Mono thiomalonates as thioester enolate equivalents—enantioselective 1,4-addition reactions to nitroolefins under mild conditions†‡
Paolo
Clerici
and
Helma
Wennemers
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, CH-4056 Basel, Switzerland. E-mail: Wennemers@unibas.ch; Fax: (+41) 61-267-0976
First published on 8th November 2011
Abstract
Mono thiomalonates (MTMs) are introduced as thioester enolate equivalents. Asymmetric organocatalyzed conjugate addition reactions to nitroolefins proceed under mild conditions to afford synthetically useful γ-nitrothioesters with excellent yields and enantioselectivities.
Introduction
Catalytic asymmetric C–C bond formations between thioesters and electrophiles remain an important challenge in organic synthesis.1 The resulting chiral thioesters are versatile building blocks since thioesters allow for further transformations into many other functional groups such as ketones, aldehydes, or amides.2 The generation of thioester enolates under mild conditions is, however, challenging due to the comparatively low acidity of the α-proton3 combined with the reactivity of thioesters towards nucleophiles.4 In recent years mild organocatalytic approaches utilizing either malonic acid half thioesters (MAHTs), dithiomalonates, or electron poor thioesters emerged.5–9 However, the tendency of MAHTs to decarboxylate without a concomitant C–C bond formation and the comparatively low reactivity of the thioester enolate precursors typically requires the use of large amounts of the catalyst (≥10 mol%), long reaction times and carefully chosen reaction conditions.6–10 Thus, there is a need for robust thioester enolate equivalents that react readily with electrophiles in the presence of small amounts of an organocatalyst to form the addition products in high yields and stereoselectivities.Herein we describe mono thiomalonates (MTMs) bearing a cleavable ester moiety as effective thioester enolate equivalents. They were envisioned to provide the addition products of thioester enolates with electrophiles after cleavage of the ester and decarboxylation (Scheme 1). We demonstrate the value of MTMs in asymmetric organocatalytic reactions with nitroolefins providing chiral γ-nitrothioesters in excellent yields and enantioselectivities. Furthermore, the synthetic versatility of the γ-nitrothioesters for subsequent transformations is highlighted.
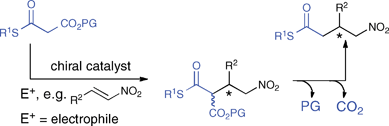 | ||
| Scheme 1 Mono thiomalonates as thioester enolate equivalents. | ||
Results and discussion
Evaluation of MTMs and catalysts
We started our investigations by preparing and evaluating the reactivity of MTM 1 bearing an acid labile p-methoxybenzyl (PMB) ester and an acid stable p-methoxyphenyl (PMP) thioester. MTM 1 was easily obtained by reacting malonic acid first with p-methoxybenzylalcohol and then p-methoxythiophenol using DCC/DMAP as coupling reagent.11 The reactivity of MTM 1 was explored by allowing it to react with nitrostyrene in the presence of 5 mol% of cinchona alkaloid urea derivatives 2–7 that had previously proven to be valuable for addition reactions of malonates and MAHTs to nitroolefins.8,12 Satisfyingly, good to excellent conversion to the conjugate addition product and stereoselectivities were observed with all of the catalysts using only a slight excess of 1.1 eq. of nitrostyrene with respect to the MTM (Table 1, entries 1–6). Also the removal of the PMB group by trifluoroacetic acid (TFA) and the subsequent base induced decarboxylation proceeded cleanly.|
|
|||||||
|---|---|---|---|---|---|---|---|
| entry | cat. | mol (%) | time (h) | solvent | T/°C | conv.a (%) | eeb (%) |
| a Estimated by TLC analysis. b Determined by chiral phase HPLC analysis, absolute configuration in brackets. c Isolated yield. | |||||||
| 1 | 2 | 5 | 24 | toluene | RT | quant. | 86 (S) |
| 2 | 3 | 5 | 6 | toluene | RT | quant. | 85 (S) |
| 3 | 4 | 5 | 6 | toluene | RT | quant. | 84 (S) |
| 4 | 5 | 5 | 24 | toluene | RT | 50 | 78 (S) |
| 5 | 6 | 5 | 6 | toluene | RT | quant. | 90 (R) |
| 6 | 7 | 5 | 6 | toluene | RT | quant. | 88 (R) |
| 7 | 6 | 5 | 24 | CH2Cl2 | RT | quant. | 84 (R) |
| 8 | 6 | 5 | 24 | Et2O | RT | quant. | 83 (R) |
| 9 | 6 | 5 | 24 | EtOH | RT | quant. | 59 (R) |
| 10 | 6 | 3 | 12 | toluene | RT | quant. | 91 (R) |
| 11 | 6 | 1 | 24 | toluene | RT | quant. | 91 (R) |
| 12 | 6 | 1 | 24 | toluene | −15 | quant. | 94 (R) |
| 13 | 6 | 1 | 24 | toluene | −50 | 95c | 98 (R) |
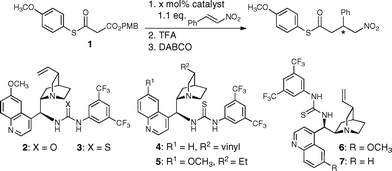
The best results in terms of reactivity and enantioselectivity were obtained with the epiquinidine thiourea derivative 6 using toluene as solvent (Table 1, entry 5). The reactivity of 6 proved to be sufficiently high to reduce the catalyst loading to as little as 1 mol% (Table 1, entry 11). Lowering the temperature significantly improved the enantioselectivity. At −50 °C the γ-nitrothioester was obtained with an enantioselectivity of 98% ee and a yield of 95% (Table 1, entry 13).13
Substrate scope
With these reaction parameters defined, the scope of reactions of MTM 1 with a variety of nitroolefins was explored. To our delight, the conjugate addition reaction products were obtained in high yields and excellent enantioselectivities for a range of different β-substituted nitroolefins (Table 2). The best reactivities were observed with electron poor aromatic nitroolefins (Table 2, entries 2–8), but also electron rich aromatic nitroolefins and aliphatic nitroolefins reacted readily (Table 2, entries 9–15). The latter typically required higher amounts of catalyst (3–20 mol%) and in some cases longer reaction times, but provided the products also in enantioselectivities of up to 99% ee.|
|
||||
|---|---|---|---|---|
| entry | R1 | mol (%) | yielda (%) | eeb (%) |
| a Isolated yield. b Determined by chiral phase HPLC analysis. c Nitroolefin was not fully soluble. d Reaction took 36 h for complete conversion. e 2 eq. of nitroolefin with respect to 1 was used. | ||||
| 1 | Ph | 1 | 95 | 98 |
| 2 | C6H4-2-Cl | 1 | 96 | 99 |
| 3 | C6H4-4-Cl | 1 | 92 | 98 |
| 4 | C6H3-2,4-Cl2 | 1 | >98 | 99 |
| 5 | C6H4-4-Br | 1 | >98 | 98 |
| 6 | C6H4-4-F | 1 | >98 | 94 |
| 7c | C6H4-4-NO2 | 5 | 82 | 97 |
| 8 | C6H4-2-CF3 | 1 | >98 | 99 |
| 9d | β-naphthyl | 5 | >98 | >99 |
| 10 | α-naphthyl | 3 | 96 | 97 |
| 11 | 2-thiophene | 1 | 98 | 98 |
| 12 | C6H4-4-OCH3 | 3 | 85 | 98 |
| 13 | C6H3-2,4-(OCH3)2 | 5 | 98 | 91 |
| 14d,e | n-hexyl | 20 | >98 | 91 |
| 15d,e | c-hexyl | 20 | 91 | 94 |
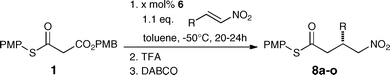
These results demonstrate that in comparison to MAHTs that provide the same products when reacted with nitroolefins,8 the MTMs are significantly more reactive, and thereby allow for the use of significantly lower amounts of the catalyst, near equimolar amounts of the reactants and shorter reaction times. Furthermore, the products were obtained in significantly higher stereoselectivities and fewer side reactions occur. These features outweigh the lower atom economy of the MTMs compared to MAHTs and demonstrate their value as chemically robust yet sufficiently reactive thioester enolate equivalents.
Initial studies were performed to evaluate the mechanism of the reaction. The nucleophilic nature of the MTM stems from the acidity of the α-proton that is higher compared to not only that of the corresponding thioesters but also malonates.3 Consequently, the α-proton is also easily abstracted within the initially formed conjugate addition product of MTM 1 and the nitroolefins. This is easily monitored by an exchange of the proton by deuterium in the presence of e.g. deuterated methanol. As a result, the stereochemistry at this stereogenic center is scrambled which has, however, no consequence for the overall enantioselectivity of the reaction since the stereogenic information at this center is lost upon decarboxylation.
Reactions performed in the presence of only a urea derivative did not provide product and in the presence of only a base (e.g. NEt3 or DABCO) or a base and a non-covalently linked urea derivative product formation was significantly slower. Thus, the bifunctional nature of the catalyst is crucial for the catalytic efficiency. Within the catalyst structure the relative orientation of the base and the thiourea moiety are important since other bifunctional molecules bearing these functional groups (e.g. Takemoto catalyst14) exhibit lower catalytic efficiency. More detailed studies on e.g. the strength and geometry of intermolecular interactions between the MTM, the nitroolefin and the catalyst are ongoing.
Derivatisation of γ-nitrothioesters
Finally, we evaluated the versatility of the γ-nitrothioesters for transformations into other compounds and explored various different functionalization strategies (Scheme 2).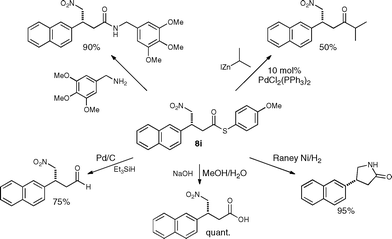 | ||
| Scheme 2 Synthetic versatility of γ-nitrothioesters. | ||
For example, reduction of the nitro group of γ-nitrothioester 8i led to the formation of γ-butyrolactams, reduction of the thioester allowed for access to γ-nitroaldehydes or by a Fukuyama coupling to γ-nitroketones.2 In all of these transformations the stereochemistry at the stereogenic center remained intact. These examples demonstrate that a multitude of different building blocks that can be further modified are easily accessible from the chiral γ-nitrothioesters.
Conclusions
In summary we have established mono thiomalonates as thioester enolate equivalents and demonstrated their value in asymmetric conjugate addition reactions with nitroolefins. Under mild organocatalytic conditions synthetically versatile γ-nitrothioesters were obtained in excellent yields and stereoselectivities. Mono thiomalonate derivatives bearing cleavable esters that are not sensitive to acid but to e.g. light or hydrogenation are expected to extend the usefulness of these thioester enolate equivalents further. We are currently exploring the scope of the mono thiomalonates in catalytic asymmetric addition reactions with other electrophiles such as imines and aldehydes.Experimental section
General aspects and materials
Materials and reagents were of the highest commercially available grade and used without further purification. Reactions were monitored by thin layer chromatography using Merck silica gel 60 F254 plates. Compounds were visualized by UV and KMnO4. Flash chromatography was performed using Merck silica gel 60, particle size 40–63 μm. 1H and 13C NMR spectra were recorded on a Bruker DPX 400 spectrometer. Chemical shifts are reported in ppm using TMS or the residual solvent peak as a reference. HPLC analyses were performed on an analytical HPLC with a diode array detector from Shimadzu. A Bruker Esquire 3000 Plus was used for electrospray ionisation (ESI) mass spectrometry. The 1H and 13C NMR spectra as well as HPLC chromatograms for all new compounds are provided in the supporting information‡.Synthesis of monothiomalonate 1
Malonic acid (100.0 mmol, 10.4 g, 2.0 eq) was dissolved in anhydrous acetonitrile (75 mL) in an argon atmosphere followed by addition of 4-(dimethylamino)-pyridine (DMAP) (10 mmol, 1.2 g, 0.2 eq) and 4-methoxybenzyl alcohol (50 mmol, 6.9 g, 1.0 eq). The solution was cooled to 0 °C and a solution of N,N′-dicyclohexylcarbodiimide (DCC) (75 mmol, 15.5 g, 1.5 eq) in anhydrous acetonitrile (25 ml) was added dropwise over 30 min. The reaction mixture was stirred at 0 °C for 30 min and then for additional 2 h at room temperature. The mixture was then filtered to remove the dicyclohexylurea, and volatiles were removed at reduced pressure. The crude mixture was re-dissolved in a mixture of CH2Cl2 (100 mL) and saturated aqueous NaHCO3 (100 mL). The two phases were separated and the aqueous phase was washed twice with CH2Cl2. The pH of the aqueous phase was then adjusted to pH 3 by addition of an aqueous solution of HCl (10%) and reextracted with CH2Cl2 (3 × 80 ml each). The combined organic phases were dried over MgSO4 and after a filtration, all volatiles were removed at reduced pressure to yield a solid that was used without further purification.The solid was dissolved in anhydrous CH2Cl2 (75 mL) and 4-methoxythiophenol (60.0 mmol, 8.4 g 1.2 eq) was added. The solution was cooled to 0 °C and a solution of DCC (75.0 mmol, 15.5 g, 1.5 eq) in CH2Cl2 (25 mL) was added dropwise within 30 min. The reaction mixture was stirred for 30 min at 0 °C and then at r.t. for two hours. The formed urea was removed by filtration and all volatiles were removed at reduced pressure. The crude compound was then purified by column chromatography using a gradient of CH2Cl2/pentane from 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to pure CH2Cl2 to yield MTM 1 (13.8 g, 80%) as a white solid.
:3 to pure CH2Cl2 to yield MTM 1 (13.8 g, 80%) as a white solid.
1H NMR (400 MHz, CDCl3, 25 °C): δ = 7.32–7.29 (m, 4H), 6.95 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 5.13 (s, 2H), 3.82 (s, 3H), 3.81 (s, 3H), 3.66 (s, 2H); 13C NMR (100 MHz, CDCl3): δ = 191.0, 166.2, 161.4, 160.2, 136.6, 130.7, 127.7, 117.9, 115.4, 114.4, 67.7, 55.8, 55.7, 49.3; IR (KBr) ν 2937, 2840, 1733, 1695, 1250 cm−1; MS (ESI): 369 (M + Na); Elemental analysis calcd (%) for C18H18O5S: C 62.41, H 5.24; found: C 62.56, H 5.20.
General procedure for the conjugate addition reactions
The nitroolefin (0.11 mmol, 1.1 equiv), MTM 1 (35.0 mg, 0.10 mmol), and the catalyst (0.001 mmol) were dissolved in toluene (1 mL) in a capped vial at −50 °C. After stirring the resulting solutions for 24 h, all volatiles were removed at reduced pressure. The oily residue was then dissolved in a solution of CH2Cl2 and TFA (2:1, 1 mL) and the mixture was stirred for 2 h. After removal of all the volatiles at reduced pressure, the residue was dissolved in CH2Cl2 (0.5 mL) and DABCO (0.1 mol, 1 eq) was added. The mixture was stirred for 1 h and then purified by column chromatography on silica gel (gradient of pentane/EtOAc 4:1 to 3:1; in the case of the aliphatic compounds a gradient of pentane/EtOAc of 10:1 to 5:1 was used).
Analytical data of conjugate addition product (3R)-4-methoxyphenyl-4-nitro-3-phenylbutanethioate (8a)
1H NMR (400 MHz, CDCl3, 25 °C): δ = 7.33 (m, 3H), 7.22 (m, 4H), 6.91 (d, J = 8.9 Hz, 2H), 4.75 (dd, J = 6.7 Hz, 12.7 Hz, 1H), 4.66 (dd, J = 8.2 Hz, 12.7 Hz, 1H), 4.05 (m, J = 7.5 Hz, 1H), 3.81 (s, 3H), 3.07 (d, J = 7.4 Hz, 2H); 13C NMR (100 MHz, CDCl3, 25 °C): δ = 195.9, 160.7, 137.7, 135.9, 129.2, 128.0, 127.4, 117.4, 114.8, 78.9, 55.3, 45.8, 40.2; MS (ESI): 332 (M + H); HPLC conditions: Chiracel OD-H column with n-hexane/iPrOH (1:1, 40 °C) at 0.5 mL min−1, UV detection λ = 254 nm: tR: (S) = 26.5 min, (R) = 31.1 min (98% ee). The data is in agreement with that reported (ref. 8).13
Analytical data of conjugate addition product (3R)-4-methoxyphenyl-3-(2-chlorophenyl)-4-nitrobutanethioate (8b)
1H NMR (400 MHz, CDCl3, 25 °C) δ = 7.47–7.42 (m, 1H), 7.31–7.21 (m, 5H), 6.98–6.91 (m, 2H), 4.86 (dd, J = 10.8, 5.1 Hz, 1H), 4.82 (dd, J = 10.8, 4.4 Hz, 1H), 4.55 (m, J = 7.0 Hz, 1H), 3.84 (s, 3H), 3.24–3.20 (m, 2H). 13C NMR (100 MHz, CDCl3) δ = 196.4, 161.3, 136.4, 135.4, 134.2, 130.9, 129.7, 128.8, 127.8, 117.8, 115.4, 77.5, 55.8, 44.5, 37.6. MS (ESI) (%): 366 (M(35Cl) + H) (100), 368 (M(37Cl) + H) (33). Elemental analysis calcd (%) for C17H16ClNO4S: C 55.81, H 4.41, N 3.83; found: C 55.79, H 4.34, N 3.83. HPLC: Chiracel OD-H column, n-hexane/i-PrOH (1:1, 40 °C) at 0.5 ml min−1, UV detection λ = 254 nm: tR: (S) = 20.5 min, (R) = 22.5 min (99% ee).
For the analytical data of the conjugate addition products 8c–8o see the supporting information.
Acknowledgements
This work was supported by Bachem, the Swiss National Science Foundation and the RTN REVCAT. H.W. is grateful to Bachem for an endowed professorshipNotes and references
- M. Benaglia, M. Cinquini and F. Cozzi, Eur. J. Org. Chem., 2000, 563–572 CrossRef CAS.
- T. Miyazaki, X. Han-ja, H. Tokuyama and T. Fukuyama, Synlett, 2004, 477–480 CAS.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- For selected examples of metal-based approaches see: (a) M. Otsuka, M. Yoshida, S. Kobayashi and M. Ohno, Tetrahedron Lett., 1981, 22, 2109–2112 CrossRef CAS; (b) C. Gennari, M. G. Beretta, A. Bernardi, G. Moro, C. Scolastico and R. Todeschini, Tetrahedron, 1986, 42, 893–909 CrossRef CAS; (c) S. Nagayama and S. Kobayashi, J. Org. Chem., 1997, 62, 232–233 CrossRef; (d) D. A. Evans, C. S. Burgey, M. C. Kozlowski and S. W. Tregay, J. Am. Chem. Soc., 1999, 121, 686–699 CrossRef CAS; (e) G. P. Howell, S. P. Fletcher, K. Geurts, B. ter Horst and B. L. Feringa, J. Am. Chem. Soc., 2006, 128, 14977–14985 CrossRef CAS.
- For examples of stoichiometric reactions, see: (a) S. J. Sauer, M. R. Garnsey and D. M. Coltart, J. Am. Chem. Soc., 2010, 132, 13997–13999 CrossRef CAS; (b) G. Zhou, J. M. Yost, S. J. Sauer and D. M. Coltart, Org. Lett., 2007, 9, 4663–4665 CrossRef CAS.
- (a) D. A. Alonso, S. Kitagaki, N. Utsumi and C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 4588–4591 CrossRef CAS; (b) M. C. Kohler, J. M. Yost, M. R. Garnsey and D. M. Coltart, Org. Lett., 2010, 12, 3376–3379 CrossRef CAS; (c) N. Utsumi, S. Kitagaki and C. F. Barbas III, Org. Lett., 2008, 10, 3405–3408 CrossRef CAS; (d) S. Rossi, M. Benaglia, F. Cozzi, A. Genoni and T. Benincori, Adv. Synth. Catal., 2011, 353, 848–854 CrossRef CAS.
- (a) A. Ricci, D. Pettersen, L. Bernardi, F. Fini, M. Fochi, R. Perez Herrera and V. Sgarzani, Adv. Synth. Catal., 2007, 349, 1037–1040 CrossRef CAS; (b) Y. Pan, C. W. Kee, Z. Jiang, T. Ma, Y. Zhao, Y. Yang, H. Xue and C.-H. Tan, Chem.–Eur. J., 2011, 17, 8363–8370 CrossRef CAS.
- J. Lubkoll and H. Wennemers, Angew. Chem., Int. Ed., 2007, 46, 6841–6844 CrossRef CAS.
- (a) M. Hatano, K. Moriyama, T. Maki and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 3823–3826 CrossRef CAS; (b) Z. Jiang, Y. Yang, Y. Pan, Y. Zhao, H. Liu and C.-H. Tan, Chem.–Eur. J., 2009, 15, 4925–4930 CrossRef CAS; (c) J. Xu, X. Fu, R. Low, Y.-P. Goh, Z. Jiang and C.-H. Tan, Chem. Commun., 2008, 5526–5528 RSC; (d) W. Ye, Z. Jiang, Y. Zhao, S. L. M. Goh, D. Leow, Y.-T. Soh and C.-H. Tan, Adv. Synth. Catal., 2007, 349, 2454–2458 CrossRef CAS.
- For studies on the reactivity of MAHTs see: (a) M. Furutachi, S. Mouri, S. Matsunaga and M. Shibasaki, Chem.–Asian J., 2010, 5, 2351–2354 CrossRef CAS; (b) N. Blaquiere, D. G. Shore, S. Rousseaux and K. Fagnou, J. Org. Chem., 2009, 74, 6190–6198 CrossRef CAS; (c) K. C. Fortner and M. D. Shair, J. Am. Chem. Soc., 2007, 129, 1032–1033 CrossRef CAS; (d) N. Sordé and S. Matile, Molecules, 2001, 6, 845–851 CrossRef; see also: (e) J. Blanchet, J. Baudoux, M. Amere, M.-C. Lasne and J. Rouden, Eur. J. Org. Chem., 2008, 5493–5506 CrossRef CAS; (f) H. Brunner, J. Müller and J. Spitzer, Monatsh. Chem., 1996, 127, 845–858 CrossRef CAS.
- A MTM with a t-Bu instead of the PMB ester was also prepared and proved to react readily with nitroolefins in the presence of the cinchona alkaloids. However, generally the enantioselectivity was lower by ∼10% ee compared to that of the PMB protected MTM 1 that was therefore used for all further studies. For details, see the supporting information‡.
- (a) J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC; (b) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS; (c) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS.
- The absolute configuration was determined as described in the supporting information‡ by X-ray crystallography and polarimetric measurements which are in agreement with an earlier report ( R. E. Zelle, Synthesis, 1991, 1023 CrossRef CAS ). Note that the absolute configuration reported in ref. 8 was based on a comparison of our data with an inaccurately reported absolute configuration.
- T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Alfredo Ricci. |
| ‡ Electronic supplementary information (ESI) available: Experimental details on the synthesis and analyses of the presented compounds. CCDC reference number 820405. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ob06638b |
| This journal is © The Royal Society of Chemistry 2012 |