Electronic effects in 1,3-dipolar cycloaddition reactions of N-alkyl and N-benzyl nitrones with dipolarophiles†
Andrei
Bădoiu
and
E. Peter
Kündig
*
Department of Organic Chemistry, University of Geneva, 30 Quai Ernest Ansermet, CH-1211, Geneva 4, Switzerland. E-mail: peter.kundig@unige.ch; Fax: (+)41-22-379-3215
First published on 28th October 2011
Abstract
1,3-Dipolar cycloadditions afforded fast access to isoxazolidines bearing N-alkyl or N-benzyl substituents. The electronic properties of the substituents in the nitrones define the activity of the dipoles and modulate diastereoselectivity in the non-catalyzed reactions. Using a chiral one-point binding ruthenium Lewis acid catalyst, products were obtained in good yields and with excellent regio-, diastereo-, and enantioselectivity.
Introduction
2010 was a special year for the 1,3-dipolar cycloadditions (1,3-DCs). Along with 50 years of continuous evolution and expansion in the field, we celebrated the 90th anniversary of the father of this chemistry, Prof. Rolf Huisgen.1Cycloadditions have always attracted the interest of the scientific community through the apparent practical simplicity that hides a complex mechanism, the elegant end efficient access to cyclic compounds, and the versatility of both starting materials and products.2 Recent developments in asymmetric catalysis have further emphasized the value of 1,3-DCs as fast and clean reactions towards functionalized, enantiopure N,O-heterocyclic compounds.3We have previously reported efficient and selective homogeneous chiral catalysts for the Diels–Alder reactions of enals4,5 and enones6 with dienes, intramolecular Diels–Alder reactions,7 as well as 1,4-additions of thiols to enones.8 The catalysts that were developed are monocationic, one-point binding Cp-complexes of iron(II), and Cp- and indenyl-complexes of ruthenium(II) that bear electron-poor diphosphinite ligands to enhance the Lewis acidity and control the chiral environment around the metal.
Exploring the versatility of these chiral Lewis acid catalysts, we turned our attention to 1,3-DCs and provided the first examples of asymmetric metal-catalyzed reactions of nitrones9 and nitrile oxides10 with enals. This field has seen rapid development with efficient metal-based catalysts11 and organocatalysts12 being reported in the literature.
In this context, we studied in depth the reactions with diarylnitrones and showed that regioselectivity is a function of the substituents on the nitrone.13N-Alkyl and N-benzyl nitrones are less reactive dipoles when compared to diaryl- and cyclic nitrones.14 However, modularity, ease of synthesis, and stability are the key characteristics of these dipoles and explain their widespread use in synthesis (Fig. 1).
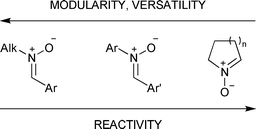 | ||
| Fig. 1 Properties and reactivity of common nitrone classes. | ||
Asymmetric catalytic 1,3-DCs have entered the field resolutely.15,16 The seminal work of MacMillan and coworkers on using organocatalysts allowed for the use of simpler and more versatile monodentate dipolarophiles.12a Since then, several other organic12 and metal-based catalysts11g–o have been found to successfully catalyze 1,3-DC reactions with N-“alkyl” nitrones.
In the present article, we extend our initial findings in the Ru-catalyzed 1,3-DC of N-alkyl and N-benzyl nitrones with enals17 and investigate intriguing selectivity aspects observed for the non-catalyzed reactions with the same dipoles.
Results and discussion
Non-catalyzed reactions
The cycloaddition reactions were carried out with a 50% excess of the enal (with respect to the nitrone), in CH2Cl2, at r. t. At the end of the reaction (checked by TLC, 72 h reaction time on average), the unreacted nitrones were precipitated with pentane and the crude product was filtered through a plug of cotton.
Signals for the endo and exo diastereomers were assigned by 1H NMR analysis. Analysis was complicated by signal broadening in the case of the endo diastereomers. This phenomenon is known to occur due to inversion at the nitrogen atom taking place on the 1H NMR-timescale.19 NOE analysis was inconclusive for the assignment of the signals corresponding to the two diastereomers. However, based on an analogy of chemical shifts for isoxazolidines previously obtained from diarylnitrones,13 we could distinguish and assign 1H NMR shifts to each of the two diastereomers.
Low to moderate yields reflect the long reaction times needed in the case of nitrones bearing electron donating groups (EDGs). The reactions were frequently accompanied by decomposition of the dipole and/or polymerization of methacrolein (Table 1, entries 1–4).
|
|
||||
|---|---|---|---|---|
| Entry | Ar | Nitrone | Yield (%)b | endo/exoc |
| a All reactions were carried out under N2, using 1a–n (0.5 mmol) and 3 (1 mmol), in 1 mL of dry solvent. b Isolated yield. c Determined by 1H NMR analysis. | ||||
| 1 | 4-NMe2-C6H4- | 1a | 40 | 17/83 |
| 2 | 4-OMe-C6H4- | 1b | 65 | 31/69 |
| 3 | 4-Me-C6H4- | 1c | 75 | 37/63 |
| 4 | 4-H-C6H4- | 1d | 65 | 40/60 |
| 5 | 4-F-C6H4- | 1e | 85 | 43/57 |
| 6 | 4-Cl-C6H4- | 1f | 87 | 49/51 |
| 7 | 4-Br-C6H4- | 1g | 90 | 52/48 |
| 8 | 4-CF3-C6H4- | 1h | 92 | 62/38 |
| 9 | 4-CN-C6H4- | 1i | 78 | 72/28 |
| 10 | 4-NO2-C6H4- | 1j | 75 | 73/27 |
| 11 | 2-F-C6H4- | 1k | 85 | 55/45 |
| 12 | C6F5- | 1l | 93 | 43/57 |
| 13 | η6-C6H4-Cr(CO)3- | 1m | 95 | 86/14 |
| 14 | 2-Py- | 1n | 94 | 95/5 |
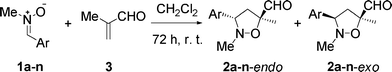
Interestingly, both the endo and the exo diastereomers of the 3,5-substituted regioisomer were obtained in ratios varying with the electronic properties of the substituents on the nitrone. Thus, EDGs on the nitrone α-aryl substituent led to a mixture of products with the exo product being the major diastereoisomer, while in the cases where electron withdrawing groups (EWGs) are placed on the nitrone α-aryl substituent, it is the endo product that becomes the major diastereoisomer. Similar observations are reported in the literature.20
In order to evaluate the effects of substitution on reaction rate for the non-catalyzed and the ruthenium-catalyzed 1,3-DCs, the EWG-substituted aryl was replaced with other EWG-like aromatic moieties. Nitrones 1m and 1n reacted smoothly at r. t. in CH2Cl2 (5 days) with methacrolein (3) to give the expected isoxazolidines in quantitative yield. Nitrone 1m, bearing a Cr(CO)3 moiety, was previously prepared in this laboratory.21Nitrone 1m led to a mixture of diastereomers 2m (endo major). On the other hand, nitrone 1n,22 bearing a 2-pyridyl fragment as the EWG aromatic part, proved to be particularly reactive in its reaction with methacrolein (3). Isoxazolidine 2n was isolated in quantitative yield and with excellent regio- and diastereoselectivity (endo).
Keeping only the 4-substituted derivatives (steric factors and/or more complex effects occur in the case of 2-F-Ph and the pentafluoro examples) and ordering the diastereomeric ratios according to the Hammett electronic parameter, a linear correlation between diastereoselectivity and substitution can be observed for the series.23 The correlation can be quantified by plotting the σP+ electronic parameter24 as a function of log[(endo-R/exo-R)/(endo-H/exo-H)] (Fig. 2).25 A correlation factor (R2) of 0.9354 was obtained, suggesting a significant degree of linearity of the dependence.
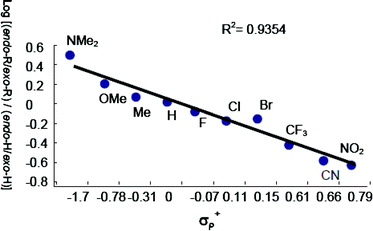 | ||
| Fig. 2 Hammett plot showing the linear correlation between the electronic parameter of the substituents on the nitrones and the endo/exo ratio of the products obtained in the non-catalyzed 1,3-DCs of N-Me, α-aryl nitrones 1a–n with methacrolein (3). | ||
No such effects were observed for the non-catalyzed reactions of methacrolein (3) with diarylnitrones carried out in the same conditions.13b However, in the case of the Ru-catalyzed asymmetric 1,3-DC of methacrolein (3) with substituted diarylnitrones, the regioselectivity was found to vary with the electronic properties of the nitrone.13 Interestingly, in the case of the N-Me nitrones, in the absence of a catalyst this trend is observed in the diastereoselectivity. The variation is not as extreme as for the diarylnitrones, but shows how small variations can have important effects on the outcome of the cycloaddition reaction.
The standardized trendline assigned to this semilogarithmic equation in Fig. 2 gives ρ (the reaction constant) equal to −0.11 (0.93 correlation factor). The very small value of the reaction constant indicates that there is little, if any, charge transfer in the transition state (TS). Moreover, the moderate correlation factor for the linear dependence does not allow for a clear picture of the reaction mechanism.
In this series, the N-i-Pr and -t-Bu nitrones 1o and 1p, respectively, gave the products in moderate yields despite long reaction times (up to 2 weeks at r. t., Table 2, entries 1 and 2) and a two-fold excess of methacrolein (3). On the other hand, the N-Bn, -PMB, and -DPM-substituted nitrones 1q, 1r, and 1s, respectively, proved to be more reactive, giving stable 3,5-substituted isoxazolidines in good yields (entries 3–5).
|
|
||||
|---|---|---|---|---|
| Entry | R | Nitrone | Yield (%)b | endo/exoc |
| a All reactions were carried out under N2, using 1o–s (0.5 mmol) and 3 (1 mmol), in 1 mL of dry solvent. b Isolated yield, full conversion of the nitrone (up to 2 weeks). c Determined by 1H NMR analysis. i-Pr: iso-propyl; t-Bu: tert-butyl; Bn: benzyl; PMB: para-methoxy-benzyl; DPM: diphenylmethyl. | ||||
| 1 | i-Pr | 1o | 65 | >95/5 |
| 2 | t-Bu | 1p | 75 | 93/7 |
| 3 | Bn | 1q | 65 | 84/16 |
| 4 | PMB | 1r | 85 | 79/21 |
| 5 | DPM | 1s | 87 | 93/7 |
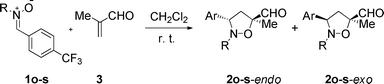
Good yields and diastereoselectivities (in favor of the endo isomer) can be obtained in the non-catalyzed reaction using these dipoles. No particular trend is observed when changing substitution and solely the 3,5-substituted regioisomers are isolated.
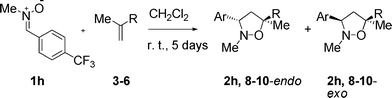
While difficult to interpret, the observed correlation between the endo/exo ratio of the products and the electronic character of the substituents confirms that for a type II 1,3-DC both types of frontier molecular orbital interactions are important in the TS. Minute modifications of the electronic properties of either of the reaction partners appear to strongly influence the TS and thus the outcome of the reaction.
The observed variation of diastereoselectivity with the electronic properties of the dipole is rather rare.20 In the past, we have shown how electronic factors can influence regioselectivity (with diarylnitrones)13 and enantioselectivity (with aryl nitrile oxides).10 Interestingly, no catalyst is employed in this case and the reaction is carried out under mild conditions. Although a number of groups have attempted to rationalize regio- and diastereoselectivity through computational studies, no clear picture has emerged so far.20a,26
Two factors should be taken into account when considering diastereoselectivity: the structure of the reaction partners (i.e. E/Z isomerism), and the interactions in the TS. For this specific transformation, the general opinion is that E/Z isomerism is responsible for the observed selectivities. However, with few exceptions,27 all examples use high temperatures, making indeed nitrone isomerization a possiblity.28 Our results and recent studies suggest that at r. t. the nitrones are found exclusively in the Z-configuration.29 Thus, we can conclude that the geometry of the reaction partners is not responsible for the observed selectivity inversion.
Information about the reaction mechanism can be extracted from the data presented above. While the Hammett plot does not show a perfectly linear correlation, it suggests an asynchronous concerted mechanism with little or no charge transfer in the TS (confirmed by computational studies).30,31 In addition, the lack of solvent effects reinforces the argument for a concerted transformation.2,32–33 Interestingly, a similar inversion of diastereoselectivity is observed when varying the electronic properties in the dipolarophile (Table 3); this is typical for type II 1,3-DCs, where HOMO–LUMO interactions of both reacting partners are possible and can influence the outcome of the reaction.2 Finally, increasing the bulk on the dipole has little influence on the selectivity (as shown in Table 2), suggesting that steric factors do not play a role in the diastereoselection process.
Thus, subtle variations in the electronic properties of either reaction partners lead to important changes in the ratio of the endo/exo isomers formed, possibly through secondary orbital interactions that occur in the TS. Unfortunately, computational studies so far have not provided more answers.
Ru-catalyzed reactions
The catalyst of choice for this transformation is (R,R)-11, a robust and easy to prepare monocationic ruthenium complex5 that has proved active and selective for a number of transformations.5–11,13,17Electron-rich dipoles 1a–c proved unreactive and could be recovered quantitatively at the end of the reaction, together with the intact catalyst. Applying harsher reaction conditions or increasing the catalyst loading did not improve the results. Conversely, nitrones 1d–l, bearing EWG substituents, led to formation of 3,5-substituted isoxazolidines with very good diastereo- and enantioselectivity. The best selectivity was obtained when using the N-Me, α-(4-Br-Ph) nitrone 1g, with 97.7% ee.17
Within the series, two dipoles stood apart: while reaction with nitro-substituted nitrone 1j proved sluggish and provided the product in moderate yield and with decreased diastereoselectivity, reaction with the nitrile-substituted nitrone 1i led to no product whatsoever. This reinforces our previous findings on substrates bearing Lewis-basic groups that lead to competitive binding to the Lewis acid, thus reducing efficiency or even shutting down the catalytic cycle.13
Less common EWG-like aromatic moieties can also be used with good results. The authors have a long-standing interest in the chemistry of chromium arene complexes.34 When attaching a Cr(CO)3 moiety on an arene, the electron density on the aromatic ring is markedly decreased, rendering the whole fragment electron-poor. Confirming this hypothesis, nitrone 1m, bearing the Cr(CO)3 moiety on the aryl part, gave excellent yield and selectivities (Scheme 1, top).
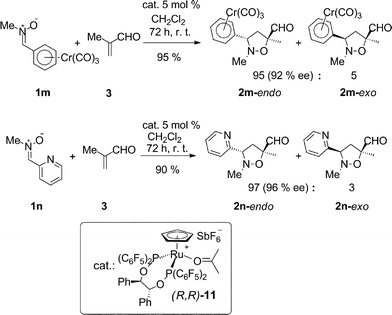 | ||
| Scheme 1 Ru-catalyzed 1,3-DC reactions of methacrolein (3) with N-Me, α-aryl nitrones 1m–n (the reactions were carried out under N2, using (R,R)-11 (0.025 mmol), 1m–n (0.5 mmol) and 3 (0.75 mmol), in 1 mL of dry solvent; isolated yields reported, diastereomeric ratio determined by 1H NMR analysis and enantiomeric ratio determined by HPLC analysis). | ||
On the same principle, using a 2-pyridyl fragment as the aryl part on the nitrone led to exclusive formation of the endo-3,5-substituted adduct 2n with excellent enantioselectivity (Scheme 1, bottom).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R (nitrone) | T/°C | Yield (%)b | endo/exoc | ee (%) endod |
| a All reactions were carried out under N2, using (R,R)-11 (5 mol%), 1h,o–s (0.5 mmol) and 3 (1 mmol), in 1 mL of dry CH2Cl2. b Isolated yield. c Determined by 1H NMR analysis. d Determined by HPLC analysis of the corresponding primary alcohol. i-Pr: iso-propyl; t-Bu: tert-butyl; Bn: benzyl; PMB: para-methoxy-benzyl; DPM: diphenylmethyl. e Full conversion not reached. | |||||
| 117 | Me (1h) | −5 | 85 | 94/6 | 92 |
| 2 | i-Pr (1o) | r. t. | 50e | 95/5 | 79 |
| 3 | t-Bu (1p) | r. t. | 19e | 95/5 | 59 |
| 4 | Bn (1q) | +5 | 95 | >95/5 | 87 |
| 5 | PMB (1r) | +5 | 80 | >95/5 | 90 |
| 6 | DPM (1s) | +10 | 94 | >95/5 | 69 |
As in the case of the non-catalyzed reactions, the N-i-Pr (1o) and -t-Bu (1p) nitrones reacted very slowly at r. t. and decomposed during the long reaction time; despite the good endo selectivity, enantioselectivities were moderate with these dipoles (entries 2 and 3) when compared with the N-Me analogue (entry 1).
Nitrones bearing benzyl groups on the nitrogen atom are extremely valuable for applications in synthesis as these moieties act as versatile protecting groups. We were pleased to find that the N-Bn (1q) and -PMB (1r) nitrones afforded the expected endo-3,5-substituted isoxazolidines 2q–r with high yields and enantioselectivities (entries 4 and 5).
Maruoka and coworkers recently provided new examples of nitrones bearing removable groups at the nitrogen atom of the nitrone, moving from the N-Bn to the N-DPM nitrones. Contrary to the N-Bn nitrones (which give the endo-3,5-substituted adducts), the endo-3,4-isoxazolidines were obtained exclusively with good yields and enantioselectivity. Moreover, the DPM group could be easily removed through oxidation, leaving the isoxazolidine ring intact.11n–o We tested an analogous nitrone (1s) with our catalytic system; unfortunately, not only the nitrone reacts slowly, but the endo-3,5-substituted isoxazolidine 2s is obtained with moderate enantioselectivity (entry 6). Too much steric bulk on the nitrone appears to slow down the catalytic cycle and leads to an erosion of the enantioselectivity by formation of racemic product through background (thermal) reaction.
The absolute configuration thus determined corresponds to C1(S), C3(S) and is the one expected from a top-endo approach of the Z-nitrone to the more accessible Cα-SiCC face of the double bond of methacrolein (3) coordinated in the chiral pocket of the [RuCp(R,R-BIPHOP-F)(acetone)][SbF6] complex (R,R)-11. This can be visualized by means of the X-ray-based models (Fig. 3).
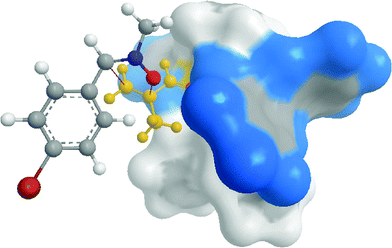 | ||
| Fig. 3 Model showing the approach of N-Me, α-(4-Br-Ph) nitrone (1g) to the accessible Cα-Si face of the C–C double bond of methacrolein (3) coordinated in the chiral pocket of the catalyst (R,R)-11.17 | ||
The same results were previously obtained when using diarylnitrones13 and nitrile oxides10 as dipoles. Diastereo- and enantioselectivity are exclusively under catalyst control. Regioselectivity in this case is under dipole control and appears to be directed by the substituent at the N of the nitrone.
Conclusions
The study of the scope and versatility of the 1,3-DCs with N-alkyl and N-benzyl nitrones with enals exposed intriguing and unprecedented trends in diastereoselectivity in the non-catalyzed reactions.On the other hand, the Ru-catalyzed reactions proved to be robust, efficient, and selective for a whole range of substrates. Electronic effects are key to activating the dipoles for the cycloaddition reaction. Synthetically relevant isoxazolidines bearing the methyl and benzyl groups at the N could thus be obtained in good yields and with high regio-, diastereo-, and enantioselectivity.
Experimental section—selected examples33
General experimental procedure for non-catalyzed reactions
In a 50 mL Schlenk tube equipped with a magnetic stirring bar, a solution of the nitrone (0.5 mmol, 1 equiv.) in CH2Cl2 (2 mL) is stirred at r. t. to give a clear solution. Methacrolein (1 mmol, 2 equiv.) is added dropwise, by syringe, at r. t. The reaction mixture is stirred at r. t. until TLC analysis (SiO2, AcOEt/cyclohexane 2/3 or CH2Cl2) shows no unreacted nitrone. Addition of dry pentane (10 mL), filtration of the precipitated nitrone through a plug of cotton in a Pasteur pipette and in vacuo removal of solvents leads to an oil. Purification by a quick filtration through a SiO2 plug (Hdry = 5 cm, Φe = 1 cm) with CH2Cl2 gives viscous, clear oils that solidify at −30 °C. Diastereomeric ratios are determined by 1H NMR of the crude mixture. Clear signals for the racemic mixture can be observed in the HPLC analysis of the corresponding primary alcohols obtained by a standard NaBH4 reduction in ethanol (CHIRACEL OD, Grad. 99+1–90+10, 0.75 mL min−1, 100 min, 254 nm or CHIRALPACK AD, Grad 99+1–85+15, 0.5 mL min−1, 80 min, 254); ν values are given in cm−1, δ values are given in ppm, J values are given in Hz, tR (retention times) are given in minutes. Some products proved too unstable for MS analysis; data for the corresponding primary alcohols is available in the ESI.†General procedure for reactions catalyzed by (R,R)-11
In a 50 mL Schlenk tube equipped with a magnetic stirring bar, the catalyst (36 mg, 0.025 mmol, 5 mol%) is loaded and CH2Cl2 (1 mL) is added. The solution is stirred at the appropriate temperature and methacrolein (62 μL, 0.75 mmol, 1.5 equiv.) is added. The mixture is stirred for further 20 min before addition of the corresponding nitrone (0.5 mmol, 1 equiv.) as a solid and in one portion. The extent of the reaction is followed by TLC analysis (SiO2, AcOEt/cyclohexane 2/3 or CH2Cl2) until no traces of nitrone are observed. Pentane is added to precipitate the catalyst and most of the unreacted nitrone, and the reaction mixture is passed through a plug of Celite 545 (P3-frit, Celite 545, Hdry = 1.5 cm, Φe = 2 cm) followed by in vacuo removal of volatiles. Purification by a quick filtration through a SiO2 plug (Hdry = 5 cm, Φe = 1 cm) with CH2Cl2 gives viscous, clear oils that solidify at −30 °C. Diastereomeric ratios are determined by 1H NMR of the crude mixture. Enantiomeric excess is determined by HPLC analysis of the corresponding primary alcohols obtained by a standard NaBH4 reduction in ethanol (CHIRACEL OD, Grad. 99+1–90+10, 0.75 mL min−1, 100 min, 254 nm or CHIRALPACK AD, Grad 99+1–85+15, 0.5 mL min−1, 80 min, 254). Given data is for the 3,5-endo isomer in the mixture. Some products proved too unstable for MS analysis; data for the corresponding primary alcohols is available in the ESI.†Acknowledgements
We would like to thank the Swiss National Science Foundation for financial support and Marlène Berthod for synthesis of intermediates. We are grateful to Prof. Jérôme Lacour, Prof. Jiri Mareda and Daniel Emery for helpful discussions.Notes and References
- (a) R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 565 CrossRef; (b) J. I. Seeman, Helv. Chim. Acta, 2005, 88, 1145 CrossRef CAS; (c) C. Rüchardt, J. Sauer and R. Sustmann, Helv. Chim. Acta, 2005, 88, 1154 CrossRef; (d) K. N. Houk, Helv. Chim. Acta, 2010, 93, 1241 CrossRef CAS.
- (a) R. Huisgen in 1,3-dipolar cycloaddition chemistry, A. Padwa, Ed.; Wiley: New York, Vol. 1, p. 1, 1984 Search PubMed; (b) A. Padwa in Comprehensive Organic Synthesis, B. M. Trost, I. Flemming, ed.; Pergamon Press: Oxford, Vol. 4, p. 1069, 1991 Search PubMed; (c) P. A. Wade in Comprehensive Organic Synthesis, B. M. Trost, I. Flemming, ed.; Pergamon Press: Oxford, Vol. 4, p. 1111, 1991 Search PubMed; (d) R. Huisgen, The Adventure Playground of Mechanisms and Novel Reactions. In Profiles Pathways, and DreamsJ. I. Seeman, Ed.; American Chemical Society: Washington, DC, 1994 Search PubMed; (e) K. V. Gothelf and K. A. Jørgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS; (f) N. T. Patil and Y. Yamamoto, Chem. Rev., 2008, 108, 3395 CrossRef CAS.
- (a) X.-F. Hu, Y.-Q. Feng and X.-F. Li, Chin. J. Org. Chem., 2005, 25, 1 Search PubMed; (b) H. Pellissier, Tetrahedron, 2007, 63, 3235 CrossRef CAS; (c) S. M. Lait, D. A. Rankic and B. A. Keay, Chem. Rev., 2007, 107, 767 CrossRef CAS; (d) L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887 CrossRef CAS; (e) C. Nájera and J. M. Sansano, Org. Biomol. Chem., 2009, 7, 4567 RSC; (f) M. Kissane and R. Maguire, Chem. Soc. Rev., 2010, 39, 845 RSC; (g) C. Nájera, J. M. Sansano and J. M. Yus, J. Braz. Chem. Soc., 2010, 21, 377 CrossRef.
- M. E. Bruin and E. P. Kündig, Chem. Commun., 1998, 2635 RSC.
- (a) E. P. Kündig, C. M. Saudan and G. Bernardinelli, Angew. Chem., Int. Ed., 1999, 38, 1220 Search PubMed; (b) E. P. Kündig, C. M. Saudan, V. Alezra, F. Viton and G. Bernardinelli, Angew. Chem., Int. Ed., 2001, 40, 4481 CrossRef; (c) E. P. Kündig, C. M. Saudan and F. Viton, Adv. Synth. Catal., 2001, 343, 51 CrossRef; (d) P. G. Anil Kumar, P. S. Pregosin, M. Vallet, G. Bernardinelli, R. F. Jazzar, F. Viton and E. P. Kündig, Organometallics, 2004, 23, 5410 CrossRef CAS; (e) V. Alezra, G. Bernardinelli, C. Corminboeuf, U. Frey, E. P. Kündig, A. E. Merbach, C. M. Saudan, F. Viton and J. Weber, J. Am. Chem. Soc., 2004, 126, 4843 CrossRef CAS.
- J. Rickerby, M. Vallet, G. Bernardinelli, F. Viton and E. P. Kündig, Chem.–Eur. J., 2007, 13, 3354 CrossRef CAS.
- (a) S. Thamapipol, G. Bernardinelli, C. Besnard and E. P. Kündig, Org. Lett., 2010, 12, 5604 CrossRef CAS; (b) S. Thamapipol and E. P. Kündig, Chimia, 2011, 65, 268 CrossRef CAS.
- A. Bădoiu, G. Bernardinelli, C. Besnard and E. P. Kündig, Org. Biomol. Chem., 2010, 8, 193 Search PubMed.
- F. Viton, E. P. Kündig and G. Bernardinelli, J. Am. Chem. Soc., 2002, 124, 4968 CrossRef CAS.
- Y. Brinkmann, J. M. Reniguntala, R. Jazzar, G. Bernardinelli and E. P. Kündig, Tetrahedron, 2007, 63, 8413 CrossRef CAS.
- (a) T. Mita, N. Ohtsuki, T. Ikeno and T. Yamada, Org. Lett., 2002, 4, 2457 CrossRef CAS; (b) T. Nagata, Y. Yorozu, T. Yamada and T. Mukaiyama, Angew. Chem., Int. Ed. Engl., 1995, 34, 2145 CrossRef CAS; (c) S. Kezuka, N. Ohtsuki, T. Mita, Y. Kogami, T. Ashizawa, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2003, 76, 2197 CrossRef CAS; (d) N. Ohtsuki, S. Kezuka, Y. Kogami, T. Mita, T. Ashizawa, T. Ikeno and T. Yamada, Synthesis, 2003, 9, 1462 Search PubMed; (e) M. Shirahase, S. Kanemasa and Y. Oderaotoshi, Org. Lett., 2004, 6, 675 CrossRef CAS; (f) M. Shirahase, S. Kanemasa and M. Hasegawa, Tetrahedron Lett., 2004, 45, 4061 CrossRef CAS; (g) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, L. A. Oro, A. I. Balana, F. J. Lahoz, T. Tejero, P. Merino, S. Franco and I. Montesa, J. Am. Chem. Soc., 2004, 126, 2717 CrossRef; (h) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, L. A. Oro, F. J. Lahoz, A. I. Balana, T. Tejero and P. Merino, J. Am. Chem. Soc., 2005, 127, 13386 CrossRef CAS; (i) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, T. Fischer, F. J. Lahoz, I. T. Dobrinovitch and L. A. Oro, Adv. Synth. Catal., 2007, 349, 1751 CrossRef CAS; (j) D. Carmona, M. P. Lamata, F. Viguri, J. Ferrer, N. Garcia, F. J. Lahoz, M. L. Martin and L. A. Oro, Eur. J. Inorg. Chem., 2006, 3155 CrossRef CAS; (k) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, F. J. Lahoz and L. A. Oro, Chem.–Eur. J., 2007, 13, 9746 CrossRef CAS; (l) D. Carmona, M. P. Lamata, F. Viguri, R. Rodriguez, F. J. Lahoz, M. J. Fabra and L. A. Oro, Tetrahedron: Asymmetry, 2009, 20, 1197 CrossRef CAS; (m) T. Kano, T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2005, 127, 11927 Search PubMed; (n) T. Hashimoto, M. Omote, T. Kano and K. Maruoka, Org. Lett., 2007, 9, 4805 CrossRef CAS; (o) T. Hashimoto, M. Omote, Y. Hato, T. Kano and K. Maruoka, Chem.–Asian J., 2008, 3, 407 CrossRef CAS; (p) Y. W. Wang, J. Wolf, P. Zavalij and M. P. Doyle, Angew. Chem., Int. Ed., 2008, 47, 1439 CrossRef CAS.
- (a) W. S. Jen, J. J. M. Wiener and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 9874 CrossRef CAS; (b) S. Karlsson and H.-E. Hogberg, Tetrahedron: Asymmetry, 2002, 13, 923 CrossRef CAS; (c) S. Karlsson and H.-E. Hogberg, Eur. J. Org. Chem., 2003, 2782 CrossRef CAS; (d) A. Puglisi, M. Benaglia, M. Cinquini, F. Cozzi and G. Celentano, Eur. J. Org. Chem., 2004, 567 CrossRef CAS; (e) M. Lemay, J. Trant and W. W. Ogilvie, Tetrahedron, 2007, 63, 11644 CrossRef CAS; (f) S. S. Chow, M. Nevalainen, C. A. Evans and C. W. Johannes, Tetrahedron Lett., 2007, 48, 277 CrossRef CAS; (g) Ł. Weseliński, P. Stępniak and J. Jurczak, Synlett, 2009, 2261 Search PubMed.
- (a) A. Bădoiu, Y. Brinkmann, F. Viton and E. P. Kündig, Pure Appl. Chem., 2008, 5, 1013 CrossRef; (b) A. Bădoiu, G. Bernardinelli, J. Mareda, E. P. Kündig and F. Viton, Chem.–Asian J., 2008, 3, 1298 CrossRef; (c) A. Bădoiu, G. Bernardinelli, J. Mareda, E. P. Kündig and F. Viton, Chem.–Asian J., 2009, 4, 1021 CrossRef (this article corrects: Chem.–Asian J., 2008, 3, 1298).
- J. Hamer and A. Macaluso, Chem. Rev., 1964, 64, 473 CrossRef CAS.
- (a) T. Shimizu, M. Ishizaki and N. Nitada, Chem. Pharm. Bull., 2002, 50, 908 CrossRef CAS; (b) H. Suga, T. Nakajima, K. Itoh and A. Kakehi, Org. Lett., 2005, 7, 1431 CrossRef CAS.
- (a) S. Kanemasa, Y. Oderatoshi, J. Tanaka and E. Wada, J. Am. Chem. Soc., 1998, 120, 12355 CrossRef CAS; (b) K. Hori, H. Kodama, T. Ohta and I. Furukawa, J. Org. Chem., 1999, 64, 5017 CrossRef CAS; (c) K. V. Gothelf and K. A. Jørgensen, Chem. Commun., 2000, 1449 RSC; (d) S. Kanemasa, Nippon Kagaku Kaishi, 2000, 3, 155 CrossRef; (e) H. Suga, A. Kakehi, S. Ito and H. Sugimoto, Bull. Chem. Soc. Jpn., 2003, 76, 327 CrossRef CAS.
- A. Bădoiu, G. Bernardinelli and E. P. Kündig, Synthesis, 2010, 2207 Search PubMed.
- (a) Organic Functional Group Preparations , S. R. Sandler and W. Karo, Academic Press: New York, 1986, Vol. 3 Search PubMed; (b) I. Brüning, R. Grashey, R. Hauck, R. Huisgen and H. Seidl, Org. Synth., Coll. Vol. 5, p. 1124 Search PubMed; (c) W. G. Hollys, P. L. Smith, D. K. Hood and S. M. Cook, J. Org. Chem., 1994, 59, 3485 CrossRef.
- E. Tyrrell, J. Allen, K. Jones and R. Beauchet, Synthesis, 2005, 14, 2393 CrossRef.
- (a) M. Joucla, F. Tonnard, D. Gree and J. Hamelin, J. Chem. Res. (M), 1978, 2901 Search PubMed; (b) H. Agirbas and S. Guner, Heterocycl. Commun., 2000, 6, 557 CrossRef CAS; (c) K. Pihlaja, P. Tahtinen, R. Shaikhutdinov, H. Hartikainen and V. Ovcharenko, J. Heterocycl. Chem., 2004, 41, 741 CrossRef CAS; (d) H. Agirbas, S. Guner, F. Budak, S. Keceli, F. Kandemirli, N. Shvets, V. Kovalishyn and A. Dimoglo, Bioorg. Med. Chem., 2007, 15, 2322 CrossRef CAS.
- R. Arvai, Thesis No 3695, University of Geneva, Geneva, 2005.
- (a) E. Dagne and N. Castagnoli Jr, J. Med. Chem., 1972, 15, 356 CrossRef CAS; (b) T. Shimizu, M. Ishizaki and N. Nitada, Chem. Pharm. Bull., 2002, 50, 908 CrossRef CAS; (c) A. B. Lysenko, K. V. Domasevitch, E. Peralta-Perez, F. Lopez-Ortiz, J. W. Steed and R. D. Lampeka, Polyhedron, 2002, 21, 769 CrossRef CAS; (d) P. Merino, T. Tejero, M. Laguna, E. Cerrada, A. Moreno and J. A. Lopez, Org. Biomol. Chem., 2003, 1, 2336 RSC; (e) E. Colacino, P. Nun, F. M. Colacino, J. Martinez and F. Lamaty, Tetrahedron, 2008, 64, 5569 CrossRef CAS; (f) X. Li, R. Wang, Y. Wang, H. Chen, Z. Li, C. Ba and J. Zhang, Tetrahedron, 2008, 64, 9911 CrossRef CAS.
- (a) T. G. Wallis, N. A. Porter and C. K. Bradsher, J. Org. Chem., 1973, 38, 2917 CrossRef CAS; (b) R. Huisgen in 1,3-Dipolar Cycloaddition Chemistry, A. Padwa, Ed.; Wiley: New York, 1984, Vol. 1, p. 1 Search PubMed; (c) M. Cushman and E. J. Madaj, J. Org. Chem., 1987, 52, 907 CrossRef CAS.
- (a) C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS; (b) H. H. Jaffe, Chem. Rev., 1953, 53, 191 CrossRef CAS; (c) P. R. Wells, Chem. Rev., 1963, 63, 171 CrossRef CAS.
- (a) C. G. Swain and E. C. Lupton Jr, J. Am. Chem. Soc., 1968, 90, 4328 CrossRef CAS; (b) D. H. McDaniel and H. C. Brown, J. Org. Chem., 1958, 23, 420 CrossRef CAS; (c) B. Wang and K. Lammertsma, J. Am. Chem. Soc., 1994, 116, 10486 CrossRef CAS; (d) E. Albrecht, J. Mattay and S. Steenken, J. Am. Chem. Soc., 1997, 119, 11605 CrossRef CAS; (e) C.-M. Che, W.-Y. Yu, P.-M. Chan, W.-C. Cheng, S.-M. Peng, K.-C. Lau and W.-K. Li, J. Am. Chem. Soc., 2000, 122, 11380 CrossRef CAS; (f) T. G. Wallis, N. A. Porter and T. K. Bradsher, J. Org. Chem., 1973, 38, 2917 CrossRef CAS; (g) T. K. Bradsher, G. L. B. Carlson, N. A. Porter, I. J. Westerman and T. G. Wallis, J. Org. Chem., 1978, 43, 822 CrossRef; (h) M. Cushman and E. J. Madaj, J. Org. Chem., 1987, 52, 907 CrossRef CAS; (i) W. Adam, H. M. Harrer, W. M. Nau and K. Peters, J. Org. Chem., 1994, 59, 3786 CrossRef CAS; (j) G. Belanger, F. Levesque, J. Paquet and G. Barbe, J. Org. Chem., 2005, 71, 291 CrossRef.
- (a) Sk. Ali Asrof and M. I. M. Wazeer, J. Chem. Soc., Perkin Trans. 2, 1986, 1789 Search PubMed; (b) A. Padwa, D. N. Kline, K. F. Koehler, M. Matzinger and M. K. Venkatramanan, J. Org. Chem., 1987, 52, 3909 CrossRef CAS; (c) U. A. R. Al-Timari, L. Fisera, P. Ertl, I. Goljer and N. Pronayova, Monatsh. Chem., 1992, 123, 999 CrossRef CAS; (d) L. Fisera, U. A. R. Al-Timari, P. Ertl and N. Pronayova, Monatsh. Chem., 1993, 124, 1019 CrossRef CAS; (e) L. Jaroskova, L. Fisera, I. Matejkova, P. Ertl and N. Pronayova, Monatsh. Chem., 1994, 125, 1413 CrossRef CAS; (f) K. V. Gothelf, R. G. Hazell and K. A. Jørgensen, J. Org. Chem., 1996, 61, 346 CrossRef CAS; (g) A. Hassan, M. I. M. Wazeer, M. T. Saeed, M. N. Siddiqui and Sk. Asrof Ali, J. Phys. Org. Chem., 2000, 13, 443 CrossRef CAS; (h) M. Carda, R. Portoles, J. Murga, S. Uriel, J. A. Marco, L. R. Domingo, R. J. Zaragoza and H. Roper, J. Org. Chem., 2000, 65, 7000 CrossRef CAS; (i) A. Baranski, R. Jasinski and M. Bujak, Polish. J. Chem., 2002, 76, 145 CAS; (j) P. Merino, J. A. Mates, J. Revuelta, T. Tejero, U. Chiaccio, G. Romeo, D. Innazzo and R. Romeo, Tetrahedron: Asymmetry, 2002, 13, 173 CrossRef CAS; (k) P. Perez, L. R. Domingo, M. J. Aurell and R. Contreras, Tetrahedron, 2003, 59, 3117 CrossRef CAS; (l) L. R. Domingo, M. J. Aurell, M. Arno and J. A. Saez, THEOCHEM, 2007, 811, 125 CrossRef CAS; (m) G. Wagner, T. N. Danks and B. Desai, Tetrahedron, 2008, 64, 477 CrossRef CAS; (n) B. Avijit, A. Nivedita and B. Pizush Kanti, J. Indian Chem. Soc., 2009, 86, 63 Search PubMed; (o) B. Avijit and A. Nivedita, J. Indian Chem. Soc., 2009, 86, 1068 Search PubMed; (p) A. Nivedita, B. Avijit, B. Manas and D. K. Tapas, Indian J. Chem., Section A, 2009, 48A, 1627 Search PubMed; (q) A. K. Nacereddine, W. Yahia, S. Bouacha and A. Djerourou, Tetrahedron Lett., 2010, 51, 2617 CrossRef CAS; (r) L. Xu, C. E. Doubleday and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 3029 CrossRef CAS.
- (a) D. Cristina, C. DeMicheli and R. Gandolfi, J. Chem. Soc., Perkin Trans. 1, 1979, 2891 RSC; (b) T. Yakura, M. Nakazawa, T. Takino and M. Ikeda, Chem. Pharm. Bull., 1992, 40, 2014 CAS; (c) K. V. Gothelf and K. A. Jørgensen, J. Org. Chem., 1994, 59, 5687 CrossRef CAS; (d) K. V. Gothelf and K. A. Jørgensen, Acta Chem. Scand., 1996, 50, 652 CrossRef CAS; (e) Sk. Ali Asrof, A. Hassan and M. I. M. Wazeer, J. Chem. Soc., Perkin Trans. 2, 1996, 1479 Search PubMed; (f) A. Hassan, M. I. M. Wazeer, H. P. Perzanowski and Sk. Ali Asrof, J. Chem. Soc., Perkin Trans. 2, 1997, 411 RSC; (g) A. Hassan, M. I. M. Wazeer and Sk. Ali Asrof, J. Chem. Soc., Perkin Trans. 2, 1998, 393 RSC.
- (a) L. W. Boyle, M. J. Peagram and G. H. Witham, J. Chem. Soc. B, 1971, 1728 RSC; (b) U. Chiaccio, A. Liguori, G. Romeo, G. Sindona and N. Uccella, Tetrahedron, 1992, 48, 9473 CrossRef; (c) U. Chiaccio, A. Liguori, G. Romeo, G. Sindona and N. Uccella, Heterocycles, 1993, 36, 799 CrossRef; (d) H. Agirbas and S. Guner, Heterocycl. Commun., 2000, 6, 557 CrossRef CAS; (e) N. K. Girdhar and M. P. S. Ishar, Tetrahedron Lett., 2000, 41, 7551 CrossRef CAS; (f) F. Heaney, O. Rooney, D. Cunningham and P. McArdle, J. Chem. Soc., Perkin Trans. 2, 2001, 373 RSC; (g) K. Pihlaja, P. Tahtinen, R. Shaikhutdinov, H. Hartikainen and V. Ovcharenko, J. Heterocycl. Chem., 2004, 41, 741 CrossRef CAS; (h) Y. Durust, C. Altug, J. Sinkkonen, O. Martiskainen and K. Pihlaja, J. Heterocycl. Chem., 2006, 43, 1267 CrossRef CAS; (i) H. Agirbas, S. Guner, F. Budak, S. Keceli, F. Kandemirli, N. Shvets, V. Kovalishyn and A. Dimoglo, Bioorg. Med. Chem., 2007, 15, 2322 CrossRef CAS.
- (a) K. Folting, W. N. Lipscomb and B. Jerslev, Acta Chem. Scand., 1963, 17, 238 CrossRef; (b) K. Koyano and H. Suzuki, Tetrahedron Lett., 1968, 15, 1859 CrossRef; (c) K. Koyano and H. Suzuki, Bull. Chem. Soc. Jpn., 1969, 42, 3306 CrossRef CAS; (d) J. S. Splitter, T.-M. Su, H. Ono and M. Calvin, J. Am. Chem. Soc., 1971, 93, 4075 CrossRef CAS; (e) D. P. Boyd, M. E. Stubbs, N. J. Thompson, H. J. C. Yeh, D. M. Jerina and R. E. Wasylichen, Org. Magn. Reson., 1980, 14, 528 CrossRef CAS; (f) P. DeShong, C. M. Dicken, R. R. Staib, A. J. Freyer and S. M. Weinreb, J. Org. Chem., 1982, 47, 4397 CrossRef CAS; (g) Y. Inouye, K. Takaya and H. Kakisawa, Magn. Reson. Chem., 1985, 23, 101l CrossRef; (h) S. Kanemasa, T. Uemura and E. Wada,, Tetrahedron Lett., 1992, 33, 7889 CrossRef CAS.
- (a) Y. D. Samuilov, S. E. Soloveva and A. I. Konovalov, Zh. Obsh. Khim., 1980, 50, 138 CAS; (b) A. Padwa, L. Fisera, K. F. Koehler, A. Rodriguez and G. S. K. Wong, J. Org. Chem., 1984, 49, 276 CrossRef CAS.
- (a) E. Stephan, Tetrahedron, 1975, 31, 1623 CrossRef CAS; (b) K. N. Houk, A. Bimanand, D. Mukherjee, J. Sims, Y. M. Chang, D. C. Kaufman and L. N. Domelsmith, Heterocycles, 1977, 7, 293 CrossRef CAS.
- R. Huisgen, Angew. Chem., Int. Ed. Engl., 1963, 2, 633 CrossRef.
- See the ESI† for general experimental information, data on solvent and temperature optimization, full experimental procedures and characterization data for all compounds..
- (a) E. P. Kündig and P. L. Timms, J. Chem. Soc., Chem. Commun., 1977, 912 RSC; (b) E. P. Kündig, Pure Appl. Chem., 1985, 57, 1855 CrossRef; (c) E. P. Kündig and M. Uemura, Kagakudojin, 1985, 105, 175 Search PubMed; (d) E. P. Kündig, N. P. Do Thi, P. Paglia, D. P. Simmons, S. Spichiger and E. Wenger in Organometallics in Organic Synthesis, A. de Meijere, H. E. tom Dieck, ed.; Springer Verlag: Berlin, 1987, p. 265 Search PubMed; (e) A. R. Pape, K. P. Kaliappan and E. P. Kündig, Chem. Rev., 2000, 100, 2917 CrossRef CAS; (f) E. P. Kündig and S. H. Pache in Science of Synthesis, T. Imamoto, Ed.; Thieme: Stuttgart/New York, 2008, Vol. 2, p. 155 Search PubMed; (g) Topics in Organometallic Chemistry, E. P. Kündig, Ed.; Springer: Berlin, 2004, Vol. 7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental information, data on solvent and temperature optimization, full experimental procedures and characterization data for all compounds. See DOI: 10.1039/c1ob06144e |
| This journal is © The Royal Society of Chemistry 2012 |