects. The metabolism of 6-TG is straightforward. Following transporter-mediated uptake into cells, it is salvaged by HPRT, the first enzyme in the purine salvage pathway, and is converted directly into 6-TG nucleotides (TGN), which are precursors for incorporation into DNA. Azathioprine is first non-enzymatically cleaved to liberate 6-MP, which is also a substrate for HPRT. A subsequent series of enzymatic conversions culminate in the formation of TGN (Fig. 2). Competing catabolic reactions, catalysed by thiopurine methyltransferase and xanthine oxidase, inactivate part of the 6-MP that is formed, and the therapeutic effectiveness of azathioprine depends on the balance between TGN production and the destruction of their precursors. The process(es) by which the thiopurines selectively inhibit the clonal expansion of tumor or immune effector cells and exert their anti-cancer or immunosuppressive effects remain incompletely understood. Possible mechanisms include cytotoxicity viaDNA 6-TG-mediated triggering of the DNA mismatch repair system and interference with de novopurine biosynthesis (this is reviewed in more detail in ref. 2).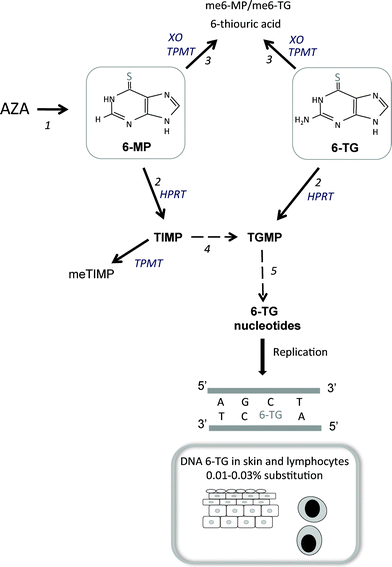 | ||
| Fig. 2 Metabolism of thiopurines : 1 Azathioprine is non-enzymatically converted to 6-MP. 2 Both 6-MP and 6-TG are salvaged by hypoxanthine-guanine phosphoribosyltransferase (HPRT) and converted into thioinosine monophosphate (TIMP) and thioguanosine monophosphate (TGMP), respectively. 3 In competing catabolic reactions, thiopurine methyltransferase (TMPT) inactivates 6-MP and 6-TG by S-methylation and 6-MP is degraded by xanthine oxidase (XO). 4TIMP derived from 6-MP that escapes catabolism by TPMT is further metabolized to TGMPvia a series of enzymatic reactions (hatched arrow) to TGMP. 5 The sequential action of kinases and reductases generates thioguanine deoxyribonucleoside triphosphate (often referred to as thioguanine nucleotides, TGN). This is a substrate for the incorporation of 6-TG into replicating DNA (as shown). | ||
Because TGN are good substrates for incorporation into DNA, patients treated with 6-MP or azathioprine accumulate DNA 6-TG in all renewing cell populations. Around 0.01–0.03% of the DNA guanine of circulating peripheral blood lymphocytes27 and skin of patients taking azathioprine is replaced by 6-TG.28 These levels of DNA 6-TG are not directly harmful to cells that are not part of the immune system. The ability of DNA 6-TG to act as a UVA photosensitizer means that even these low levels constitute a potential hazard.
Thiopurine photochemistry
The substitution of the oxygen atom of canonical guanine with sulphur greatly enhances its chemical reactivity and, of particular importance in the context of this themed issue, has considerable impact upon its photochemical properties. The canonical DNA bases all absorb UVR maximally in the UVC spectral region. In contrast, thiopurines are significant UVA chromophores, and both 6-MP and 6-TG have an absorbance maximum at approximately 340 nm. In the following sections, we describe some of the photochemical reactions of DNA 6-TG that may contribute to the accumulation of DNA damage. Most of these data come from studies on cultured human cells grown in the presence of low concentrations of 6-TG to introduce a low level of DNA substitution. The cells are subsequently exposed to non-toxic and non-mutagenic doses of UVA—comparable to normal day-to-day exposure. The doses of UVA are generally ≤ 10 kJ m−2—around one-tenth of the doses normally used to investigate the effects of UVA alone in cultured cells. To put this into perspective, on a sunny summer's day around midday in south-east England, skin is exposed to around 20 W m−2UVA–so exposed skin receives 10 kJ m−2UVA in less than 10 min. UVA irradiation and DNA 6-TG are synergistically cytotoxic. The effect is pronounced and depending on genotype, cells containing non-toxic levels of DNA 6-TG can be killed by UVA doses as low as 0.5 kJ m−2.In aqueous solution, both 6-TG and 6-MP are Type I and Type II UVA photosensitisers,29 and generate a variety of highly damaging ROS.2 When 6-TG has been incorporated into DNA, excitation by UVA results in the transfer of energy to triplet molecular oxygen in a Type II reaction to generate singlet oxygen (1O2)—a relatively long-lived form of ROS. The generation of ROS within DNA is potentially extremely harmful. In the following section, we describe some of the UVA photoproducts of DNA 6-TG.
Photochemical consequences of DNA 6-TG and UVA interaction
The ROS generated by the interaction between UVA and DNA 6-TG are predominantly localized to the nucleus.28 They are the likely mediators of the following reactions.The second type of protein damage, photochemical DNA-protein crosslinking, occurs via the reactive thiol of DNA 6-TG. UVA irradiation of mixtures of the free 6-TG base and low molecular weight thiol compounds such as cysteine, β-mercaptoethanol, N-acetylcysteine or glutathione (GSH) generates addition products.31 These reactions are dependent on oxygen and, in the case of GSH at least, proceed by nucleophilic substitution involving an oxidized 6-TG intermediate (GSO3). Analogous reactions occur at 6-TG in DNA. Incubation of oligoeotides containing GSO3 with peptides containing a single cysteine results in efficient crosslinking between the oligonucleotide and oligopeptide.32 The high efficiency of the reaction suggested that DNA-protein crosslinks might be significant UVA photoproducts in cells containing DNA 6-TG. This has been verified experimentally, and extensive DNA-protein crosslinking occurs in 6-TG/UVA treated cells.32 Importantly, the crosslinking reaction is very favourable, and these lesions are detectable after UVA irradiation of cultured cells with low levels of DNA 6-TG that approximate those in the skin of patients taking azathioprine.
In cultured cells containing DNA 6-TG, DNA replication and transcription are inhibited very shortly after UVA irradiation.33,36 The Chk1 protein is phosphorylated, indicating activation of the ATR-related cell cycle checkpoint. At higher UVA doses, neither replication nor transcription recovers significantly over the subsequent 24 h. This is accompanied by degradation of the Chk1 protein and checkpoint abrogation, probably as a consequence of the persistence of unrepaired DNA lesions.37
In addition to DNA 6-TG itself, canonical bases are also at risk of oxidation by these ROS, and 6-TG or azathioprine treatment combined with UVA increases the level of DNA 8-oxoguanine, a hallmark of oxidative stress.38 These same experiments provided evidence that DNA 6-TG is not the only source of photochemically-generated ROS but that the pool of nucleotide precursors may also contribute.
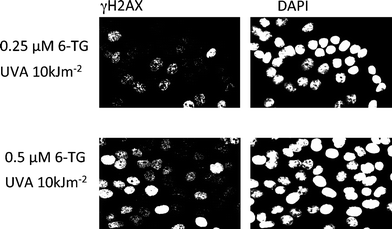 | ||
| Fig. 3 DNA replication -blocking lesions and double-strand breakage in cells treated with 6-TG and UVA : HCT116 cells that had been treated with the concentrations of 6-TG shown to introduce DNA 6-TG were irradiated with 10 kJ m−2UVA. Fixed cells were stained with anti-γH2AX antibody (left panels) and with DAPI to reveal all nuclei. There is a 6-TG dose-dependent increase in the fraction of cells exhibiting focal (light) or pan-nuclear (heavy) staining for H2AX. These represent DNA double-strand breaks and inhibited replication, respectively. No H2AX staining was observed in cells treated with 6-TG or UVA alone. | ||
DNA repair of 6-TG/UVA DNA lesions
6-TG is not a natural DNA constituent. As a consequence, there has never been selective pressure to evolve solutions to problems posed by photochemical 6-TG DNA damage. A notable feature of the iatrogenic DNA lesions introduced by UVA-activated DNA 6-TG is the likely challenge that most of them pose to a cell's repair systems. With the exception of DNA 8-oxoguanine, which is efficiently removed, and DNA single-strand breaks that can be rejoined quickly and efficiently, most photochemical DNA 6-TG lesions are likely to be difficult to repair. NER-defective xeroderma pigmentosum cells are no more sensitive to 6-TG/UVA than repair proficient cells,28 which suggests that NER does not excise the predominant cytotoxic lesions. Among the 6-TG photoproducts, DNA GSO2 and GSO3—which significantly destabilize the DNA helix and would be likely candidate for removal by NER—are not actively repaired and remain in DNA for at least 24 h after radiation.34DNA-protein crosslinks are probably difficult for DNA repair systems to deal with. The majority of photochemical DNA-protein crosslinks persist for many hours32—although the possible selective removal of individual crosslinked protein species has not yet been addressed. A high density of DNA-bound proteins is likely to impede normal DNA functions by preventing access to important replication, transcription and repair enzymes. The MSH2 and MSH6 DNA mismatch repair, and the XPA nucleotide excision repair proteins are among those identified in the DNA-complexed fraction from 6-TG/UVA treated cells.32 This suggests that sequestration of key DNA repair factors might threaten DNA integrity by directly reducing DNA repair capacity.DNA interstrand crosslinks require complex processing that involves cooperation among several different DNA repair pathways. The necessity to incise both DNA strands means that DSBs are an inevitable intermediates in this processing. These occur in the S phase with a high probability of subsequent detrimental effects. The precise fate of DNA–protein crosslinks is not well established. Crosslinking between proteins and DNA may occur naturally during the normal course of DNA metabolism. If so, these reactions are likely to be infrequent and any systems that have evolved to counteract their effects are likely to be overwhelmed by the abrupt introduction of large numbers of substrate DNA lesions.
How is this relevant to skin cancer?
The IARC designation of azathioprine as a carcinogen in man40 is based largely on the incidence of sun-related skin cancer (particularly SCC) in organ transplant patients. Epidemiological data for shorter intermittent thiopurine exposure, for example in inflammatory bowel disease, are rather less compelling but do indicate that persistent azathioprine use (> 1 year) also increases skin cancer risk.41The studies of the UVA photochemistry of DNA 6-TG described above reveal how potentially hazardous it is. The chronic formation of different types of DNA damage (summarised in Fig. 4), some of which are poorly repaired and are likely to result in replication-related DNA strand breakage, is effectively a form of genomic instability. What is the evidence that these molecular events constitute a real hazard for patients taking azathioprine? The UVA skin sensitivity of these patients indicate that they do sustain some or all of the DNA damage we have described. Initial measurements in a small number of patients indicated that azathioprine treatment is associated with an increased sensitivity to erythema induction by UVA, but not by UVB. UVR induced erythema correlates well with photochemical DNA damage.42 These initial findings were confirmed and extended in a more recent study in a large group of kidney transplant patients. Replacing azathioprine by the unrelated immunosuppressant mycophenolate initiated a reduction in DNA 6-TG and concomitant decline in skin UVA sensitivity in all patients. These observations provide a clear indication that UVA sensitivity is not a consequence of immunosuppression but is instead related to the level of DNA 6-TG and the induction of photochemical DNA lesions. Further evidence of this was provided by a reduced proportion of cells with mutated p53 in biopsies from UVA-irradiated skin of the patients on mycophenolate.43
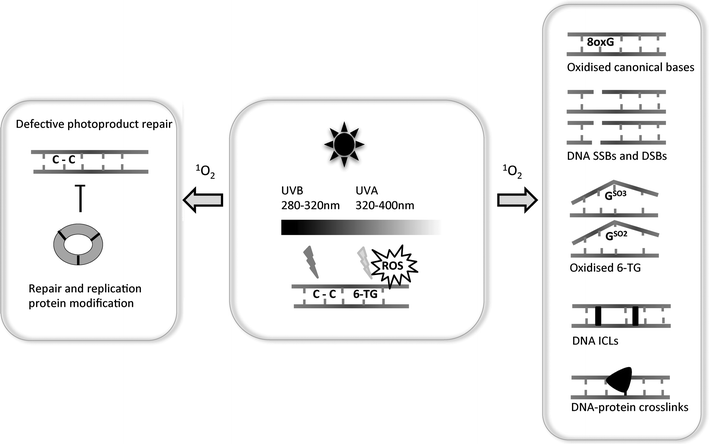 | ||
| Fig. 4 6-TG and UV-induced DNA lesions: The centre panel depicts the damaging effects of the UVB and UVA components of incident sunlight on DNA containing 6-TG. UVB induces predominantly intrastrand pyrimidine dimerization. UVA interacts with 6-TG to generate ROS—principally 1O2—which causes the multiple forms of DNA damage depicted in the right panel. In addition to DNA lesions, these ROS also cause protein modification. One example of this is depicted in the left-hand panel—the covalent crosslinking of the subunits of PCNA, an essential DNA replication and repair protein. One potential consequence of damage to DNA repair proteins is the extended persistence of potentially mutagenic UVB-induced DNA lesions. | ||
The generation of 1O2 from DNA 6-TG constitutes a clear mutational risk, although most of the photochemical 6-TG DNA lesions that have been identified are more likely to arrest DNA replication than to permit the misreplication that is required for mutagenesis. Indeed, sequence analysis of candidate genes in SCC from immunosuppressed patients provides little support for the idea of an increase in mutations due directly to oxidized DNA bases. The possible involvement of error-prone DNA polymerases in bypassing oxidized DNA 6-TG lesions or processing interstrand DNA crosslinks is a possible source of indirect mutation. The TP53 mutational spectra of transplant-related NMSC are dominated by dipyrimidine UVB signature transition mutations. This might simply reflect pre-existing UVB-induced mutations that predate immunosuppression. On the other hand, a study of PTCH mutations in transplant-related BCC (which, unlike SCC, do not have a pre-malignant precursor) also revealed an excess of UVB-like mutations with little indication of an increased contribution from miscoding by oxidized DNA bases.22 Does the absence of excess oxygen-related mutations mean that UVA-induced 6-TG DNA photoproducts do not influence mutagenesis? This seems unlikely, and recent evidence suggests that they may affect mutagenesis indirectly. Excision of UVC-induced 6-4 pyrimidine:pyrimidone (6-4) and CPD is significantly impaired when cells containing low levels of DNA 6-TG are irradiated with UVA.34 The extent of inhibition is UVA dose-dependent. Whether this effect is due to ROS-mediated damage of critical NER repair proteins, their sequestration in non-productive complexes with 6-TG photoproducts or simply their inability to locate their substrates because of too extensive DNA 6-TG photodamage remains to be determined. One consequence of this inhibition would be an enhanced mutagenicity of UVB photoproducts—consistent with the observed spectrum of mutations in transplant-related NMSC.
Concluding remarks
Over the past 5 years, we have gained considerable insight into the photochemistry of DNA 6-TG. The studies we have described have uncovered photochemical damage to DNA itself, and to some of the proteins associated with it. We believe that some or all of these reaction products are important in the initiation of skin cancer associated with long-term thiopurine exposure. Of course, it is impossible to completely disentangle their roles from those of other likely contributory factors, such as reduced immunosurveillance and oncogenic viruses. Nevertheless, in view of the critical role of sunshine exposure, a photochemical mechanism goes some way to explain why the skin cancer risk is so much higher in transplant recipients than in patients with non-pharmacological immunosuppression. We can be reasonably confident that some, or all, of the types of DNA and protein damage we have described occur in patients. 6-TG is readily detectable in patients' DNA. Their skin is selectively sensitive to erythema induction by UVA—generally considered to reflect the presence of DNA lesions. Skin photosensitivity is reversed when azathioprine is withdrawn, and this coincides with a reduction in the level of DNA 6-TG. Sun-exposed skin sustains both UVA- and UVB-induced DNA damage. The possibility that DNA-6-TG lesions increase cancer risk in organ transplant recipients by attenuating UVB photoproduct repair is the subject of ongoing research.Thiopurines are invaluable agents in the treatment of frequently complex and refractory inflammatory conditions, and a patient's quality of life is often much improved by their use. It is therefore likely that physicians will continue to prescribe these drugs. In view of our increased understanding of DNA 6-TG photochemistry, it is essential that all prescribing doctors are aware of potential sun-related side effects and discuss with their patients the need for vigilant photoprotection against UVA as well as UVB. For patients, such as organ transplant recipients, who are known to be at particularly high risk, early contact with a dermatologist is critical and this is now generally a routine aspect of management. It seems prudent to extend this sun protection advice and dermatological screening to all patients who take thiopurines for extended periods.
Acknowledgements
We thank all our past and present laboratory colleagues whose work has contributed to the development of the ideas described here. We are particularly grateful to Drs. Jane McGregor, Catherine Harwood and Reto Brem for their comments on the manuscript and to Reto for his permission to use the unpublished data in Figure 3. This work was supported by Cancer Research UK.References
- G. B. Elion, The purine path to chemotherapy, Science, 1989, 244, 41–47 CAS.
- P. Karran and N. Attard, Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer, Nat. Rev. Cancer, 2008, 8, 24–36 CrossRef CAS.
- R. T. Bustami, A. O. Ojo, R. A. Wolfe, R. M. Merion, W. M. Bennett, S. V. McDiarmid, A. B. Leichtmana, P. J. Held and F. K. Port, Immunosuppression and the risk of post-transplant malignancy among cadaveric first kidney transplant recipients, Am. J. Transplant., 2004, 4, 87–93 CrossRef.
- I. Penn, W. Hammond, L. Brettschneider and T. E. Starzl, Malignant Lymphomas in Transplantation Patients, Transplant Proc., 1969, 1, 106–112 CAS.
- A. G. Sheil, A. P. Disney, T. H. Mathew and N. Amiss, De novo malignancy emerges as a major cause of morbidity and late failure in renal transplantation, Transplant Proc., 1993, 25, 1383–1384 CAS.
- A. E. Grulich, M. T. vanLeeuwen, M. O. Falster and C. M. Vajdic, Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis, Lancet, 2007, 370, 59–67 CrossRef.
- S. Euvrard, J. Kanitakis and A. Claudy, Skin cancers after organ transplantation, N. Engl. J. Med., 2003, 348, 1681–1691 CrossRef.
- H. M. Ramsay, A. A. Fryer, C. M. Hawley, A. G. Smith and P. N. Harden, Non-melanoma skin cancer risk in the Queensland renal transplant population, Br. J. Dermatol., 2002, 147, 950–956 CrossRef CAS.
- M. M. Hartevelt, J. N. Bavinck, A. M. Kootte, B. J. Vermeer and J. P. Vandenbroucke, Incidence of skin cancer after renal transplantation in The Netherlands, Transplantation, 1990, 49, 506–509 CrossRef CAS.
- P. Jensen, S. Hansen, B. Møller, T. Leivestad, P. Pfeffer, O. Geiran, P. Fauchald and S. Simonsen, Skin cancer in kidney and heart transplant recipients and different long-term immunosuppressive therapy regimens, J. Am. Acad. Dermatol., 1999, 40, 177–186 CrossRef CAS.
- F. R. de Gruijl, UV-induced immunosuppression in the balance, Photochem. Photobiol., 2008, 82, 2–9 Search PubMed.
- G. F. L. Hofbauer, J. N. Bouwes-Bavinck and S. Euvrard, Organ transplantation and skin cancer: basic problems and new perspectives, Exp. Dermatol., 2010, 19, 473–482 CrossRef.
- J. N. B. Bavinck, M. Feltkamp, L. Struijk and J. Ter Schegget, Human papillomavirus infection and skin cancer risk in organ transplant recipients, J. Invest. Dermatol. Symp. Proc., 2001, 6, 207–211 CrossRef.
- I. Nindl and F. Rosl, Molecular concepts of virus infections causing skin cancer in organ transplant recipients, Am. J. Transplant., 2008, 8, 2199–2204 CrossRef CAS.
- M. C. Feltkamp, M. N. deKoning, J. N. B. Bavinck and J. Ter Schegget, Betapapillomaviruses: innocent bystanders or causes of skin cancer, J. Clin. Virol., 2008, 43, 353–360 CrossRef CAS.
- F. E. Ghissassi, R. Baan, K. Straif, Y. Grosse, B. Secretan, V. Bouvard, L. Benbrahim-Tallaa, N. Guha, C. Freeman, L. Galichet and V. Cogliano, on behalf of the WHO IARC Cancer Monograph Working Group, A review of human carcinogens—Part D: Radiation, Lancet Oncol., 2009, 10, 751–752 CrossRef.
- E. C. Friedberg, G. C. Walker, W. Siede, R. D. Wood, R. A. Schultz and T. Ellenberger, DNA Repair and Mutagenesis, ASM Press, Washington, 2nd edn, 2006 Search PubMed.
- G. P. Pfeifer, Y. H. You and A. Besaratinia, Mutations induced by ultraviolet light, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 571, 19–31 CrossRef CAS.
- L. Daya-Grosjean, Xeroderma pigmentosum and skin cancer, Adv. Exp. Med. Biol., 2008, 637, 19–27 CrossRef CAS.
- J. M. McGregor, R. J. M. Berkhout, M. Rozycka, J. Ter Scheget, J. N. B. Bavinck, L. Brooks and T. Crook, p53 mutations implicate sunlight in post-transplant skin cancer irrespective of human papillomavirus status, Oncogene, 1997, 15, 1737–1740 CAS.
- S. Queille, L. Luron, A. Spatz, M. F. Avril, V. Ribrag, P. Duvillard, C. Hiesse, A. Sarasin, J. P. Armand and L. Daya-Grosjean, Analysis of skin cancer risk factors in immunosuppressed renal transplant patients shows high levels of UV-specific tandem CC to TT mutations in the p53 gene, Carcinogenesis, 2007, 28, 724–731 CrossRef CAS.
- C. A. Harwood, N. R. Attard, P. O'Donovan, P. Chambers, C. M. Perrett, C. M. Proby, J. M. McGregor and P. Karran, PTCH mutations in basal cell carcinomas from azathioprine-treated organ transplant recipients, Br. J. Cancer, 2008, 99, 1276–1284 CrossRef CAS.
- P. Autier, J. F. Doré, A. M. Eggermont and J. W. Coebergh, Epidemiological evidence that UVA radiation is involved in the genesis of cutaneous melanoma, Curr. Opin. Oncol., 2011, 23, 189–196 CrossRef.
- P. J. Rochette, J. P. Therrien, R. Drouin, D. Perdiz, N. Bastien, E. A. Dobretsky and E. Sage, UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine-thymine dipyrimidines and correlate with the mutation spectrum in rodent cells, Nucleic Acids Res., 2003, 31, 2786–2794 CrossRef CAS.
- S. Mouret, C. Philippe, J. Gracia-Chantegrel, A. Banyasz, S. Karpati, D. Markovitsi and T. Douki, UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8, 1706–1711 CAS.
- E. D. Pleasance, R. K. Cheetham, P. J. Stephens, D. J. McBride, S. J. Humphray, C. D. Greenman, I. Varela, M. L. Lin, G. R. Ordóñez, G. R. Bignell, K. Ye, J. Alipaz, M. J. Bauer, D. Beare, A. Butler, R. J. Carter, L. Chen, A. J. Cox, S. Edkins, P. I. Kokko-Gonzales, N. A. Gormley, R. J. Grocock, C. D. Haudenschild, M. M. Hims, T. James, M. Jia, Z. Kingsbury, C. Leroy, J. Marshall, A. Menzies, L. J. Mudie, Z. Ning, T. Royce, O. B. Schulz-Trieglaff, A. Spiridou, L. A. Stebbings, L. Szajkowski, J. Teague, D. Williamson, L. Chin, M. T. Ross, P. J. Campbell, D. R. Bentley, P. A. Futreal and M. R. Stratton, A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2010, 463, 191–196 CrossRef CAS.
- C. Cuffari, D. Y. Li, J. Mahoney, J. Y. Barnes and T. M. Bayless, Peripheral blood mononuclear cell DNA 6-thioguanine metabolite levels correlate with decreased interferon-g production in patients with Crohn's disease on AZA therapy, Dig. Dis. Sci., 2004, 49, 133–137 CrossRef CAS.
- P. O'Donovan, C. M. Perrett, X. Zhang, B. Montaner, Y.-Z. Xu, C. A. Harwood, J. M. McGregor, S. L. Walker, F. Hanaoka and P. Karran, Azathioprine and UVA light generate mutagenic oxidative DNA damage, Science, 2005, 309, 1871–1874 CrossRef CAS.
- V. J. Hemmens and D. E. Moore, Photochemical sensitization by azathioprine and its metabolites–I. 6-Mercaptopurine, Photochem. Photobiol., 1986, 43, 247–255 CrossRef CAS.
- B. Montaner, P. O'Donovan, O. Reelfs, C. M. Perrett, X. Zhang, Y.-Z. Xu, X. Ren, P. Macpherson, D. Frith and P. Karran, Reactive oxygen-mediated damage to a human DNA replication and repair protein, EMBO Rep., 2007, 8, 1074–1079 CrossRef CAS.
- X. Ren, Y.-Z. Xu and P. Karran, Photooxidation of 6-thioguanine by UVA: the formation of addition products with low molecular weight thiol compounds, Photochem. Photobiol., 2010, 86, 1038–1045 CrossRef CAS.
- Q. Gueranger, A. Kia, D. Frith and P. Karran, Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA, Nucleic Acids Res., 2011, 39, 5057–5066 CrossRef CAS.
- X. Zhang, G. Jeffs, X. Ren, P. O'Donovan, B. Montaner, C. M. Perrett, P. Karran and Y.-Z. Xu, Novel DNA lesions generated by the interaction between therapeutic thiopurines and UVA light, DNA Repair, 2007, 6, 344–354 CrossRef CAS.
- F. Li, DNA damage and UVA-induced oxidation of DNA 6-thioguanine, PhD Thesis, University of London, 2010 Search PubMed.
- R. Brem, I. Daehn and P. Karran, Efficent DNA interstrand crosslinking by 6-thioguanine and UVA radiation, DNA Repair, 2011, 10, 869–876 CrossRef CAS.
- R. Brem, F. Li and P. Karran, Reactive oxygen species generated by thiopurine/UVA cause irreparable transcription-blocking DNA lesions, Nucleic Acids Res., 2009, 37, 1951–1961 CrossRef CAS.
- R. Brem, F. Li, B. Montaner, O. Reelfs and P. Karran, DNA breakage and cell cycle checkpoint abrogation induced by a therapeutic thiopurine and UVA radiation, Oncogene, 2010, 29, 3953–3963 CrossRef CAS.
- M. S. Cooke, T. L. Duarte, D. Cooper, J. Chen, S. Nandagopal and M. D. Evans, Combination of azathioprine and UVA irradiation is a major source of cellular 8-oxo-7,8-dihydro-2′-deoxyguanosine, DNA Repair, 2008, 7, 1982–1989 CrossRef CAS.
- L. H. Thompson and J. M. Hinz, Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2009, 668, 54–72 CrossRef CAS.
- Y. Grosse, R. Baan, K. Straif, B. Secretan, F. E. Ghissassi, V. Bouvard, L. Benbrahim-Tallaa, N. Guha, L. Galichet and V. Cogliano, on behalf of the WHO IARC Cancer Monograph Working Group. A review of human carcinogens, Part A: Pharmaceuticals, Lancet Oncol., 2009, 10, 13–14 CrossRef.
- M. D. Long, H. H. Herfarth, C. A. Pipkin, C. Q. Porter, R. S. Sandler and M. D. Kappelman, Increased risk for non-melanoma skin cancer in patients with inflammatory bowel disease, Clin. Gastroenterol. Hepatol., 2010, 8, 268–274 CrossRef.
- H. Hacham, S. E. Freeman, R. W. Gange, D. J. Maytum, J. C. Sutherland and B. M. Sutherland, Do pyrimidine dimer yields correlate with erythema induction in human skin irradiated in situ with ultraviolet light (275–365 nm)?, Photochem. Photobiol., 1991, 53, 559–563 CrossRef CAS.
- G. F. L. Hofbauer, N. R. Attard, C. A. Harwood, J. M. McGregor, P. Dziunycz, G. Iotzova-Weiss, G. Straub, R. Meyer, Y. Kamenisch, M. Berneburg, L. E. French, R. P. Wuthrich, P. Karran and A. L. Serra, Reversal of UVA skin photosensitivity and DNA damage in kidney transplant patients by replacing azathioprine, Am. J. Transplant., 2011 Search PubMed , in press.
Footnotes |
| † Contribution to the themed issue on the biology of UVA. |
| ‡ Present address: Department of Dermatology, The Royal London Hospital, London, E1 1BB, UK |
| This journal is © The Royal Society of Chemistry and Owner Societies 2012 |