Barton radical reactions of 2-C-branched carbohydrates†
Tukaram M.
Pimpalpalle
,
Jian
Yin‡
and
Torsten
Linker
*
Department of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: linker@uni-potsdam.de; Fax: +49 331 9775076; Tel: +49 331 9775212
First published on 1st September 2011
Abstract
Barton esters have been introduced into the side chain of carbohydrates with high yields in only a few steps from easily available glycals. Their radical reactions afford 2-C-methyl and 2-C-bromomethyl hexoses, pentoses and disaccharides in good yields in analytically pure form. Since the Barton esters have been synthesized by an oxidative radical addition and their transformations by reductive radical processes, our results demonstrate the power of such reactions in carbohydrate chemistry.
Introduction
Radical reactions are of current interest in organic synthesis and have found many applications in natural product synthesis, and particularly in carbohydrate chemistry.1 Although the generation of radicals from alkyl halides in the presence of tin hydrides1 or silanes2 is still the most common procedure, carboxylic acids 1 can serve as radical precursors as well. For instance, electrochemical oxidation of the corresponding carboxylates 2 (Kolbe electrolysis) provides alkyl radicals 3 after decarboxylation,3 and the same radicals 3 are also obtained by photolysis of thiohydroxamate esters 4 under cleavage of N–O and C–C bonds under mild conditions (Barton reaction) (Scheme 1).4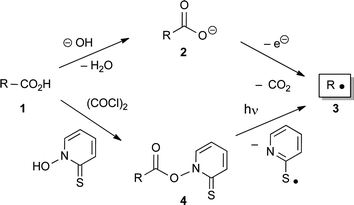 | ||
| Scheme 1 Generation of radicals 3 from carboxylic acids 1. | ||
Although there are numerous examples of tin hydride radical reactions in carbohydrate chemistry,1 the Barton reaction was only applied in this field in the total synthesis of keto-deoxy-octulosonic acids (KDO),5 to generate radicals at the anomeric center6 or with carbohydrates as chiral auxiliaries.7 Herein we describe the first Barton reactions of 2-C-branched saccharides, which allow the reduction and further functionalization of the side-chain of carbohydrates.
Results and discussion
During our studies on transition-metal-mediated radical reactions in carbohydrate chemistry,8 we developed an easy entry to carboxylic acids 1a.9 For the synthesis of the corresponding Barton esters 4a, the reaction conditions had to be carefully optimized (Table 1). Thus, thionyl or oxalyl chloride, which are often used in the preparation of Barton esters4via the acid chlorides and 2-mercaptopyridine-N-oxide sodium salt (R = Na) failed (entries 1 and 2). The starting material gluco-1a decomposed even at 0 °C, due to the acidic reaction conditions. Next, we investigated N,N′-dicyclohexyl-carbodiimide (DCC) in combination with the free 2-mercaptopyridine-N-oxide (R = H), which has been used recently for the synthesis of Barton esters.10 Although incomplete conversion was observed, the product gluco-4a was isolated with 65% yield (entry 3).|
|
|||||
|---|---|---|---|---|---|
| Entry | Config. | Activator | R | Conv. (%) | 4a (%)b |
| a For procedure see experimental section. b Yield of analytically pure products, isolated by column chromatography. c Decomposition of starting material 1a. | |||||
| 1 | gluco | SOCl2 | Na | >97 | <5c |
| 2 | gluco | (COCl)2 | Na | >97 | <5c |
| 3 | gluco | DCC | H | 70 | 65 |
| 4 | gluco | EDCI | H | >97 | 90 |
| 5 | galacto | EDCI | H | >97 | 93 |
| 6 | xylo | EDCI | H | >97 | 91 |
| 7 | arabino | EDCI | H | >97 | 89 |
| 8 | malto | EDCI | H | >97 | 81 |
| 9 | lacto | EDCI | H | >97 | 83 |
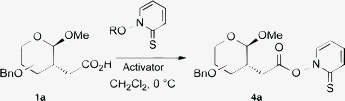
The best conditions were finally found with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDCI) as activator for the acid group.11 Thus, Barton estergluco-4a was isolated in 90% yield (entry 4). We were able to successfully apply these conditions for other hexoses, pentoses and even disaccharides (Table 1, entries 5–9). Although Barton esters are quite sensitive compounds, all products 4a were isolated in good to high yields in analytically pure form, including correct elemental analysis (experimental section). Accordingly, we found a convenient entry to such radical precursors in only a few steps from easily available glycals.
To establish the potential of Barton esters 4a in the synthesis of 2-C-branched saccharides, we first investigated reductive decarboxylations (Table 2). This is a very common transformation in radical chemistry, and it requires only t-butanethiol as hydrogen atom donor and no toxic tin hydrides.4 However, due to the lability of carbohydrates, the conditions for the initiation had to be carefully optimized with Barton estergluco-4a. Thus, simple thermolysis in benzene afforded the 2-C-methyl glycoside gluco-5 with only 30% yield besides decomposition of the starting material (entry 1). Therefore, we investigated the initiation of the Barton reaction by photolysis, which is attractive in terms of lower temperatures and milder conditions.4c However, sodium lamp irradiation gave no conversion (entry 2). With a tungsten lamp, which was used in natural product synthesis via Barton esters very recently,12 the yield was increased to 56% (entry 3). Finally, the best results were obtained with a 250 W low-pressure mercury lamp, which afforded 2-C-methyl glycoside gluco-5 with 78% yield (entry 4). We were again able to apply these conditions for other hexoses, pentoses and disaccharides (Table 2, entries 5–9) and all products 5 were isolated with moderate to good yields in analytically pure form (experimental section). Interestingly, our synthetic approach is based on a combination of oxidative (CAN-mediated addition of malonates to glycals) and reductive (Barton decarboxylation) radical reactions, demonstrating the power of such transformations in carbohydrate chemistry. Thus, we found a new entry to 2-C-methyl saccharides, which are available by cyclopropane-opening only as anomeric mixtures.13
|
|
|||||
|---|---|---|---|---|---|
| Entry | Config. | Temp. (°C) | Light source | Conv. (%) | 5 (%)b |
| a For procedure see experimental section. b Yield of analytically pure products, isolated by column chromatography. c Decomposition of starting material 4a. | |||||
| 1 | gluco | 80 | — | >97 | 30c |
| 2 | gluco | 25 | Na lamp | <5 | <5 |
| 3 | gluco | 25 | W lamp | 70 | 56 |
| 4 | gluco | 25 | Hg lamp | >97 | 78 |
| 5 | galacto | 25 | Hg lamp | >97 | 79 |
| 6 | xylo | 25 | Hg lamp | >97 | 77 |
| 7 | arabino | 25 | Hg lamp | >97 | 71 |
| 8 | malto | 25 | Hg lamp | >97 | 68 |
| 9 | lacto | 25 | Hg lamp | >97 | 64 |
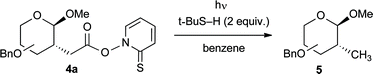
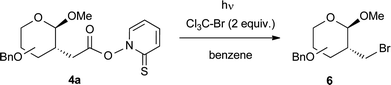
To introduce functional groups by the Barton reaction, the decarboxylative halogenation is an attractive choice, since long radical chains and mild conditions are advantageous.4 Again, we employed a 250 W low-pressure mercury lamp for initiation, this time in the presence of bromotrichloromethane as a cheap bromine source (Table 3). Indeed, the 2-C-bromomethyl glycosides 6 were isolated with moderate to good yields with gluco-, galacto-, xylo-, arabino-, malto- and lacto-configurations. Although similar compounds are available by cyclopropane-opening,13a,14 our method is applicable for various saccharides and provides sole diastereomers and no anomeric mixtures. Furthermore, the bromides 6 might serve as precursors for SN2 or radical reactions in carbohydrate chemistry.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Config. | Temp. (°C) | Light Source | Conv. (%) | 6 (%)b |
| a For procedure see experimental section. b Yield of analytically pure products, isolated by column chromatography. | |||||
| 1 | gluco | 25 | Hg lamp | >97 | 76 |
| 2 | galacto | 25 | Hg lamp | >97 | 74 |
| 3 | xylo | 25 | Hg lamp | >97 | 71 |
| 4 | arabino | 25 | Hg lamp | >97 | 73 |
| 5 | malto | 25 | Hg lamp | >97 | 70 |
| 6 | lacto | 25 | Hg lamp | >97 | 69 |
Conclusions
In conclusion, we have synthesized Barton esters of 2-C-branched carbohydrates for the first time. The method is applicable for hexoses, pentoses and disaccharides and affords analytically pure products with high yields. The Barton esters are suitable precursors for radical reductions and brominations. Thus, 2-C-methyl and 2-C-bromomethyl saccharides are easily available. Our studies demonstrate the power of radical reactions in carbohydrate chemistry, since products were obtained by a sequence of oxidative malonate additions and reductive decarboxylations. The bromide groups of the 2-C-branched saccharides are suitable precursors for further transformations, which are currently under investigation in our lab.Experimental section
General methods
All reactions requiring anhydrous conditions were performed under a positive pressure of argon using oven-dried glassware (110 °C), which was cooled under argon. Solvents for anhydrous reactions were dried according to Perrinet al.15Benzene and dichloromethane were distilled from calcium hydride and stored over molecular sieves. All commercial reagents were obtained from Sigma-Aldrich, Acros or Fluka Chemical Co. Progress of the reactions was monitored by tlc. Column chromatographies were performed on silica gel 60–120/100–200/230–400 mesh obtained from ACROS Organics Belgium. Typical syringe and cannula techniques were used to transfer air- and moisture-sensitive reagents. IR spectra were recorded on a Perkin–Elmer infrared spectrometer model 599-B and model 1620 FT-IR. NMR spectra were recorded either on a Bruker Avance 300 or Avance 500 or Avance 600 instrument using deuterated chloroform solvent. Elemental analyses were performed on a Vario EL III analyzer (Elementar Analysensysteme GmbH, Hanau, Germany). Optical rotations were measured on a JASCO P-1020 digital polarimeter at 589 nm, melting points on an Electrothermal MEL-TEMP apparatus (uncorrected).General procedure for the synthesis of Barton esters 4a
The sugar carboxylic acid 1a (2.0 mmol) was dissolved in 30 mL of dry dichloromethane and was protected from light with an aluminium foil at 0 °C. 2-Mercaptopyridine N-oxide (390 mg, 2.5 mmol), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide (EDCI) (620 mg, 4.0 mmol) and a catalytic amount of 4-(dimethylamino)pyridine (DMAP) (20 mg) were added and the mixture was stirred at 0 °C for 1–2 h until tlc showed complete conversion of the starting material. Then a saturated solution of sodium bicarbonate was added, and the mixture was extracted with dichloromethane (3 × 10 mL). The combined organic extracts were dried using sodium sulfate, filtered and concentrated under reduced pressure at 30 °C. The desired product was isolated by flash chromatography in analytically pure form.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); [α]20D = +25.6 (c = 1.02 in CHCl3); IR (film): v = 2925, 1806, 1607, 1525, 1446, 1410, 1362, 1281, 1207, 1050 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.26 (ddt, J = 11.0, 8.7, 6.0 Hz, 1H, 2-H), 2.77 (d, J = 6.0 Hz, 2H, 7-H), 3.40 (dt, J = 9.3, 5.2 Hz, 1H, 5-H), 3.42 (s, 3H, OMe), 3.50 (dd, J = 11.0, 8.8, Hz, 1H, 3-H), 3.64 (dd, J = 9.3, 8.8 Hz, 1H, 4-H), 3.68 (d, J = 5.2 Hz, 2H, 6-H), 4.25 (d, J = 8.7 Hz, 1H, 1-H), 4.47 (d, J = 11.9 Hz, 1H, CH2Ph), 4.52 (d, J = 10.9 Hz, 1H, CH2Ph), 4.57 (d, J = 11.9 Hz, 1H, CH2Ph), 4.58 (d, J = 11.9 Hz, 1H CH2Ph), 4.71 (d, J = 11.0, Hz, 1H, CH2Ph), 4.91 (d, J = 11.0 Hz, 1H, CH2Ph), 6.24 (dt, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 6.96 (dt, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 7.00 (dd, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 7.06-7.28 (m, 15H, arom. H), 7.51 (dd, J = 7.0, 1.5 Hz, 1H thiopyr. C-H); 13C NMR (125 MHz, CDCl3): δ = 30.2 (t, C-7), 44.7 (d, C-2), 56.8 (q, OMe), 68.4 (t, C-6), 73.3, 74.5, 74.7 (3t, CH2Ph), 74.9, 79.5, 81.8 (3d, C-3, C-4, C-5), 102.9 (d, C-1), 112.1 (d, thiopyr. N-C-H), 127.5, 127.6, 127.7, 127.9, 128.2, 128.3 (15d, arom. C-H), 133.4, 136.8, 137.5 (3d, thiopyr. C-H), 137.6, 137.7, 137.8 (3 s, arom. C-CH2O), 167.2 (s, COOR), 175.5 (NC
:1); [α]20D = +25.6 (c = 1.02 in CHCl3); IR (film): v = 2925, 1806, 1607, 1525, 1446, 1410, 1362, 1281, 1207, 1050 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.26 (ddt, J = 11.0, 8.7, 6.0 Hz, 1H, 2-H), 2.77 (d, J = 6.0 Hz, 2H, 7-H), 3.40 (dt, J = 9.3, 5.2 Hz, 1H, 5-H), 3.42 (s, 3H, OMe), 3.50 (dd, J = 11.0, 8.8, Hz, 1H, 3-H), 3.64 (dd, J = 9.3, 8.8 Hz, 1H, 4-H), 3.68 (d, J = 5.2 Hz, 2H, 6-H), 4.25 (d, J = 8.7 Hz, 1H, 1-H), 4.47 (d, J = 11.9 Hz, 1H, CH2Ph), 4.52 (d, J = 10.9 Hz, 1H, CH2Ph), 4.57 (d, J = 11.9 Hz, 1H, CH2Ph), 4.58 (d, J = 11.9 Hz, 1H CH2Ph), 4.71 (d, J = 11.0, Hz, 1H, CH2Ph), 4.91 (d, J = 11.0 Hz, 1H, CH2Ph), 6.24 (dt, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 6.96 (dt, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 7.00 (dd, J = 7.0, 1.5 Hz 1H thiopyr. C-H), 7.06-7.28 (m, 15H, arom. H), 7.51 (dd, J = 7.0, 1.5 Hz, 1H thiopyr. C-H); 13C NMR (125 MHz, CDCl3): δ = 30.2 (t, C-7), 44.7 (d, C-2), 56.8 (q, OMe), 68.4 (t, C-6), 73.3, 74.5, 74.7 (3t, CH2Ph), 74.9, 79.5, 81.8 (3d, C-3, C-4, C-5), 102.9 (d, C-1), 112.1 (d, thiopyr. N-C-H), 127.5, 127.6, 127.7, 127.9, 128.2, 128.3 (15d, arom. C-H), 133.4, 136.8, 137.5 (3d, thiopyr. C-H), 137.6, 137.7, 137.8 (3 s, arom. C-CH2O), 167.2 (s, COOR), 175.5 (NC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S); Elemental analysis(%) calcd for C35H37NO7S: C 68.27, H 6.06, N 2.27, S 5.21; found: C 68.29, H 6.09, N 2.23, S 5.26; Mass (ESI-MS); m/z 638.41(M + Na)+.
:1); [α]20D = +17.9 (c = 1.02 in CHCl3); IR (film): v = 2924, 1807, 1607, 1524, 1444, 1909, 1363, 1281, 1207, 1051 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.72 (ddt, J = 10.8, 8.5, 6.0 Hz, 1H, 2-H), 2.76 (dd, J = 14.6, 6.0 Hz, 1H, 7-H), 2.85 (dd, J = 14.6, 6.0 1H 7′-H), 3.40 (s, 3H, OMe), 3.46 (dd, J = 10.8, 2.2, Hz, 1H, 3-H), 3.53 (dd, J = 6.9, 5.8 Hz, 1H, 6-H), 3.56 (ddd, J = 8.8, 5.8, 0 Hz, 1H, 5-H), 3.63 (dd, J = 8.8, 6.9 Hz, 1H, 6′-H), 3.93 (d, J = 2.2 Hz, 1H, 4-H), 4.26 (d, J = 8.5 Hz, 1H 1-H), 4.35 (d, J = 11.1 Hz, 1H, CH2Ph), 4.39 (d, J = 11.7 Hz, 1H, CH2Ph), 4.43(d, J = 11.6 Hz, 1H, CH2Ph), 4.55 (d, J = 11.7 Hz, 1H, CH2Ph), 4.65 (d, J = 11.1 Hz, 1H, CH2Ph), 4.81 (d, J = 11.6 Hz, 1H, CH2Ph), 6.28 (dt, J = 6.8, 1.6 Hz 1H thiopyr. C-H), 7.01 (dd, J = 6.8, 1.5 Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.8, 1.5 Hz 1H thiopyr. C-H), 7.15–7.21 (m, 15H, arom. H), 7.55 (dd, J = 6.8, 1.6 Hz, 1H thiopyr. C-H);13C NMR (150 MHz, CDCl3): δ = 30.30 (t, C-7), 40.1 (d, C-2), 56.7 (q, OMe), 68.7 (t, C-6), 70.6, 73.5, 80.6 (3d, C-3, C-4, C-5), 71.5, 73.6, 74.4 (3t, CH2Ph), 103.5 (C-1), 112.1 (d, thiopyr. N-C-H), 127.5, 127.8, 127.9, 128.0, 128.1, 128.2 (15d, arom. C-H), 133.3, 137.1, 137.7 (3d, thiopyr. C-H), 137.2, 137.8, 138.5 (3 s, arom. C-CH2O), 167.5 (s, COOR), 175.9 (NCS); Elemental analysis(%) calcd for C35H37NO7S: C 68.27, H 6.06, N 2.27, S 5.21; found: C 68.23, H 6.03, N 2.29, S 5.29; Mass (ESI-MS); m/z 638.44(M + Na)+.
:1); [α]20D = +32.7 (c = 1.01 in CHCl3); IR (film): v = 3063, 3030, 2924, 1812, 1722, 1607, 1496, 1465, 1374, 1205, 1157, 1069, 1028, 995 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.18 (dddd, J = 10.8, 8.3, 6.8, 5.3 Hz 1H, 2-H), 2.75 (dd, J = 15.5, 6.8 Hz 1H, 6-H), 2.85 (dd, J = 15.5, 5.3 Hz, 1H, 6′-H), 3.21 (dd, J = 11.7, 9.4 Hz, 1H 5-H), 3.40 (s, 3H, OMe), 3.47 (dd, J = 10.8, 8.2, Hz, 1H, 3-H), 3.62 (ddd, J = 9.4, 8.2, 5.0 Hz, 1H, 4-H), 3.98 (dd, J = 11.7, 5.0 Hz, 1H, 5′-H), 4.23 (d, J = 8.3 Hz, 1H, 1-H), 4.56 (d, J = 10.9 Hz, 1H, CH2Ph) 4.57 (d, J = 11.7 Hz, 1H, CH2Ph), 4.61 (d, J = 11.7 Hz, 1H, CH2Ph), 4.96 (d, J = 10.9 Hz, 1H, CH2Ph), 6.24 (dt, J = 6.8, 1.8 Hz 1H thiopyr. C-H), 6.90 (dd, J = 6.8, 1.8, Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.8, 1.8 Hz 1H thiopyr. C-H), 7.21–7.28 (m, 10H, arom. H), 7.55 (dd, J = 6.8, 1.8 Hz, 1H thiopyr. C-H); 13C NMR (150 MHz, CDCl3): δ = 30.8 (t, C-6), 43.8 (d, C-2), 56.8 (q, OMe), 63.5 (t, C-5), 72.7, 74.6, (2t, CH2Ph), 79.5, 80.5 (2d, C-3, C-4), 103.4 (d, C-1), 112.0 (d, thiopyr. N-C-H), 127.8, 127.8, 127.9, 128.3, 128.5, 128.5 (10d, arom. C-H), 133.4, 137.1, 137.7 (3d, thiopyr. C-H), 137.8, 138.1, (2 s, arom. C-CH2O), 167.4 (s, COOR), 175.8 (NCS); Elemental analysis(%) calcd for C27H29NO6S: C 65.44, H 5.90, N 2.83, S 6.47; found C 65.39, H 5.93, N 2.86, S 6.43; Mass (ESI-MS); m/z 518.24(M + Na)+.
:1); [α]20D = +19.7 (c = 1.01 in CHCl3); IR (film): v = 3067, 3034, 2922, 1816, 1723, 1602, 1496, 1468, 1374, 1204, 1152, 1064, 1028, 995 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.70 (dddd, J = 11.0, 8.5, 6.6, 5.6 Hz 1H, 2-H), 2.83 (dd, J = 14.5, 5.6 Hz 1H, 6-H), 2.83 (dd, J = 14.5, 6.6 Hz, 1H, 6′-H), 3.29 (dd, J = 12.9, 3.6, Hz, 1H 5-H), 3.43 (s, 3H, OMe), 3.50 (dd, J = 11.0, 2.8 Hz, 1H, 3-H), 3.68 (ddd, J = 3.6, 2.8, 2.1 Hz, 1H, 4-H), 4.13 (dd, J = 12.9, 2.1 Hz, 1H, 5′-H), 4.24 (d, J = 8.5 Hz, 1H, 1-H), 4.26 (d, J = 11.3 Hz, 1H, CH2Ph), 4.49 (d, J = 11.3 Hz, 1H, CH2Ph), 4.58 (d, J = 12.3 Hz, 1H, CH2Ph), 4.72 (d, J = 12.3 Hz, 1H, CH2Ph), 6.32 (dt, J = 6.9, 1.8 Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.9, 1.8, Hz 1H thiopyr. C-H), 7.05 (dt, J = 6.9, 1.8 Hz 1H thiopyr. C-H), 7.19–7.27 (m, 10H, arom. H), 7.32 (dd, J = 6.9, 1.8 Hz, 1H thiopyr. C-H); 13C NMR (150 MHz, CDCl3): δ = 30.3 (t, C-6), 40.3 (d, C-2), 56.8 (q, OMe), 63.5 (t, C-5), 69.3, 70.8 (2t, CH2Ph), 71.0, 78.7 (2d, C-3, C-4), 103.7 (d, C-1), 112.0 (d, thiopyr. N-C-H), 127.7, 127.9, 128.3, 128.8, 128.5, 133.5 (10d, arom. C-H), 137.0, 137.4, 137.7 (3d, thiopyr. C-H), 137.7, 137.8 (2 s, arom. C-CH2O), 167.6 (s, COOR), 176.2 (NCS); Elemental analysis(%) calcd for C27H29NO6S: C 65.44, H 5.90, N 2.83, S 6.47; found C 65.37, H 5.99, N 2.82, S 6.44; Mass (ESI-MS); m/z 518.24(M + Na)+.
:1); [α]20D = +26.2 (c = 1.02 in CHCl3); IR (film): v = 3032, 2941, 1832, 1751, 1648, 1497, 1480, 1215, 1155 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.42 (ddt, J = 10.9, 8.6, 6.0 Hz 1H, 2-H), 2.71 (dd, J = 15.9, 6.0 Hz 1H, 13-H), 2.85 (dd, J = 15.9, 6.0 Hz, 1H, 13′-H), 3.42 (dd, J = 9.5, 3.9 1H 12-H), 3.42 (ddd, J = 9.5, 3.9, 3.0, 1H, 11-H), 3.43 (s, 3H, OMe), 3.44 (dd, J = 7.9, 4.3 Hz, 1H, 6-H), 3.50 (dd J = 7.9, 2.8 Hz, 1H 6′-H), 3.56 (dd, J = 9.5, 3.0 Hz, 1H, 12′-H), 3.63 (dd, J = 10.9, 8.1 Hz, 1H, 3-H), 3.70 (dd, J = 11.3, 10.8 Hz, 1H, 9-H), 3.78 (dd, J = 9.0, 4.3, 2.8 Hz, 1H, 5-H), 3.83 (dd, J = 10.8, 9.5 Hz, 1H, 10-H), 3.88 (d, J = 11.3, 3.5 Hz, 1H, 8-H), 4.06 (t, J = 9.0, 8.0 Hz, 1H, 4-H), 4.2 (d, J = 8.6 Hz, 1H, 1-H), 4.29 (d, J = 12 Hz, 1H, CH2Ph), 4.40 (d, J = 11.0 Hz, 1H, CH2Ph), 4.46 (d, J = 12.0 Hz, 1H, CH2Ph), 4.47 (d, J = 11.5 Hz, 1H, CH2Ph), 4.48 (d, J = 11.0 Hz, 1H, CH2Ph), 4.49 (d, J = 11.2 Hz 1H CH2Ph), 4.50 (d, J = 11.2 Hz, 1H, CH2Ph), 4.52 (d, J = 11.5, Hz, 1H, CH2Ph), 4.66 (d, J = 11.0 Hz, 1H, CH2Ph), 4.73 (d, J = 10.3 Hz, 1H, CH2Ph), 4.76 (d, J = 10.3 Hz, 1H, CH2Ph), 4.97 (d, J = 11.0 Hz, 1H, CH2Ph), 5.33 (d, J = 3.5 1H, 7-H), 6.27 (dt, J = 7.0, 1.6 Hz 1H thiopyr. C-H), 6.90 (dd, J = 7.0, 1.6, Hz 1H thiopyr. C-H), 7.01 (dt, J = 7.0, 1.6 Hz 1H thiopyr. C-H), 7.03–7.29 (m, 30H, arom. H), 7.53 (dd, J = 7.0, 1.6 Hz, 1H thiopyr. C-H);13C NMR (125 MHz, CDCl3): δ = 31.5 (t, C-13), 44.2 (d, C-2), 57.7 (q, OMe), 69.3, 70.23 (2t, C-6, C-12), 72.2, 72.9, 75.9, 76.3, 78.7, 80.7 (6t, CH2Ph), 72.9, 74.2, 74.2, 74.3, 74.3, 76.0, 76.4 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 98.0, 104.0 (2d, C-1, C-7), 113.1 (d, thiopyr. N-C-H), 128.4, 128.5, 128.5, 128.6, 128.7, 128.7, 128.8, 129.2, 129.9, 129.4 (30d arom. C-H), 138.0, 138.3, 138.6 (3d, thiopyr. C-H), 138.8, 138.9, 139.3, 139.3, 139.3, 139.6 (6 s, arom. C-CH2O), 168.4 (s, COOR), 176.8 (NCS).
:1); [α]20D = +13.2 (c = 1.01 in CHCl3); IR (film): v = 3035, 2941, 1836, 1752, 1647, 1497, 1480, 1335, 1210, 1151 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.24 (dddd, J = 11.2, 8.5, 6.3, 5.4 Hz, 1H, 2-H), 2.80 (dd, J = 15.3, 6.3 Hz, 1H, 13-H), 2.87 (dd, J = 15.5, 5.4 Hz, 1H, 13′-H), 3.1 (m, 4H, 5-H, 11-H), 3.42 (s, 3H, OMe), 3.44 (dd, J = 11.0, 8.8 Hz, 2H, 12-H), 3.65 (dd, J = 9.5, 7.9, Hz, 2H, 6-H), 3.81 (dd, J = 11.2, 10.1 Hz, 1H, 3-H), 3.81 (dd, J = 9.0, 3.8 Hz, 1H, 9-H), 4.00 (t, J = 9.0, 9.0 Hz, 1H, 8-H), 4.18 (d, J = 11.5 Hz, 1H, CH2Ph), 4.27 (d, J = 12.2 Hz, 1H, CH2Ph), 4.27 (d, J = 12.2 Hz, CH2Ph), 4.28 (d, J = 8.5 Hz, 1H, 1-H), 4.33 (d, J = 12.2 Hz, 1H, CH2Ph), 4.37 (d, J = 9.0 Hz, 1H, 7-H), 4.43 (d, J = 11.5 Hz, 1H, CH2Ph), 4.44 (d, J = 12.0 Hz, 1H, CH2Ph), 4.53 (d, J = 12.0 Hz, 1H, CH2Ph), 4.62 (d, J = 10.1, 2.2 Hz, 1H, 4-H), 4.62 (dd, J = 3.8, 2.5 Hz 1H 10-H), 4.87 (d, J = 12.0 Hz, 1H, CH2Ph), 5.14 (d, J = 10.2 Hz, 1H, CH2Ph), 6.20 (dt, J = 7.0, 1.8 Hz 1H thiopyr. C-H), 6.87 (dd, J = 7.0, 1.8, Hz, 1H thiopyr. C-H), 7.01 (dt, J = 7.0, 1.8 Hz 1H thiopyr. C-H), 7.01–7.29 (m, 30H, arom. H), 7.52 (dd, J = 7.0, 1.8 Hz, 1H thiopyr. C-H);13C NMR (125 MHz, CDCl3): δ = 30.6 (t, C-13), 44.4 (d, C-2), 56.8 (q, OMe), 68.0, 68.1 (2t, C-6, C-12), 72.60, 73.0, 73.6, 74.3, 74.5, 75.2 (6t, CH2Ph), 73.0, 73.3, 75.4, 76.8, 80.0, 80.3, 82.3 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 102.6, 103.0 (2d, C-1, C-7), 112.0 (d, thiopyr. N-C-H), 127.2, 127.3, 127.3, 127.4, 127.5, 127-5, 127.6, 127.8, 128.0, 128.1, 128.2, 128.3 (30d, arom. C-H), 137.0, 137.3, 137.8 (3d, thiopyr. C-H), 138.0, 138.3, 138.5, 138.6, 138.7, 139.0 (6 s, arom. C-CH2O), 167.4 (s, COOR), 175.8 (NCS).
S); Elemental analysis(%) calcd for C35H37NO7S: C 68.27, H 6.06, N 2.27, S 5.21; found: C 68.29, H 6.09, N 2.23, S 5.26; Mass (ESI-MS); m/z 638.41(M + Na)+.
:1); [α]20D = +17.9 (c = 1.02 in CHCl3); IR (film): v = 2924, 1807, 1607, 1524, 1444, 1909, 1363, 1281, 1207, 1051 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.72 (ddt, J = 10.8, 8.5, 6.0 Hz, 1H, 2-H), 2.76 (dd, J = 14.6, 6.0 Hz, 1H, 7-H), 2.85 (dd, J = 14.6, 6.0 1H 7′-H), 3.40 (s, 3H, OMe), 3.46 (dd, J = 10.8, 2.2, Hz, 1H, 3-H), 3.53 (dd, J = 6.9, 5.8 Hz, 1H, 6-H), 3.56 (ddd, J = 8.8, 5.8, 0 Hz, 1H, 5-H), 3.63 (dd, J = 8.8, 6.9 Hz, 1H, 6′-H), 3.93 (d, J = 2.2 Hz, 1H, 4-H), 4.26 (d, J = 8.5 Hz, 1H 1-H), 4.35 (d, J = 11.1 Hz, 1H, CH2Ph), 4.39 (d, J = 11.7 Hz, 1H, CH2Ph), 4.43(d, J = 11.6 Hz, 1H, CH2Ph), 4.55 (d, J = 11.7 Hz, 1H, CH2Ph), 4.65 (d, J = 11.1 Hz, 1H, CH2Ph), 4.81 (d, J = 11.6 Hz, 1H, CH2Ph), 6.28 (dt, J = 6.8, 1.6 Hz 1H thiopyr. C-H), 7.01 (dd, J = 6.8, 1.5 Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.8, 1.5 Hz 1H thiopyr. C-H), 7.15–7.21 (m, 15H, arom. H), 7.55 (dd, J = 6.8, 1.6 Hz, 1H thiopyr. C-H);13C NMR (150 MHz, CDCl3): δ = 30.30 (t, C-7), 40.1 (d, C-2), 56.7 (q, OMe), 68.7 (t, C-6), 70.6, 73.5, 80.6 (3d, C-3, C-4, C-5), 71.5, 73.6, 74.4 (3t, CH2Ph), 103.5 (C-1), 112.1 (d, thiopyr. N-C-H), 127.5, 127.8, 127.9, 128.0, 128.1, 128.2 (15d, arom. C-H), 133.3, 137.1, 137.7 (3d, thiopyr. C-H), 137.2, 137.8, 138.5 (3 s, arom. C-CH2O), 167.5 (s, COOR), 175.9 (NCS); Elemental analysis(%) calcd for C35H37NO7S: C 68.27, H 6.06, N 2.27, S 5.21; found: C 68.23, H 6.03, N 2.29, S 5.29; Mass (ESI-MS); m/z 638.44(M + Na)+.
:1); [α]20D = +32.7 (c = 1.01 in CHCl3); IR (film): v = 3063, 3030, 2924, 1812, 1722, 1607, 1496, 1465, 1374, 1205, 1157, 1069, 1028, 995 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.18 (dddd, J = 10.8, 8.3, 6.8, 5.3 Hz 1H, 2-H), 2.75 (dd, J = 15.5, 6.8 Hz 1H, 6-H), 2.85 (dd, J = 15.5, 5.3 Hz, 1H, 6′-H), 3.21 (dd, J = 11.7, 9.4 Hz, 1H 5-H), 3.40 (s, 3H, OMe), 3.47 (dd, J = 10.8, 8.2, Hz, 1H, 3-H), 3.62 (ddd, J = 9.4, 8.2, 5.0 Hz, 1H, 4-H), 3.98 (dd, J = 11.7, 5.0 Hz, 1H, 5′-H), 4.23 (d, J = 8.3 Hz, 1H, 1-H), 4.56 (d, J = 10.9 Hz, 1H, CH2Ph) 4.57 (d, J = 11.7 Hz, 1H, CH2Ph), 4.61 (d, J = 11.7 Hz, 1H, CH2Ph), 4.96 (d, J = 10.9 Hz, 1H, CH2Ph), 6.24 (dt, J = 6.8, 1.8 Hz 1H thiopyr. C-H), 6.90 (dd, J = 6.8, 1.8, Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.8, 1.8 Hz 1H thiopyr. C-H), 7.21–7.28 (m, 10H, arom. H), 7.55 (dd, J = 6.8, 1.8 Hz, 1H thiopyr. C-H); 13C NMR (150 MHz, CDCl3): δ = 30.8 (t, C-6), 43.8 (d, C-2), 56.8 (q, OMe), 63.5 (t, C-5), 72.7, 74.6, (2t, CH2Ph), 79.5, 80.5 (2d, C-3, C-4), 103.4 (d, C-1), 112.0 (d, thiopyr. N-C-H), 127.8, 127.8, 127.9, 128.3, 128.5, 128.5 (10d, arom. C-H), 133.4, 137.1, 137.7 (3d, thiopyr. C-H), 137.8, 138.1, (2 s, arom. C-CH2O), 167.4 (s, COOR), 175.8 (NCS); Elemental analysis(%) calcd for C27H29NO6S: C 65.44, H 5.90, N 2.83, S 6.47; found C 65.39, H 5.93, N 2.86, S 6.43; Mass (ESI-MS); m/z 518.24(M + Na)+.
:1); [α]20D = +19.7 (c = 1.01 in CHCl3); IR (film): v = 3067, 3034, 2922, 1816, 1723, 1602, 1496, 1468, 1374, 1204, 1152, 1064, 1028, 995 cm−1; 1H NMR (600 MHz, CDCl3): δ = 2.70 (dddd, J = 11.0, 8.5, 6.6, 5.6 Hz 1H, 2-H), 2.83 (dd, J = 14.5, 5.6 Hz 1H, 6-H), 2.83 (dd, J = 14.5, 6.6 Hz, 1H, 6′-H), 3.29 (dd, J = 12.9, 3.6, Hz, 1H 5-H), 3.43 (s, 3H, OMe), 3.50 (dd, J = 11.0, 2.8 Hz, 1H, 3-H), 3.68 (ddd, J = 3.6, 2.8, 2.1 Hz, 1H, 4-H), 4.13 (dd, J = 12.9, 2.1 Hz, 1H, 5′-H), 4.24 (d, J = 8.5 Hz, 1H, 1-H), 4.26 (d, J = 11.3 Hz, 1H, CH2Ph), 4.49 (d, J = 11.3 Hz, 1H, CH2Ph), 4.58 (d, J = 12.3 Hz, 1H, CH2Ph), 4.72 (d, J = 12.3 Hz, 1H, CH2Ph), 6.32 (dt, J = 6.9, 1.8 Hz 1H thiopyr. C-H), 7.03 (dt, J = 6.9, 1.8, Hz 1H thiopyr. C-H), 7.05 (dt, J = 6.9, 1.8 Hz 1H thiopyr. C-H), 7.19–7.27 (m, 10H, arom. H), 7.32 (dd, J = 6.9, 1.8 Hz, 1H thiopyr. C-H); 13C NMR (150 MHz, CDCl3): δ = 30.3 (t, C-6), 40.3 (d, C-2), 56.8 (q, OMe), 63.5 (t, C-5), 69.3, 70.8 (2t, CH2Ph), 71.0, 78.7 (2d, C-3, C-4), 103.7 (d, C-1), 112.0 (d, thiopyr. N-C-H), 127.7, 127.9, 128.3, 128.8, 128.5, 133.5 (10d, arom. C-H), 137.0, 137.4, 137.7 (3d, thiopyr. C-H), 137.7, 137.8 (2 s, arom. C-CH2O), 167.6 (s, COOR), 176.2 (NCS); Elemental analysis(%) calcd for C27H29NO6S: C 65.44, H 5.90, N 2.83, S 6.47; found C 65.37, H 5.99, N 2.82, S 6.44; Mass (ESI-MS); m/z 518.24(M + Na)+.
:1); [α]20D = +26.2 (c = 1.02 in CHCl3); IR (film): v = 3032, 2941, 1832, 1751, 1648, 1497, 1480, 1215, 1155 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.42 (ddt, J = 10.9, 8.6, 6.0 Hz 1H, 2-H), 2.71 (dd, J = 15.9, 6.0 Hz 1H, 13-H), 2.85 (dd, J = 15.9, 6.0 Hz, 1H, 13′-H), 3.42 (dd, J = 9.5, 3.9 1H 12-H), 3.42 (ddd, J = 9.5, 3.9, 3.0, 1H, 11-H), 3.43 (s, 3H, OMe), 3.44 (dd, J = 7.9, 4.3 Hz, 1H, 6-H), 3.50 (dd J = 7.9, 2.8 Hz, 1H 6′-H), 3.56 (dd, J = 9.5, 3.0 Hz, 1H, 12′-H), 3.63 (dd, J = 10.9, 8.1 Hz, 1H, 3-H), 3.70 (dd, J = 11.3, 10.8 Hz, 1H, 9-H), 3.78 (dd, J = 9.0, 4.3, 2.8 Hz, 1H, 5-H), 3.83 (dd, J = 10.8, 9.5 Hz, 1H, 10-H), 3.88 (d, J = 11.3, 3.5 Hz, 1H, 8-H), 4.06 (t, J = 9.0, 8.0 Hz, 1H, 4-H), 4.2 (d, J = 8.6 Hz, 1H, 1-H), 4.29 (d, J = 12 Hz, 1H, CH2Ph), 4.40 (d, J = 11.0 Hz, 1H, CH2Ph), 4.46 (d, J = 12.0 Hz, 1H, CH2Ph), 4.47 (d, J = 11.5 Hz, 1H, CH2Ph), 4.48 (d, J = 11.0 Hz, 1H, CH2Ph), 4.49 (d, J = 11.2 Hz 1H CH2Ph), 4.50 (d, J = 11.2 Hz, 1H, CH2Ph), 4.52 (d, J = 11.5, Hz, 1H, CH2Ph), 4.66 (d, J = 11.0 Hz, 1H, CH2Ph), 4.73 (d, J = 10.3 Hz, 1H, CH2Ph), 4.76 (d, J = 10.3 Hz, 1H, CH2Ph), 4.97 (d, J = 11.0 Hz, 1H, CH2Ph), 5.33 (d, J = 3.5 1H, 7-H), 6.27 (dt, J = 7.0, 1.6 Hz 1H thiopyr. C-H), 6.90 (dd, J = 7.0, 1.6, Hz 1H thiopyr. C-H), 7.01 (dt, J = 7.0, 1.6 Hz 1H thiopyr. C-H), 7.03–7.29 (m, 30H, arom. H), 7.53 (dd, J = 7.0, 1.6 Hz, 1H thiopyr. C-H);13C NMR (125 MHz, CDCl3): δ = 31.5 (t, C-13), 44.2 (d, C-2), 57.7 (q, OMe), 69.3, 70.23 (2t, C-6, C-12), 72.2, 72.9, 75.9, 76.3, 78.7, 80.7 (6t, CH2Ph), 72.9, 74.2, 74.2, 74.3, 74.3, 76.0, 76.4 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 98.0, 104.0 (2d, C-1, C-7), 113.1 (d, thiopyr. N-C-H), 128.4, 128.5, 128.5, 128.6, 128.7, 128.7, 128.8, 129.2, 129.9, 129.4 (30d arom. C-H), 138.0, 138.3, 138.6 (3d, thiopyr. C-H), 138.8, 138.9, 139.3, 139.3, 139.3, 139.6 (6 s, arom. C-CH2O), 168.4 (s, COOR), 176.8 (NCS).
:1); [α]20D = +13.2 (c = 1.01 in CHCl3); IR (film): v = 3035, 2941, 1836, 1752, 1647, 1497, 1480, 1335, 1210, 1151 cm−1; 1H NMR (500 MHz, CDCl3): δ = 2.24 (dddd, J = 11.2, 8.5, 6.3, 5.4 Hz, 1H, 2-H), 2.80 (dd, J = 15.3, 6.3 Hz, 1H, 13-H), 2.87 (dd, J = 15.5, 5.4 Hz, 1H, 13′-H), 3.1 (m, 4H, 5-H, 11-H), 3.42 (s, 3H, OMe), 3.44 (dd, J = 11.0, 8.8 Hz, 2H, 12-H), 3.65 (dd, J = 9.5, 7.9, Hz, 2H, 6-H), 3.81 (dd, J = 11.2, 10.1 Hz, 1H, 3-H), 3.81 (dd, J = 9.0, 3.8 Hz, 1H, 9-H), 4.00 (t, J = 9.0, 9.0 Hz, 1H, 8-H), 4.18 (d, J = 11.5 Hz, 1H, CH2Ph), 4.27 (d, J = 12.2 Hz, 1H, CH2Ph), 4.27 (d, J = 12.2 Hz, CH2Ph), 4.28 (d, J = 8.5 Hz, 1H, 1-H), 4.33 (d, J = 12.2 Hz, 1H, CH2Ph), 4.37 (d, J = 9.0 Hz, 1H, 7-H), 4.43 (d, J = 11.5 Hz, 1H, CH2Ph), 4.44 (d, J = 12.0 Hz, 1H, CH2Ph), 4.53 (d, J = 12.0 Hz, 1H, CH2Ph), 4.62 (d, J = 10.1, 2.2 Hz, 1H, 4-H), 4.62 (dd, J = 3.8, 2.5 Hz 1H 10-H), 4.87 (d, J = 12.0 Hz, 1H, CH2Ph), 5.14 (d, J = 10.2 Hz, 1H, CH2Ph), 6.20 (dt, J = 7.0, 1.8 Hz 1H thiopyr. C-H), 6.87 (dd, J = 7.0, 1.8, Hz, 1H thiopyr. C-H), 7.01 (dt, J = 7.0, 1.8 Hz 1H thiopyr. C-H), 7.01–7.29 (m, 30H, arom. H), 7.52 (dd, J = 7.0, 1.8 Hz, 1H thiopyr. C-H);13C NMR (125 MHz, CDCl3): δ = 30.6 (t, C-13), 44.4 (d, C-2), 56.8 (q, OMe), 68.0, 68.1 (2t, C-6, C-12), 72.60, 73.0, 73.6, 74.3, 74.5, 75.2 (6t, CH2Ph), 73.0, 73.3, 75.4, 76.8, 80.0, 80.3, 82.3 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 102.6, 103.0 (2d, C-1, C-7), 112.0 (d, thiopyr. N-C-H), 127.2, 127.3, 127.3, 127.4, 127.5, 127-5, 127.6, 127.8, 128.0, 128.1, 128.2, 128.3 (30d, arom. C-H), 137.0, 137.3, 137.8 (3d, thiopyr. C-H), 138.0, 138.3, 138.5, 138.6, 138.7, 139.0 (6 s, arom. C-CH2O), 167.4 (s, COOR), 175.8 (NCS).
General procedure for the reduction of Barton esters 4a
The Barton ester 4a (2.0 mmol) was dissolved in 20 mL of dry benzene and was protected from light with aluminium foil under argon atmosphere. tert-Butanethiol (360 mg, 4.0 mmol) was added at 25 °C. The reaction mixture was subsequently exposed to light using a 250 W low-pressure mercury lamp. After completion of the reaction (approximately 1–2 h), the crude reaction mixture was concentrated and the residue was purified by flash chromatography, affording the products 5.:1); [α]20D = +36.4 (c = 1.01 in CHCl3); IR (film): v = 2925, 1496, 1453, 1361, 1214, 1090, 1026, 907 cm−1;1H NMR (300 MHz, CDCl3): δ = 0.97 (d, J = 6.4, Hz, 3H, CH3), 1.70 (ddq, J = 10.6, 8.6, 6.4 Hz, 1H, 2-H), 3.16 (dd, J = 10.6, 8.7 Hz, 1H, 3-H), 3.38 (ddd, J = 9.6, 4.5, 2.4 Hz, 1H, 5-H), 3.43 (s, 3H, OMe), 3.51 (dd, J = 9.6, 8.7, Hz, 1H, 4-H), 3.67 (dd, J = 10.8, 4.5 Hz, 1H, 6-H), 3.68 (dd, J = 10.8, 2.5 Hz, 1H, 6′-H), 3.93 (d, J = 8.6 Hz, 1H, 1-H), 4.49 (d, J = 11.6 Hz, 2H, CH2Ph), 4.57 (d, J = 11.4 Hz, 1H, CH2Ph), 4.72 (d, J = 10.9 Hz, 1H, CH2Ph), 4.80 (d, J = 10.9 Hz, 1H, CH2Ph), 7.10–7.29 (m, 15H, arom. H); 13C NMR (75 MHz, CDCl3): δ = 12.5 (q, CH3), 42.6 (d, C-2), 56.7 (q, OMe), 69.2 (t, C-6), 73.4, 74.7, 75.1 (3t, CH2Ph), 75.2, 79.4, 85.2 (3d, C-3, C-4, C-5), 105.5 (d, C-1), 127.6, 127.7, 127.8, 127.8, 128.3, 128.3 (15d, arom. C-H), 138.1, 138.2. 138.4 (3 s, arom. C-CH2O); Elemental analysis(%); calcd for: C29H34O5 C 75.30, H 7.41,; found: C 75.33, H 7.47; Mass (ESI-MS); m/z 485.28(M + Na)+.
:1); [α]20D = +31.4 (c = 1.02 in CHCl3); IR (film): v = 3030, 2924, 2867, 2358, 1718, 1496, 1454, 1363, 1206, 1153, 1078, 1028 cm−1; 1H NMR (300 MHz, CDCl3): δ = 0.95 (d, J = 6.6 Hz, 3H, CH3), 2.11 (ddq, J = 11.0, 8.6, 6.6 Hz, 1H, 2-H), 3.03 (d, J = 11.0, 2.6 Hz, 1H, 3-H), 3.39 (s, 3H, OMe), 3.44 (dd, J = 7.4, 5.4 Hz, 1H, 5-H), 3.57 (dd, J = 9.2, 5.4 Hz, 1H, 6-H), 3.57 (dd, J = 9.2. 7.4 Hz, 1H, 6′-H), 3.80 (d, J = 2.5 Hz, 1H, 4-H), 3.87 (d, J = 8.6 Hz, 1H, 1-H), 4.35 (d, J = 11.6 Hz, 1H, CH2Ph), 4.37 (d, J = 11.6 Hz, 1H, CH2Ph), 4.42 (d, J = 11.8 Hz, 1H, CH2Ph), 4.53 (d, J = 11.7, 1H, CH2Ph), 4.62 (d, J = 11.7 Hz, 1H, CH2Ph), 4.80 (d, J = 11.8 Hz, 1H, CH2Ph), 7.15–7.28 (m, 15H, arom. H); 13C NMR (75 MHz, CDCl3): δ = 12.3 (q, CH3), 37.3(d, C-2), 56.6 (q, OMe), 69.3 (t, C-6), 71.6, 73.5, 74.1 (3t, CH2Ph), 70.6, 73.6, 83.0 (3d, C-3, C-4, C-5), 106.2 (d, C-1), 127.4, 127.7, 127.8, 127.8, 128.1, 128.3 (15d, arom. C-H), 138.0, 138.0, 138.8 (3 s, arom. C-CH2O); Elemental analysis(%) calcd for: C29H34O5 C 75.30, H 7.41; found: C 75.33, H 7.48; Mass (ESI-MS); m/z 485.28(M + Na)+.
:1); [α]20D = +28.4 (c = 1.02 in CHCl3); IR (film): v = 2917, 2849, 1496, 1454, 1367, 1204, 1175, 1091, 1072, 1028 cm−1; 1H NMR (300 MHz, CDCl3): δ = 0.97 (d, J = 6.6 Hz, 3H, CH3), 1.61 (ddq, J = 11.6, 8.4, 6.6 Hz, 1H, 2-H), 3.12 (ddd, J = 9.7, 8.5, 1.8 Hz, 2H, 5-H), 3.38 (s, 3H, OMe), 3.54 (ddd, J = 9.7, 8.5, 5.1, Hz, 1H, 4-H), 3.89 (d, J = 8.4 Hz, 1H, 1-H), 3.94 (dd, J = 11.5, 5.1 Hz, 1H, 3-H), 4.55 (d, J = 11.5 Hz, 1H, CH2Ph), 4.57 (d, J = 11.0 Hz, 1H, CH2-Ph), 4.62 (d, J = 11.5 Hz, 1H, CH2Ph), 4.85 (d, J = 11.0 Hz, 1H, CH2Ph), 7.20–7.27 (m, 10H, arom. H); 13C NMR (75 MHz, CDCl3): δ = 12.8 (q, CH3), 41.7 (d, C-2), 56.5 (q, OMe), 63.6 (t, C-5), 72.6, 74.8, (2t, CH2Ph), 79.1, 83.4 (3d, C-3, C-4), 106.0 (d, C-1), 127.5, 127.6, 127.8, 128.0, 128.3 (10d, arom. C-H), 138.2, 138.5, (2 s, arom. C-CH2O); Elemental analysis(%) calcd for: C21H26O4 C 73.66, H 7.65; found: C 73.61, H 7.80; Mass (ESI-MS); m/z 365.56(M + Na)+
:1); [α]20D = +13.9 (c = 1.02 in CHCl3); IR (film): v = 2912, 2846, 1494, 1451, 1362, 1204, 1178, 1093, 1072, 1021 cm−1; 1H NMR (300 MHz, CDCl3): δ = 0.95 (d, J = 6.6 Hz, 3H, CH3), 2.11 (ddq, J = 10.6, 8.2, 6.6 Hz, 1H, 2-H), 3.0 (dd, J = 10.6, 3.0 1H, 3-H), 3.18 (d, J = 12.8 1H, 5-H), 3.38 (s, 3H, OMe), 3.53 (dd, J = 3.0, 2.6 Hz, 1H, 4-H), 3.81 (d, J = 8.2 Hz, 1H. 1-H), 4.06 (dd, J = 12.8, 2.6 Hz, 1H, 5′-H), 4.24 (d, J = 11.9 Hz, 1H, CH2Ph), 4.46 (d, J = 11.9 Hz, 1H, CH2-Ph), 4.52 (d, J = 12.6 Hz, 1H, CH2Ph), 4.68 (d, J = 12.6 Hz, 1H, CH2Ph), 7.13–7.30 (m, 10H, arom. H); 13C NMR (75 MHz, CDCl3): δ = 12.5 (q, CH3), 37.4 (d, C-2), 56.4 (q, OMe), 63.0 (t, C-5), 70.7, 70.7, (2t, CH2Ph), 69.6, 80.6 (3d, C-3, C-4), 106.1 (d, C-1), 127.4, 127.5, 127.6, 127.8, 128.2 (10d, arom. C-H), 138.0, 138.3 (2 s, arom. C-CH2O); Elemental analysis(%) calcd for: C21H26O4 C 73.66, H 7.65; found: C 73.56, H 7.78; Mass (ESI-MS); m/z 365.54(M + Na)+.
:1); [α]20D = +36.9 (c = 1.02 in CHCl3); IR (film): v = 3032, 2943, 1833, 1753, 1649, 1495, 1483, 1339, 1211, 1153 cm−1; 1H NMR (500 MHz, CDCl3): δ = 1.00 (d, J = 4.1, Hz, 3H, CH3), 1.68 (ddq, J = 9.8, 8.6, 4.1, Hz, 1H, 2-H), 3.07 (dd, J = 9.8, 8.5 Hz 1H, 3-H), 3.27 (dd, J = 6.8, 4.2 Hz, 1H, 6-H), 3.28 (dd, J = 6.8, 3.8 Hz, 1H, 6′-H), 3.29 (dd, J = 7.6, 5.2 Hz, 1H, 12-H), 3.30 (dd, J = 7.6, 3.0 Hz, 1H, 12′-H), 3.40 (s, 3H, OMe), 3.44 (dd, J = 9.8, 8.6 Hz, 1H, 9-H), 3.67 (ddd J = 8.4, 4.2, 3.8 Hz, 1H, 5-H), 3.69 (ddd, J = 7.8, 5.2, 3.6 Hz 1H, 11-H), 3.74 (dd, J = 8.4, 8.3 Hz, 1H, 4-H), 3.83 (dd J = 9.8, 7.8 Hz, 1H, 10-H), 3.89 (d, J = 2.4 Hz, 1H, 7-H), 3.92 (dd J = 8.6, 2.4 Hz, 1H, 8-H), 4.15 (d, J = 12.0 Hz, 1H, CH2Ph), 4.26 (d, J = 12.8 Hz, 1H CH2Ph), 4.33(d, J = 12.0 Hz, 1H, CH2Ph), 4.36 (d, J = 8.6 Hz, 1H, 1-H), 4.41 (d, J = 10.0 Hz, 1H, CH2Ph), 4.46 (d, J = 11.0 Hz, 1H, CH2Ph), 4.47 (d, J = 12.0 Hz, 1H, CH2Ph), 4.50 (d, J = 12.0 Hz, 1H, CH2Ph), 4.65 (d, J = 10.0 Hz, 1H, CH2Ph), 4.72 (d, J = 11.0 Hz 1H CH2Ph), 4.76 (d, J = 11.0 Hz, 1H, CH2Ph) 4.89 (d, J = 112.0 Hz, 1H, CH2Ph), 5.03,(d, J = 11.0 Hz, 1H, CH2Ph), 7.03–7.25 (m, 30H, arom. H), 13C NMR (125 MHz, CDCl3): δ = 12.6 (q, CH3), 42.1 (d, C-2), 56.5 (q, OMe), 68.2, 68.6 (2t, C-6, C-12), 72.7, 73.1, 73.4, 74.6, 74.7, 75.2, (6t, CH2Ph), 73.0, 74.0, 75.6, 77.3, 80.2, 82.6, 83.2 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 103.8, 106.7 (2d, C-1, C-7), 127.0, 127.2, 127.3, 127.4, 127.5, 127.6, 127.8, 127.9, 128.0, 128.1, 128.3(30d, arom. C-H), 138.2, 138.6, 138.6, 138.9, 139.1, 139.1 (6 s, arom. C-CH2O); Elemental analysis(%) calcd for: C56H62O10 C 75.14, H 6.98; found: C 75.27, H 7.18; Mass (ESI-MS); m/z894.54(M)+.
:1); [α]20D = +21.3 (c = 1.02 in CHCl3); IR (film): v = 3035, 2941, 1836, 1752, 1647, 1497, 1480, 1335, 1210, 1151 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.16 (d, J = 6.4, Hz, 3H, CH3), 1.84 (ddq, J = 10.6, 8.7, 6.4, Hz, 1H, 2-H), 3.23 (dd, J = 10.6, 8.7 Hz, 1H, 3-H), 3.56 (s, 3H, OMe), 3.63 (d, J = 9.8 Hz, 1H, 9-H), 3.41 (dd J = 6.5, 4.6 Hz, 1H, 6-H), 3.43 (dd, J = 7.2, 5.3 Hz, 1H, 12-H), 3.45 (dd, J = 6.5, 3.6 Hz, 1H, 6′-H), 3.48 (dd, J = 7.2, 6.0 Hz, 1H, 12′-H), 3.80 (ddd, J = 8.3, 4.6, 3.6 Hz, 1H, 5-H), 3.84 (ddd, J = 6.0, 5.3, 2.0 Hz, 1H, 11-H), 3.92 (dd, J = 9.8, 2.3 Hz, 1H, 8-H), 3.99 (d, J = 2.0 Hz, 1H, 10-H), 4.04 (dd, J = 8.7, 8.3 Hz, 1H, 4-H), 4.08 (d, J = 8.7, 3.6 Hz, 1H, 1-H), 4.29 (d, J = 11.7 Hz, 1H, CH2Ph), 4.40 (d, J = 11.7 Hz, 1H, CH2Ph), 4.48 (d, J = 11.4 Hz, 1H, CH2Ph), 4.48 (d, J = 10.2 Hz, 1H, CH2Ph), 4.57 (d, J = 10.0 Hz, 1H, CH2Ph), 4.61 (d, J = 10.0 Hz, 1H, CH2Ph), 4.66 (d, J = 10.0 Hz, 1H, CH2Ph), 4.78 (d, J = 10.5 Hz, 1H, CH2Ph), 4.87 (dd, J = 11.0 Hz, 1H, CH2Ph), 4.89 (d, J = 2.3 Hz, 1H, 7-H), (d, J = 11.0 Hz, 1H, CH2Ph), 5.05 (d, J = 11.2 Hz, 1H, CH2Ph), 5.18 (d, J = 10.5 Hz, 1H, CH2Ph), 7.17–7.41 (m, 30H, arom. H), 13C NMR (75 MHz, CDCl3): δ = 13.6 (q, CH3), 43.1 (d, C-2), 57.6 (q, OMe), 69.0, 69.5 (2t, C-6, C-12), 73.6, 74.0, 74.3, 75.6, 75.7, 76.2 (6t, CH2Ph), 73.9, 74.8, 76.5, 78.2, 81.1, 83.4, 84.1 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 103.8, 106.7, (2d, C-1, C-7), 128.1, 128.2, 128.3, 128.4, 128.4, 128.5, 128.6, 128.7, 128.9, 129.0, 129.1, 129.3, (30d, arom. C-H), 139.1, 139.5, 139.6, 139.8, 140.0, 140.1, (6 s, arom. C-CH2O); Elemental analysis(%) calcd for: C56H62O10 C 75.14, H 6.98; found: C 75.28, H 7.99; Mass (ESI-MS); m/z894.54(M)+.
General procedure for the bromination of Barton esters 4a
The Barton ester 4a (2.0 mmol) was dissolved in 20 mL of dry benzene and was protected from light with aluminium foil under argon atmosphere. Bromotrichloromethane (795 mg, 4.0 mmol) was added at 25 °C. The reaction mixture was subsequently exposed to light using a 250 W low- pressure mercury lamp. After completion of the reaction (approximately 1–2 h), the crude reaction mixture was concentrated and the residue was purified by flash chromatography, affording the products 6.:1); [α]20D = +14.6 (c = 1.02 in CHCl3); IR (film): v = 3063, 3029, 2858, 1950, 1732, 1496, 1362, 1421,1312, 1100, 1027 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.75 (dddd, J = 10.4, 8.1, 2.8, 2.4 Hz, 1H, 2-H), 3.38 (ddd, J = 9.5, 3.5, 3.0 Hz, 1H, 5-H), 3.46 (s, 3H, OMe), 3.60 (dd, J = 9.5, 8.8 Hz, 1H, 4-H), 3.67 (dd, J = 11.9, 2.8 Hz, 1H, 7-H), 3.68 (dd, J = 11.9, 2.0 Hz, 1H, 7′-H), 3.70 (dd, J = 10.5, 3.5 Hz, 1H, 6-H), 3.71 (dd, J = 10.5, 3.0 Hz, 1H, 6′-H), 3.76 (dd, J = 10.4, 8.8 Hz, 1H, 3-H), 4.32 (d, J = 8.1 Hz, 1H, 1-H), 4.46 (d, J = 12.0 Hz, 1H, CH2Ph), 4.50 (d, J = 10.7 Hz, 1H, CH2Ph), 4.56 (d, J = 12.0 Hz, 1H, CH2Ph), 4.69 (d, J = 10.8 Hz, 1H, CH2Ph), 4.71 (d, J = 10.8 Hz, 1H, CH2Ph), 4.86 (d, J = 10.7 Hz, 1H, CH2Ph), 7.08–7.28 (m, 15H, arom. H); 13C NMR (75 MHz, CDCl3): δ = 31.5 (t, C-7), 47.5 (d, C-2), 57.1 (q, OMe), 68.8 (d, C-6), 73.4, 74.7, 75.4 (3t, CH2Ph), 74.9, 79.7, 79.8 (3d, C-3, C-4, C-5), 102.15 (d, C-1), 127.5, 127.7, 127.7, 128.3, 128.4, 128.4 (15d, arom. C-H), 137.9, 138.1, 138.2 (3 s, arom. C-CH2O); elemental analysis(%) calcd for C29H33BrO5: C 64.33, H 6.14; found: C 64.39, H 6.19; Mass (ESI-MS); m/z 563.23(M + Na)+.
:1); [α]20D = +17.1 (c = 1.01 in CHCl3); IR (film): v = 3030, 2918, 1496, 1363, 1250,1100, 1086 cm−1; 1H NMR (300 MHz, CDCl3): δ = 2.20 (dddd, J = 10.8, 8.1, 2.6, 2.5 Hz, 1H, 2-H), 3.44 (s, 3H, OMe), 4.49 (ddd, J = 3.1, 2.3, 2.5 1H 5-H), 3.55 (dd, J = 12.3, 2.6 Hz, 2H, 7-H), 3.57 (dd, J = 12.3, 2.4 Hz, 2H, 7′-H), 3.63 (d, J = 10.8 1H, 3-H), 3.73 (dd, J = 10.0, 3.1 Hz, 1H, 6-H), 3.82 (dd, J = 10.0, 2.3 Hz, 1H, 6′-H), 3.88 (d, J = 2.5 Hz, 1H 4-H), 4.32 (d, J = 8.1 Hz, 1H, 1-H), 4.36 (d, J = 11.8 Hz, 1H, CH2Ph), 4.42 (d, J = 11.8 Hz, 1H, CH2Ph), 4.47 (d, J = 11.0 Hz, 1H, CH2Ph), 4.51 (d, J = 11.7 Hz, 1H, CH2Ph), 4.65 (d, J = 11.0 Hz, 1H, CH2Ph), 4.78 (d, J = 11.7 Hz, 1H, CH2Ph), 7.16–7.29 (m, 15H, arom. H);13C NMR (75 MHz, CDCl3): δ = 32.6 (dt C-7), 42.3 (d, C-2), 57.0 (q, OMe), 68.9 (d, C-6), 72.2, 73.5, 74.3 (3t, CH2Ph), 70.9, 73.4, 78.4 (C-3, C-4, C-5), 102.4 (C-1), 127.5, 127.7, 127.9, 128.0, 128.1, 128.4 (15d, arom. C-H), 37.7, 137.9, 138.6 (3 s, arom. C-CH2O); Elemental analysis(%) calcd for C29H33BrO5: C 64.33, H 6.14; found: C 64.36, H 6.21; Mass (ESI-MS); m/z 563.26(M + Na)+.
:1); [α]20D = +9.6 (c = 1.01 in CHCl3); IR (film): v = 2915, 2842, 1455, 1454, 1368, 1202, 1178, 1091, 1077, 1022 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.68 (dddd, J = 9.8, 8.0, 3.0, 2.5 Hz, 1H, 2-H), 3.15 (dd, J = 11.6, 9.8 1H, 5-H), 3.42 (s, 3H, OMe), 3.58 (ddd, J = 10.2, 9.8, 5.0 Hz, 1H, 4-H), 3.62 (dd, 1H, J = 10.2, 9.8 Hz, 3-H), 3.65 (dd, J = 10.0, 3.0 Hz, 1H, 6-H), 3.71 (dd, J = 10.0, 2.5 Hz, 1H, 6′-H), 3.92 (dd, J = 11.6, 5.0 Hz, 1H, 5′-H), 4.28 (d, J = 8.0 Hz, 1H, 1-H), 4.54 (d, J = 11.5 Hz, 1H, CH2Ph), 4.60 (d, J = 11.5 Hz, 1H, CH2-Ph), 4.66 (d, J = 10.8 Hz, 1H, CH2Ph), 4.91 (d, J = 10.8 Hz, 1H, CH2Ph), 7.17–7.26 (m, 10H, arom. H); 13C NMR (75 MHz, CDCl3): 31.7 (t, C-7), 46.9 (d, C-2), 57.0 (q, OMe), 63.5 (t, C-5), 72.8, 75.3 (2t, CH2Ph), 78.5, 79.5 (3d, C-3, C-4), 102.6 (d, C-1), 127.7, 127.8, 127.9, 128.0, 128.4, 128.4 (10d, arom. C-H), 138.0, 138.4, (2 s, arom. C-CH2O); Elemental analysis(%) calcd for: C21H25BrO4 C 59.86, H 5.98; found: C 59.95, H 6.09; Mass (ESI-MS); m/z 420.16(M)+.
:1); [α]20D = +25.2 (c = 1.02 in CHCl3); IR (film): v = 2920, 2832, 1448, 1444, 1362, 1211, 1188, 1084, 1076, 1028 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.68 (dddd, J = 12.8, 7.7, 2.3, 2.0 Hz, 1H, 2-H), 3.21 (d, J = 12.8 1H, 3-H), 3.45 (s, 3H, OMe), 3.56 (dd, J = 10.5, 1.8 Hz, 1H, 5-H), 3.58 (dd, J = 10.5, 2.0 Hz, 1H, 5′-H), 3.73 (dd, J = 10.0, 2.0 Hz, 1H, 6-H), 3.82 (dd, J = 10.0, 2.3 Hz, 1H, 6′-H), 4.06 (dd, J = 1.8, 2.0 Hz, 1H, 4-H), 4.23 (d, J = 7.8 Hz, 1H, 1-H), 4.36 (d, J = 11.0 Hz, 1H, CH2Ph), 4.48 (d, J = 11.0 Hz, 1H, CH2Ph), 4.54 (d, J = 12.30 Hz, 1H, CH2Ph), 4.69 (d, J = 12.30 Hz, 1H, CH2Ph), 7.19–7.31 (m, 10H, arom. H); 13C NMR (75 MHz, CDCl3): 32.5 (t, C-7), 42.5 (d, C-2), 56.9 (q, OMe), 63.3 (t, C-5), 71.1, 71.5 (2t, CH2Ph), 70.1, 76.5 (3d, C-3, C-4), 102.9 (d, C-1), 127.6, 127.8, 127.9, 128.0, 128.3, 128.4, (10d, arom. C-H), 137.8, 138.3 (2 s, arom. C-CH2O); Elemental analysis(%) calcd for: C21H25BrO4 C 59.86, H 5.98; found: C 59.88 H 6.01; Mass (ESI-MS); m/z420.13(M)+.
:1); [α]20D = +13.8 (c = 1.02 in CHCl3); IR (film): v = 3045, 2948, 1837, 1751, 1648, 1497, 1481, 1331, 1210, 1152 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.73(dddd, J = 8.6, 6.5, 5.1, 3.6, Hz 1H, 2-H), 3.27(dd J = 6.3, 4.5 Hz, 1H, 6-H), 3.29 (dd, J = 7.2, 5.0 Hz, 1H, 12-H), 3.30 (dd, J = 6.3, 3.6 Hz, 1H, 6′-H), 3.33(dd, J = 7.2, 5.6 Hz, 1H, 12′-H), 3.44 (s, 3H, OMe), 3.46 (dd, J = 10.2, 6.5 Hz 1H, 3-H), 3.62(ddd J = 9.6, 4.5, 3.6 Hz, 1H, 5-H), 3.65(ddd, J = 8.9, 5.6, 5.0 Hz 1H, 11-H), 3.61 (dd, J = 2.1, 1.8 Hz, 1H, 10-H), 3.70(dd J = 9.8, 1.9 Hz, 1H, 9-H), 3.79 (dd, J = 10.8, 3.6 Hz, 1H, 13-H), 3.81 (dd, J = 10.8, 5.1 Hz, 1H, 13′-H), 3.95 (dd, J = 9.8, 9.2 Hz, 1H, 8-H), 4.16(d, J = 11.8 Hz, 1H CH2Ph), 4.26(d, J = 12.6 Hz, 1H, CH2Ph) 4.28 (d, J = 10.5 Hz, 1H, CH2Ph), 4.30 (d, J = 12.0 Hz, 1H, CH2Ph), 4.49 (dd, J = 10.2, 9.6 Hz, 1H, 4-H), 4.50(d, J = 9.2 Hz, 1H, 7-H), 4.60 (d, J = 12.6 Hz, 1H, CH2Ph), 4.65(d, J = 12.0 Hz, 1H CH2Ph), 4.69(d, J = 11.0 Hz, 1H, CH2Ph), 4.73(d, J = 11.5 Hz, 1H, CH2Ph), 4.77 (d, J = 11.8 Hz, 1H, CH2Ph), 4.90 (d, J = 11.5 Hz 1H CH2Ph), 5.15(d, J = 10.0 Hz, 1H, CH2Ph), 6.99–7.25 (m, 30H, arom. H), 13C NMR (125 MHz, CDCl3): δ = 31.8 (t, C-13), 47.2 (d, C-2), 56.9 (q, OMe), 68.1, 68.2 (2t, C-6, C-12), 72.6, 73.0, 73.0, 73.3, 74.6, 75.3 (6t, CH2Ph), 73.0, 73.1 73.8, 77.3, 77.9, 80.1, 82.5 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 127.2, 127.3, 127.4, 127.5, 127.6, 127.8, 127.9, 128.1, 128.2, 128.3 (30d, arom. C-H), 138.1, 138.4, 138.5, 138.8, 138.9, 139.0 (6 s, arom. C-CH2O); Elemental analysis(%) calcd for: C56H61BrO10 C 69.06 H 6.31; found: C 69.34 H 6.38; Mass (ESI-MS); m/z 973.58(M)+.
:1); [α]20D = +36.3 (c = 1.01 in CHCl3); IR (film): v = 3035, 2941, 1836, 1752, 1647, 1497, 1480, 1335, 1210, 1151 cm−1; 1H NMR (300 MHz, CDCl3): δ = 1.73 (dddd, J = 8.6, 6.5, 5.1, 3.6, Hz 1H, 2-H), 3.27 (dd J = 6.3, 4.5 Hz, 1H, 6-H), 3.29 (dd, J = 7.2, 5.0 Hz, 1H, 12-H), 3.30 (dd, J = 6.3, 3.6 Hz, 1H, 6′-H), 3.33(dd, J = 7.2, 5.6 Hz, 1H, 12′-H), 3.44 (s, 3H, OMe), 3.46 (dd, J = 10.2, 6.5 Hz 1H, 3-H), 3.62 (ddd J = 9.6, 4.5, 3.6 Hz, 1H, 5-H), 3.65 (ddd, J = 8.9, 5.6, 5.0 Hz 1H, 11-H), 3.61 (dd, J = 2.1, 1.8 Hz, 1H, 10-H), 3.70 (dd J = 9.8, 1.9 Hz, 1H, 9-H), 3.79 (dd, J = 10.8, 3.6 Hz, 1H, 13-H), 3.81 (dd, J = 10.8, 5.1 Hz, 1H, 13′-H), 3.95 (dd, J = 9.8, 9.2 Hz, 1H, 8-H), 4.16 (d, J = 11.8 Hz, 1H CH2Ph), 4.26 (d, J = 12.6 Hz, 1H, CH2Ph), 4.28 (d, J = 10.5 Hz, 1H, CH2Ph), 4.30 (d, J = 12.0 Hz, 1H, CH2Ph), 4.49 (dd, J = 10.2, 9.6 Hz, 1H, 4-H), 4.50 (d, J = 9.2 Hz, 1H, 7-H), 4.60 (d, J = 12.6 Hz, 1H, CH2Ph), 4.65 (d, J = 12.0 Hz, 1H CH2Ph), 4.69 (d, J = 11.0 Hz, 1H, CH2Ph), 4.73 (d, J = 11.5 Hz, 1H, CH2Ph), 4.77 (d, J = 11.8 Hz, 1H, CH2Ph), 4.90 (d, J = 11.5 Hz 1H CH2Ph), 5.15(d, J = 10.0 Hz, 1H, CH2Ph), 6.99–7.25 (m, 30H, arom. H), 13C NMR (125 MHz, CDCl3): δ = 31.9 (t, C-13), 47.2 (d, C-2), 57.1 (q, OMe), 68.0, 68.2 (2t, C-6, C-12), 72.6, 73.1, 73.4, 74.7, 75.2, 75.4 (6t, CH2Ph), 73.0, 73.7, 75.2, 77.33 78.0, 80.0, 82.4 (7d, C-3, C-4, C-5, C-8, C-9, C-10, C-11), 127.3, 127.4, 127.5, 127.5, 127.6, 127.9, 128.0, 128.2, 128.31, 128.3, (30d, arom. C-H), 138.1, 138.4, 138.5, 138.7, 138.8, 139.0 (6 s, arom. C-CH2O); Elemental analysis(%) calcd for: C56H61BrO10 C 69.06 H 6.31; found: C 69.44 H 6.42; Mass (ESI-MS); m/z 973.56(M)+.
Acknowledgements
We thank the University of Potsdam for generous financial support.Notes and references
- Books: (a) B. Giese, Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, Pergamon Press, Oxford, 1986 Search PubMed; (b) D. P. Curran, N. A. Porter and B. Giese, Stereochemistry of Radical Reactions, VCH, Weinheim, 1996 Search PubMed; (c) A. J. Pearce, J.-M. Mallet and P. Sinay, in Radicals in Organic Synthesis, ed. P. Renaud and M. Sibi, Wiley-VCH, Weinheim, 2001, vol. 2, ch. 6.3, pp. 538–577 Search PubMedRecent reviews: (d) G. J. Rowlands, Tetrahedron, 2009, 65, 8603 CrossRef CAS; (e) G. J. Rowlands, Tetrahedron, 2010, 66, 1593 CrossRef CAS.
- Reviews: (a) C. Chatgilialoglu, Organosilanes in Radical Chemistry, Wiley, Chichester, 2004 CrossRef; (b) C. Chatgilialoglu, Chem.–Eur. J., 2008, 14, 2310 CrossRef CAS.
- Review: H. J. Schäfer, Top. Curr. Chem., 1990, 152, 91 CrossRef.
- Reviews: (a) D. H. R. Barton and S. Zard, Pure Appl. Chem., 1986, 58, 675 CrossRef CAS; (b) D. P. Curran, Synthesis, 1988, 489 CrossRef CAS; (c) P. I. Dalko, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. Horspool and F. Lenci, CRC Press, Boca Raton, 2nd edn, 2004, ch. 67, pp. 1–23 Search PubMed; (d) H. Togo, Advanced Free Radical Reactions for Organic Synthesis, Elsevier, 2004, ch. 8, pp. 199–213 Search PubMed; (e) M. F. Saraiva, M. R. C. Couri, M. Le Hyaric and M. V. de Almeida, Tetrahedron, 2009, 65, 3563 CrossRef CAS.
- (a) D. H. R. Barton, J. C. Jaszberenyi, W Liu and T. Sinaga, Tetrahedron, 1996, 52, 2717 CrossRef CAS; (b) D. H. R. Barton and W Liu, Tetrahedron Lett., 1997, 38, 367 CrossRef CAS; (c) D. H. R. Barton, M. V. de Almeida, V. Mauro, W. Liu, T. Shinada, J. C. Jaszberenyi, H. F. Dos Santos and M. Le Hyaric, Tetrahedron, 2001, 57, 8767 CrossRef CAS.
- (a) D. Crich and T. J. Ritchie, J. Chem. Soc., Chem. Commun., 1988, 1461 RSC; (b) D. H. R. Barton and M. Ramesh, J. Am. Chem. Soc., 1990, 112, 891 CrossRef CAS; (c) D. Crich, J.-T. Hwang and H. Yuan, J. Org. Chem., 1996, 61, 6189 CrossRef CAS.
- (a) P. Garner, R. Leslie and J. T. Anderson, J. Org. Chem., 1996, 61, 6754 CrossRef CAS; (b) P. Garner, J. T. Anderson, P. B. Cox, S. J. Klippenstein, R. Leslie and N. Scardovi, J. Org. Chem., 2002, 67, 6195 CrossRef CAS.
- (a) T. Linker, T. Sommermann and F. Kahlenberg, J. Am. Chem. Soc., 1997, 119, 9377 CrossRef CAS; (b) T. Sommermann, B. G. Kim, K. Peters, E.-M. Peters and T. Linker, Chem. Commun., 2004, 2624 RSC; (c) E. Elamparuthi and T. Linker, Org. Lett., 2008, 10, 1361 CrossRef CAS; (d) T. Linker, D. Schanzenbach, E. Elamparuthi, T. Sommermann, W. Fudickar, V. Gyóllai, L. Somsák, W. Demuth and M. Schmittel, J. Am. Chem. Soc., 2008, 130, 16003 CrossRef CAS; (e) E. Elamparuthi and T. Linker, Angew. Chem., Int. Ed., 2009, 48, 1853 CrossRef CAS; Reviews: (f) T. Linker, J. Organomet. Chem., 2002, 661, 159 CrossRef CAS; (g) E. Elamparuthi, B. G. Kim, J. Yin, M. Maurer and T. Linker, Tetrahedron, 2008, 64, 11925 CrossRef CAS.
- (a) J. Yin, J. Spindler and T. Linker, Chem. Commun., 2007, 2712 RSC; (b) J. Yin, T. Sommermann and T. Linker, Chem.–Eur. J., 2007, 13, 10152 CrossRef CAS; (c) J. Yin and T. Linker, Chem.–Eur. J., 2009, 15, 49 CrossRef CAS; (d) J. Yin and T. Linker, Tetrahedron, 2011, 67, 2447 CrossRef CAS.
- T. Ling, E. Poupon, E. J. Rueden and E. A. Theodorakis, Org. Lett., 2002, 4, 819 CrossRef CAS.
- H. Ito, S. Takeguchi, T. Kowagishi and K. Iguchi, Org. Lett., 2006, 8, 4883 CrossRef CAS.
- Z. Xu, W. Hu, Q. Liu, L. Zhang and Y. Jia, J. Org. Chem., 2010, 75, 7626 CrossRef CAS.
- (a) C. V. Ramana, R. Murali and M. Nagarajan, J. Org. Chem., 1997, 62, 7694 CrossRef CAS; (b) J. Beyer and R. Madsen, J. Am. Chem. Soc., 1998, 120, 12137 CrossRef CAS; (c) J. Beyer, P. R. Skaanderup and R. Madsen, J. Am. Chem. Soc., 2000, 122, 9575 CrossRef CAS.
- G. A. Wallace, R. W. Scott and C. H. Heathcock, J. Org. Chem., 2000, 65, 4145 CrossRef CAS.
- D. D. Perrin and W. L. F Armarego, Purification of Laboratory Chemicals, Fourth Edition, Butterworth Heinemann, Oxford, 1966 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ob06370g |
| ‡ Current address: Department of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14776 Potsdam & Freie Universität Berlin Institut für Chemie und Biochemie, Arnimallee 22, 14195 Berlin, Germany |
| This journal is © The Royal Society of Chemistry 2012 |