Luminescent lanthanide-binding peptides: sensitising the excited states of Eu(III) and Tb(III) with a 1,8-naphthalimide-based antenna†
Célia S.
Bonnet
*ab,
Marc
Devocelle
c and
Thorfinnur
Gunnlaugsson
*a
aSchool of Chemistry, Centre for Synthesis and Chemical Biology, Trinity College Dublin, Dublin, 2, Ireland. E-mail: gunnlaut@tcd.ie; Fax: +353 1 671 2826; Tel: +353 1 896 3459
bCentre de Biophysique Moléculaire, CNRS, Rue Charles Sadron, 45071, Orléans Cedex 2, France. E-mail: celia.bonnet@cnrs-orleans.fr
cCentre for Synthesis and Chemical Biology, Department of Pharmaceutical and Medicinal Chemistry, Royal College of Surgeons in Ireland, 123 St. Stephen's Green, Dublin, 2, Ireland
First published on 31st October 2011
Abstract
The investigation into the luminescence properties of a lanthanide-binding peptide, derived from the Ca-binding loop of the parvalbumin, and modified by incorporating a 1,8-naphthalimide (Naph) chromophore at the N-terminus is described. Here, the Naph is used as a sensitising antenna, which can be excited at lower energy than classical aromatic amino acids, such as tryptophan (the dodecapeptide of which was also synthesised and studied herein). The syntheses of the Naph antenna, its solid phase incorporation into the dodecapeptide, and the NMR investigation into the formation of the corresponding lanthanide complexes in solution is presented. We also show that this Naph antenna can be successfully employed to sensitize the excited states of both europium and terbium ions, the results of which was used to determined the stability constants of their formation complexes, and we demonstrated that our peptide ‘loop’ can selectively bind these lanthanide ions over Ca(II).
Introduction
The use of lanthanide(III) complexes in structural analysis,1–3 for probing supramolecular interactions,4,5 or in various biomedical applications5–9 is of great current interest in chemistry. This is in particularly due to the unique magnetic and photophysical properties that these ions possess.10Gd(III) complexes and conjugates have been employed as MRI relaxation contrast agents, while, with the exception of La(III) and Lu(III), most of the remaining lanthanides have unique luminescent properties, which have been explored in the development of various luminescent supramolecular devices. This includes the development of luminescent switches, sensors, logic gate mimics, imaging agents, etc. using ions such as Eu(III) and Tb(III), which emit in the visible ranges,11–13 whereas ions such as Nd(III), Yb(III) emit in the near infra-red; an area of particular interest for biological, and telecommunication applications.14–16 The main advantages of using these ions in the development of luminescent probes and devices is attributed to their long-lived excited states and line-like emission bands which occurs at long wavelengths. These features overcome interferences from short-lived background fluorescence and light scattering from biological matter.17 As these are Lapporte forbidden transitions, these excited states are most effectively populated by using sensitizing antennae, often embedded, or conjugated, via short spacers into macrocyclic structures.18 In particular, the use of polyaminocarboxylate based ligands, such as cyclen, has been widely investigated for such applications.19–27 In contrast, the use of peptides that can be synthetically modified with such sensitizing antennae, and possess donor moieties that can enable the binding of lanthanide ions, has been much less explored. Such systems could be of significant value, particularly for use in biological media, for instance, with the view of generating novel luminescent protein mimics, employed to investigate protein-protein or protein-substrate interactions.Due to the similar ionic radii of Ca(II), the lanthanides have been used as substitutes for Ca(II) ions in various Ca-binding peptides and proteins,28–31 or with the aim of developing luminescent peptides.32,33 For the latter, such structures usually possess amino acids with aromatic residues, such as tryptophan, tyrosine and phenylalanine, which can be used to sensitize the excited state of Tb(III).34 Recently, this area of research has attracted significant attention, particularly through the work of Imperiali et al., who has developed lanthanide-binding tags appended to various proteins (LBTs).34,35 These have been used to elucidate protein structure,3,36 detect protein-protein interactions and to phase X-ray crystallographic data.37 Some other examples within this area of research includes that of Vázquez et al.,38 who developed novel sensors for investigating protein-substrate interactions, Franklin et al.39–40 who investigated the DNA binding affinity and hydrolysis of such lanthanide based peptides. Recently, Ito et al.41 have also developed lanthanide complexes containing the cyclic peptide c(RGDfK) to visualize the α(v)β(3)-integrin-expressing tumor cells, while Delangle et al.42 have developed Gd(III)-based complexes of cyclic decapeptide with two independent faces as “regioselectively addressable functionalised templates” (RAFTs).
All the above examples have been formed by using natural occurring amino acids, and while these complexes are clearly useful for in vitro studies, there are intrinsic limitations for their use in vivo. Firstly, the stability of the lanthanide-peptide complexes formed are limited, as such peptides bear only simple binding groups such as carboxylates, phenolates, or amides. Secondly, only few of these amino acids can be employed as antennae. Consequently, modified amino acids have to be used to introduce strong donor groups for reinforcing metal complexation43,44 or to improve the luminescence properties.45,46
We have recently reported preliminary results for the use of 1,8-naphthalimide (Naph) structures to sensitize the excited states of both Eu(III) and Tb(III).47,48 This sensitizing antenna is particularly attractive as it can be excited at lower energy (345 nm), and by simple synthetic modifications, the photophysical properties can be easily tuned. Hence, we have employed this structure in our laboratory in various supramolecular systems and devices.49,50 Herein, we give full account of the use of this antenna within peptide structures designed to form stable complexes with lanthanide ions for use in biological applications.
The peptide sequence chosen for this investigation is shown in Fig. 1 as the dodecapeptide P1; designed on the Ca(II) binding loop of the parvalbumin protein, an amino acid sequence which includes three Glu and three Asp amino acids; each one able to provide a chelating carboxylate residues for binding to Ln(III). In P1, the Naph antenna, the synthesis of which is shown in Fig. 1, was incorporated at the N-terminus of the peptide. Furthermore, we also developed P2 for use in comparison studies. This system lacks the Naph antenna, but has instead, a Trp moiety that can be employed as such an antenna.
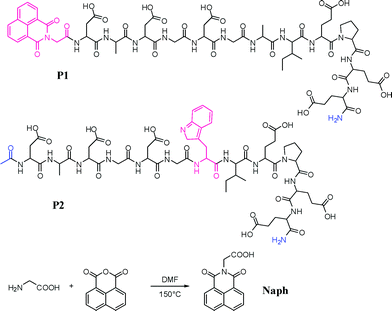 | ||
| Fig. 1 The two dodecapeptides P1 and P2 developed in this study and the synthesis of the Naph antenna that was incorporated into P1. | ||
Results and discussion
Syntheses of the dodecapeptides P1 and P2
The dodecapeptide sequence DADGDGWIEPEE of the model peptide P2 was synthesised using standard manual solid-phase synthesis based on Fmoc/tBu protection strategy.51 The peptide was cleaved off the resin and the side chains were deprotected using a mixture of TFA/TIS/H2O 95/2.5/2.5, and purified by reverse-phase HPLC, and characterised by Maldi-Tof MS (See ESI).The synthesis of the Naph derivative is shown in Fig. 1, and was formed in one step by reacting glycine (1.2 eq.) with naphthalic anhydride (1 eq.) in DMF at 150 °C over 24 h. The resulting brown oil was purified by precipitation from methanol to give a white solid in 56% yield (see Experimental section).
The peptide sequence DADGDGAIEPEE was synthesised using standard automated solid phase peptide synthesis according to the Fmoc/tBu strategy and HBTU-HOBt-DIEA coupling chemistry in NMP. The Naph antenna was then coupled manually at the N-terminus of the peptide sequence using a double coupling procedure. The resulting modified polymer-bound peptide Naph-Asp(tBu)-Ala-Asp(tBu)-Gly-Asp(tBu)-Gly-Ala-Ile-Glu(tBu)-Pro-Glu(tBu)-Glu(tBu) was then cleaved from the resin and the side chains were deprotected using TFA/thioanisole/TIS/EDT/H2O (v/v/v/v/v = 77/5.75/5.75/5.75/5.75), and P1 was finally purified by reverse-phase HPLC, and characterised by their ESMS (see ESI†).
Photophysical evaluation of P1 and P2
The photophysical properties of P1 and P2 were investigated in 10 mM HEPES buffer solution in the presence of 0.1 M NaCl, to maintain a constant ionic strength. The UV-Vis absorption, the fluorescence emission and the excitation spectra, were recorded both in the absence, as well as in the presence of one equivalent of either Eu(III) or Tb(III).The UV-vis absorption spectrum of P1 was dominated by a broad band centred at 344 nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε = 4.04) which was assigned to the n–π* transition of the Naph antenna (see Fig. S3, ESI†). Excitation into this band gave rise to a fluorescence emission typical of such Naph moieties, with λmax = 393 nm. In analogous manner, the Trp-antenna-based peptide P2 was analysed in buffered solution, and the UV-vis absorption spectrum showed a broad band with λmax at 280 nm, which had the typical Trp structure. Excitation of this band gave rise to the characteristic Trp emission centred at 334 nm.
ε = 4.04) which was assigned to the n–π* transition of the Naph antenna (see Fig. S3, ESI†). Excitation into this band gave rise to a fluorescence emission typical of such Naph moieties, with λmax = 393 nm. In analogous manner, the Trp-antenna-based peptide P2 was analysed in buffered solution, and the UV-vis absorption spectrum showed a broad band with λmax at 280 nm, which had the typical Trp structure. Excitation of this band gave rise to the characteristic Trp emission centred at 334 nm.
Upon addition of one equivalent of TbCl3 or EuCl3 to P1, no significant changes were seen in the UV-vis absorption spectra of P1, but the characteristic emission spectra of Eu(III) or Tb(III) ions were observed as shown in Fig. 2a and 2c respectively. This emission is most likely due to an energy transfer mechanism occurring from the Naph, to the Eu(III) and Tb(III) excited states. This was confirmed by recording the fluorescence excitation spectra of both systems in the presence of Eu(III) and Tb(III), by fixing the emission at 616 nm (5D4 → 7F2 transition) and 544 nm (5D0 → 7F5 transition), respectively (see ESI, Fig. S4–S5†). The excitation spectra were found to be structurally similar to the absorption spectra of these systems, confirming the successful energy transfer from these antennae to the Eu(III) and Tb(III) excited states. Additionally, in the case of Tb(III), a new band centred at 270 nm was observed and attributed to the formation of a 4f–5d transition (which could be MLCT in nature).52,53 In a similar manner, P2 was analysed and similar results were observed to those described above (see ESI, Fig. S7–S9†). As expected, P2 was also able to sensitise the Tb(III) excited state, and the relative intensity of the emission at 544 nm was significantly greater than that seen for P1. It is possible that this is partially due to closer proximity of this antenna to the lanthanide in comparison to P1, where the Naph antenna is located at the N-terminus. But there is also certainly a difference in the mechanism of the energy transfer to the Tb(III) excited state (5D4 = 20500 cm−1) which is thought to occur via the triplet state of the Trp (24830 cm−1),55 and which might occur through the singlet state of the Naph (27980 cm−1), because the triplet state is too low in energy (18540 cm−1).54 On the contrary, the relative intensity of Eu(III) luminescence at 616 nm was found to be significantly greater in the case of P1 than that seen for P2, Fig. 2a and 2b, despite the difference in the location of these two antennae within the peptide structures. Even though we did not quantify the efficiency of the energy transfer for these two systems, it is clear from these results that the Naph antenna can be successfully employed for populating the Eu(III) excited state, and that it does so more efficiently than Trp. This can be explained because the T1 energy of Naph is better suited for populating Eu(III) (5D0 = 17200 cm−1)9 than the T1 energy of Trp, and additionally in the case of Trp a non-radiatively deactivated LMCT could also be formed.
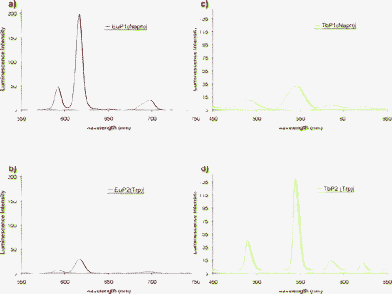 | ||
| Fig. 2 (a) Emission spectra of P1 (30 μM) in the presence of Eu(III) (17 μM) with excitation of Naph at 345 nm (slit width 5 nm for excitation and emission). (b) Emission spectra of P2 (30 μM) in the presence of Eu(III) (17 μM) with excitation of Trp at 280 nm (slit width 20 nm for excitation and 10 nm for emission). (c) Emission spectra of P1 (30 μM) in the presence of Tb(III) (17 μM) with excitation of Naph at 345 nm (slit width 20 nm for excitation and emission). (d) Emission spectra of P2 (30 μM) in the presence of Tb(III) (17 μM) with excitation of Trp at 280 nm (slit width 5 nm for excitation and 2.5 nm for emission). | ||
Photophysical titrations of P1 and P2 using Eu(III) and Tb(III)
We next investigated the formation of these lanthanide-based peptides by carrying out careful spectroscopic titration in solutions, where the fluorescence emission and the lanthanide emission spectra were monitored. A typical evolution of the time-resolved Eu(III) emission for the titration of P1, upon excitation of the antenna at 345 nm, is shown in Fig. 3. Here the characteristic line-like and narrow emission bands of the Eu(III) emission became apparent upon binding of the ion to the peptide; the binding of which was monitored by observing the changes in the Eu(III) ΔJ = 2 transition at 617 nm. The results are shown as an inset in Fig. 3. First, it should be pointed out that P1 displays the same behaviour than P2 towards Ln(III) complexation, where the metal-centred luminescence increased upon formation of the lanthanide peptide complex, within the use of one equivalent of the lanthanide ions. However, beyond the use of one equivalent, the Eu(III) emission reduced in intensity, see inset in Fig. 3. This was attributed to the formation of mononuclear and binuclear species, the latter resulting in a quenching of the luminescence intensity as previously observed.56–58 Indeed, upon further analysis of these changes using the non-linear regression analysis program SPECFIT, this hypothesis was confirmed, as these changes were best fitted to two step binding equilibria.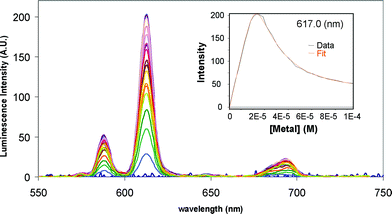 | ||
| Fig. 3 The overall changes in the Eu(III) emission upon titrating P1 (21 μM) with EuCl3 at pH 7.0 (10 mM HEPES) and in 0.1 M NaCl. Inset: corresponding titration profile at 617 nm and the best fit obtained using the non-linear regression analysis programme SPECFIT. | ||
The stability constants determined from the fitting of the changes in the Eu(III) emission of both P1 and P2 are summarized in Table 1. They show that both P1 and P2 give rise to similar binding constants for these ions. Unfortunately, due to the low solubility of the Tb2P1 species, we were unable to perform full titrations on this system, and, hence, unable to determine the Tb(III) affinity for P1. From Table 1, we can, however, conclude that the formation of the binuclear complexes is more difficult in terms of binding affinity than the formation of the mononuclear complexes. The stepwise stability constant is ca. 5 for all the systems; nearly two orders of magnitude lower than the formation of the mononuclear species. This difference can be explained by unfavourable electrostatic repulsion between the two lanthanide ions. This has previously been observed, even for systems with a large cavity, such as the one of a modified α-cyclodextrin (where the distance between the two Ln(III) ions is 5.74 Å in the solid state), and for which the stepwise formation constant of the binuclear species is nearly four orders of magnitude lower than the one of the mononuclear species.59 Concerning the mononuclear complex, the stability constants are of the same order of magnitude than the 17-amino acid peptide developed by Imperiali et al. (logβTb = 7.2) and optimised by screening methods, where the coordination sphere of Tb(III) is fully saturated, without any water molecule coordinated.35 It is also higher than the stability constants reported for natural Ca(II) binding loops in calmodulin, where the affinity was found to be 5.5 for Tb(III) and 6.2 for Eu(III).60 Finally, and possibly most importantly, the presence of the new sensitizer, the Naph antenna, does not significantly alter the affinity of the peptide sequence for Eu(III); being logβ = 6.8 for P1, compared to logβ = 6.75 for the unmodified version P2.
| Tb(III) | Eu(III) | ||
|---|---|---|---|
| P1 | logβ11 |
— | 6.8 (0.1) |
| logβ21 |
— | 11.8 (0.1) | |
| P2 | logβ11 |
6.8 (0.1) | 6.75 (0.08) |
| logβ21 |
11.9 (0.1) | 11.9 (0.1) |
In addition to explore the binding of the lanthanide ions by P1 and P2, we were also interested in assessing the selectivity of our system for these lanthanide ions with respect to endogenous cations such as Ca(II), Cu(II), and Zn(II). In order to do that, we chose the model compound P2 and we performed competition experiments starting with the Tb(III) complex Tb2P2 (typically, [Tb(III)] = 5 × [P2] at the start of the titration), and we monitored the decrease of the Tb(III) luminescence intensity upon addition of the desired cation. A complete quenching of the luminescence intensity was observed upon addition of 5 eq. of Cu2+, as is demonstrated in Fig. 4, whereas the addition of 1000 eq. of Ca(II) resulted in 60% of quenching (Fig. S10, ESI†), and the addition of 2000 eq. of Zn(II) resulted in less than 40% of quenching, making it impossible to determine a stability constant from these changes. For Cu(II), the best model to fit the data using SPECFIT involved the formation of mixed species between M(II)/Ln(III) and P2, whereas for Ca(II) only the formation of CaP2 was observed. The results are summarized in Table 2. The Ca(II) complex formed with P2 is clearly less stable than the one formed with native parvalbumin (logβCaP ∼ 8).61 This could be attributed to the modification made from the native Parvalbumin binding loop, and also from the fact that other residues, not strictly from the binding loop could contribute to the stability of the complex in the naturally occurring protein. We achieved a good selectivity for Ln(III) over Ca(II) as the stability of Ln(III) complexes is two order of magnitude higher than seen for the corresponding Ca(II) complex. Previous work in this field has shown that the Ln(III) complexes formed with the first calmodulin domain are one order of magnitude more stable than the corresponding Ca(II) complexes.60
| Ca(II) | Cu(II) | |
|---|---|---|
| logβ101 |
4.61 (0.05) | 7.53 (0.05) |
| logβ111 |
11.7 (0.1) |
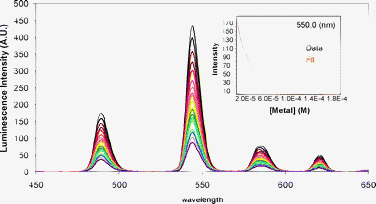 | ||
| Fig. 4 Evolution of the luminescence spectra of P2 (18.8 μM)-Tb(III) (75.2 μM) upon titration by CuCl2 and after excitation of Trp at 280 nm at pH 7.0 (10 mM HEPES) and in 0.1 M NaCl. Inset: The corresponding titration profile at 550 nm and the best fit obtained using SPECFIT. | ||
Elucidation of the structure of the metallo-peptides in solution
In order to gain a better insight into the structure of the metallo-peptides formed, we used circular dichroism experiments to monitor their formation. The CD spectra of P1, P1 with one and two eq. of Eu(III), P2, P2 with one and two eq. of Tb(III) are shown Fig. S11–S12 in ESI.† The same trend was observed for both systems upon binding to the lanthanides: a stronger negative band at 200 nm was observed for the free peptides, than for the metallo-peptides. This is characteristic of a random coil configuration in the absence of lanthanide ions.62 Moreover, additional changes were observed upon addition of the lanthanide ions such as the transition centred at 220 nm became more negative which is indicative of some folding interactions. However, we were unable to identify any typical β-sheet or α-helix signatures from these spectral changes, and no significant changes were observed that allowed us to distinguish between the mononuclear and the binuclear species in these spectra. Because of this, we thus decided to carry out NMR experiments on these systems to get more information on the structure of the resulting metallo-peptides.The NMR titration of both P1 and P2 were undertaken using the diamagnetic ion, La(III), and Eu(III) which is paramagnetic, in 90:10 H2O:D2O solutions. Selected spectra from these investigations are shown as stack-plots in Fig. 5. On all occasions, we only observed changes in the 1H NMR spectra up to the addition of 2 equivalents of metal ion added, with no significant changes occurring at higher equivalents. This confirms the formation of 1:1 and 2:1 complexes (see also Fig. S13, ESI†). In the case of P1, we could not determine the structure because in H2O/D2O solutions, the NH proton resonances were too broad, and becoming almost invisible when the pH was adjusted to 7. Although in the presence of La(III) the NH signals appear again (see Fig. S14, ESI†), they remained too broad for any further analysis. However, the NH-resonances were better resolved for P2 and its complexes, which allowed us to analyse those changes in more details. This difference between P1 and P2 is possibly due to a difference in dynamics between the two peptides in solution. For the titration of P2 with La(III) the addition of less than one equivalent of La(III) resulted in a broadening in the NMR spectra, suggesting the formation of a labile 1:1 complex, with dynamic equilibrium between the free and the bound form of P2. Upon addition of one equivalent of La(III), the spectrum sharpened significantly, making it possible to obtain a complete assignment of the resonances. To achieve this, we carried out t-ROESY experiments. A partial spectrum is shown in Fig. 6a, and demonstrates that while we did not observe enough long-range correlations to allow for a full structure determination, a correlation between the NH of Asp1 and the Hγ of Ile8 was observed. This suggests a folding of the P2peptide upon complexation with the La(III) ion, which further confirms the conformation changes observed in the CD spectra above. Further analysis of the chemical shifts of the different protons identified in these experiments, showed that the Hβ protons of the three Asp and the Hα protons of Trp and Ile were all shifted upon interaction of P2 with La(III). These results are summarised in Table 3, and the changes in the NMR spectra suggest a direct coordination of the three Asp in P2 to the La(III) ion, as well as the amide backbone of the Trp. Importantly, the coordination of the amide of the Trp in the position 7 of the Ca-binding loop is consistent with what has been observed for typical coordination of metal ions in EF hand motif for other peptide-Ln complexes both by X-ray in the solid state35 and by NMR in solution.40 We had previously determined the number of water molecules directly coordinated to the Ln(III) ion in the mononuclear complex by luminescence lifetime measurements. We found a q value of ∼3 for both P1 and P2 complexes,47 showing the coordinatively unsaturated nature of the lanthanide ions within P1 and P2. These results, together with the stability constant values confirm that the introduction of Naph does not affect the coordination sphere of the Ln(III). Moreover, these results are consistent with the direct coordination of three Asp, and suggest that possibly one or two of the Asp residues are coordinating to the ion in a bidentate manner in P2. Upon addition of a second equivalent of La(III) to P2 further shifts were observed for the side chains of two of the three Glu residues, suggesting that the second metal ion is coordinated within the Glu pocket of P2. This was further confirmed by the t-ROESY correlations, Fig. 6b, observed between the Hγ of Ile8 and the NH of Glu12, and between the Hγ of Ile8 and the C-terminal NH2. This is also an indication of the folding of the peptide at the C-terminus. It should be noted that a correlation is also observed between the NH of Glu9 and the CH3 at the N-terminus, confirming that we still have a folding of the N-terminus part of the peptide.
| P2 | P2 + 6 eq. NaOH | P2La | P2La2 | |
|---|---|---|---|---|
| Asp1 Hβ | 2.72–2.80 | 2.51–2.62 | 2.39–2.69 | 2.42–2.63 |
| Asp1 Hβ | 2.77–2.83 | 2.64 | 2.63–2.99 | 2.66–3.05 |
| Asp1 Hβ | 2.78 | 2.63 | 2.60–3.03 | 2.63–3.07 |
| Glu 9 Hβ | 1.93–2.05 | 1.88–2.25 | 1.89–2.18 | 1.77–2.20 |
| Hγ | 2.43 | |||
| Glu 11 Hβ | 1.84–2.00 | 1.95–2.22 | 2.00–2.25 | 2.03–2.24 |
| Hγ | 2.42 | 4.72 | ||
| Glu 12 Hβ | 1.93–2.09 | 1.92–2.24 | 2.20–2.41 | 2.20–2.41 |
| Hγ | 2.41 | |||
| Trp Hα | 4.60 | 4.58 | 4.90 | 4.95 |
| Ile Hα | 3.99 | 9.96 | 4.46 | 4.37 |
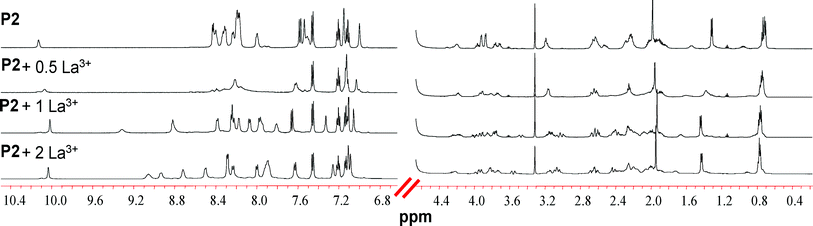 | ||
| Fig. 5 Partial 1H NMR spectra (600 MHz, 298 K) of the titration of P2 with La3+ in a 9/1 H2O/D2O solution. | ||
![Partial 1H t-ROESY spectra at 600 MHz, 298 K with (a) [P2] = [La] = 0.63 mM and (b) [P2] = [La]/2 = 0.63 mM.](/image/article/2012/OB/c1ob06567j/c1ob06567j-f6.gif) | ||
| Fig. 6 Partial 1H t-ROESY spectra at 600 MHz, 298 K with (a) [P2] = [La] = 0.63 mM and (b) [P2] = [La]/2 = 0.63 mM. | ||
Conclusions
In summary, we have developed a peptide, based on the Ca-binding loop of the parvalbumin, bearing a sensitising naphthalimide chromophore that can used to populate both the excited states Eu(III) and Tb(III). The Naph has been coupled to a glycine and then easily introduced at the N-terminus of the peptide using standard peptide solid-phase synthesis. The binding loop used and studied through the use of a similar peptide bearing a Trp chromophore does show a good selectivity for Ln(III) over Cu(II), Zn(II), and more importantly Ca(II). The presence of two coordination pockets composed of Asp and Glu, and corresponding to the formation of 1/1 and 2/1 Ln/P complexes respectively was evidenced. The introduction of the Naph antenna does not affect the stability of the lanthanide complexes formed with the peptide, and does not change the number of water molecules directly coordinated to the Ln(III) ions. This is a first step towards the optimisation of peptide- lanthanide complexes as the use of lower excitation energies than the natural amino acids can provide is compulsory for further possible in vivo applications.Experimental section
General
All chemicals were purchased at the purest grade commercially available and were used without further purification unless otherwise mentioned. Solvents used were HPLC grade unless otherwise mentioned. Dichloromethane for peptide synthesis was distilled prior to use. Water was purified with a Waters Milli-Q system to give a specific resistance <15 MΩ cm.Chromatographic analysis and purification were performed on a BioCAD SPRINT Perfusion Chromatography Workstation (PerSeptive Biosystems) using Phenomenex Gemini columns (5 Å, C18, 4.6mmd/250mmL (analytic) 100mmd:250mmL (semi-preparative), solvent A = H2O/TFA (v/v = 99.9/0.1), solvent B = CH3CN/TFA (v/v = 99.9/0.1), flow rate: 1 mL min−1 (analytical) 4.5 mL min−1 (semi-preparative). The gradient employed was 2 to 60% B in 18 column volumes with UV monitoring at 214 and 350 nm (for P1) or only 214 nm for P2.
The peptides were characterized by Matrix Assisted Laser Desorption Ionisation- Time Of Flight- Mass Spectroscopy (α-cyano-4-hydroxy-cinnamic acid matrix).
Mass spectrum of Naph was determined using electrospray on a Micromass LCT spectrometer. High-resolution mass spectra were determined relative to a standard of leucine enkephaline.
Synthesis
13C NMR (DMSO-d6, 150 MHz), δ (ppm): 169.2 (COOH), 163.0 (CO), 134.9, 131.4, 131.1, 127.36, 127.33, 121.5 (Car), 41.1 (CH2). ESI-MSm/z: [L + Na]+ calculated for C14H9NO4Na: 278.04; Found, 278.05.
Preparation of liquid samples
The peptide concentration was systematically determined by measuring the UV-absorption of the tryptophan residue for P2 (λmax = 280 nm, εmax = 5690 cm−1 M−1), and the Naph residue for P1 (λmax = 345 nm, εmax = 11500 cm−1 M−1). Metal stock solutions were prepared by dissolving LnCl3·6H2O or LnOTf3 in H2O or D2O. Their exact Ln3+ concentrations were systematically determined by colorimetric titration in acetate buffer (pH = 4.5) using standardized H2Na2edta solution and xylenol orange as an indicator,63 except for Ca(II) solutions, which were titrated at pH 12 using Calcon as an indicator.
Luminescence titrations
Solutions were all made at pH = 7.0 in 10 mM HEPES buffer, 0.1 M NaCl. UV-Vis spectra were recorded on a Varian UV-Vis spectrophotometer from 250 to 500 nm using excitation and emission slit width of 1 nm with a medium scan speed. Luminescence and lifetimes were measured on a Varian Cary Eclipse Fluorescence spectrophotometer following an excitation at 345 nm for P1, and at 280 nm for P2. The following settings were as follow: for fluorescence mode: PMT detector: 600 mV, scan speed: 600 nm min−1, averaging time: 0.1 s, data interval: 1 nm. For phosphorescence mode: delay time: 0.1 ms, flash count: 1, PMT detector: 1000 mV, data interval: 1 nm, total decay time: 0.02 s, gate time: 10 ms.Spectra were analysed by using the program SPECFIT, which employs a singular value decomposition algorithm and refines the data according to a global least-squares analysis procedure.64,65 The values obtained are the average of at least three titrations.
Terbium and europium luminescence lifetimes were measured by recording the decay of the emission intensity at 545 nm and 616 nm, respectively, after excitation at 345 nm for P1 and 280 nm for P2. It was also checked that direct excitation at 368 nm and 396 nm give the same results. The lifetimes were measured in such conditions that only the 1/1 complex is present in solution. The signals were analyzed as single-exponential decays. As the peptide is highly hydrated, the lifetimes were measured for different H2O/D2O ratio ranging from 0.2/0.8 to 1/0, and the lifetime in D2O was extrapolated from the measured values.
NMR experiments
All the NMR experiments were performed on a Bruker Avance II 600 MHz spectrometer equipped with a 5 mm TCI cryoprobe. The spectra were recorded at 298 K in H2O/D2O (v/v, 95/5) using watergate W5 when necessary66 with a bandwidth of 7200 Hz and a power of 4.05 dB for the Watergate pulse. 2D NMR spectra were acquired in phase-sensitive mode with TPPI for quadrature detection in the indirect dimension using 2048×128 matrices. TOCSY experiments were performed by using a MLEV-17 spin-lock sequence with a mixing time of 70 ms. Off-resonance ROESY experiments were recorded with a mixing time of 300 ms (5000 Hz spin-lock). The spectra of P2 were recorded at 0.63 mM. A titrated solution of NaOH was added and spectra were recorded up to 6 equivalents of NaOH added with respect to P2. A titrated solution of Ln(III) (La or Eu) was then added and spectra were recorded up to 4 equivalents of metal ion with respect to P2. The same procedure was followed with P1 and the corresponding spectra were recorded with a concentration of P1 of 0.20 mM.Circular dichroism
CD spectra were recorded at 25 °C on a JASCO's J-810 dichrograph in a cell with a 0.1 cm path length. The peptide concentration was 100 μM in 10 mM HEPES, NaCl 0.1 M at pH 7.4. All spectra were obtained from 260 to 180 nm at intervals of 1 nm, a band width of 2 nm. The contribution of the buffer was always subtracted and three spectra were averaged for each sample. CD spectra are reported in molar ellipticity per α-amino acid residue.Acknowledgements
We particularly like to thank Science Foundation Ireland (SFI) for RFP 2008, RFP 2009 funding and SFI (grant 06/RFP/CHO024/EC07) for the acquisition of the peptide synthetizer, and PRTLI for Cycle 4 funding. We would like to thank particularly Dr John O'Brien (TCD), Dr Dilip Rai (CSCB, UCD), and Céline Petit and Stéphane Desgranges (CSCB, RCSI) for their help.Notes and references
- L. Di Bari and P. Salvadori, Coord. Chem. Rev., 2005, 249, 2854 CrossRef CAS.
- E. Pazos, O. Vazquez, J. L. Mascarenas and M. Eugenio Vazquez, Chem. Soc. Rev., 2009, 38, 3348 RSC.
- J. Wohnert, K. J. Franz, M. Nitz, B. Imperiali and H. Schwalbe, J. Am. Chem. Soc., 2003, 125, 13338 CrossRef.
- B. R. Sculimbrene and B. Imperiali, J. Am. Chem. Soc., 2006, 128, 7346 CrossRef CAS.
- C. S. Bonnet and E. Toth, Am. J. Neuroradiol., 2009, 31, 401 CrossRef.
- (a) C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS; (b) P. Caravan, J. J. Ellison, T. J. McMurry and R. B. Lauffer, Chem. Rev., 1999, 99, 2293 CrossRef CAS.
- A. E. Merbach and E. Toth, The chemistry of contrast agents, Wiley, New York, 2001 Search PubMed.
- S. Pandya, J. H. Yu and D. Parker, Dalton Trans., 2006, 2757 RSC.
- J. C. G. Bunzli, Acc. Chem. Res., 2006, 39, 53 CrossRef.
- (a) J. P. Leonard, C. B. Nolan, F. Stomeo and T. Gunnlaugsson, Top. Curr. Chem., 2007, 281, 1 CrossRef CAS; (b) T. Gunnlaugsson and J. P. Leonard, Chem. Commun., 2005, 3114 RSC; (c) D. Parker, Chem. Soc. Rev., 2004, 33, 156 RSC.
- (a) J. Massue, S. J. Quinn and T. Gunnlaugsson, J. Am. Chem. Soc., 2008, 130, 6900 CrossRef CAS; (b) C. S. Bonnet, J. Massue, S. J. Quinn and T. Gunnlaugsson, Org. Biomol. Chem., 2009, 7, 3074 RSC; (c) N. S. Murray, S. P. Jarvis and T. Gunnlaugsson, Chem. Commun., 2009, 4959 RSC.
- (a) C. S. Bonnet and T. Gunnlaugsson, New J. Chem., 2009, 33, 1025 RSC; (b) S. J. A. Pope and R. H. Laye, Dalton Trans., 2006, 3108 RSC; (c) S. J. A. Pope, B. P. Burton-Pye, R. Berridge, T. Khan, P. J. Skabara and S. Faulkner, Dalton Trans., 2006, 2907 RSC.
- C. P. Montgomery, B. S. Murray, E. J. New, R. Pal and D. Parker, Acc. Chem. Res., 2009, 42, 925 CrossRef CAS.
- L. Pellegatti, J. Zhang, B. Drahos, S. Villette, F. Suzenet, G. Guillaumet, S. Petoud and E. Toth, Chem. Commun., 2008, 6591 RSC.
- M. Andrews, J. E. Jones, L. P. Harding and S. J. A. Pope, Chem. Commun., 2011, 47, 206 RSC.
- J. C. G. Bunzli and S. V. Eliseeva, J. Rare Earths, 2010, 28, 824 CrossRef CAS.
- C. M. G. dos Santos, A. J. Harte, S. J. Quinn and T. Gunnlaugsson, Coord. Chem. Rev., 2008, 252, 2512 CrossRef CAS.
- J. C. G. Bunzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048 RSC.
- (a) S. E. Plush and T. Gunnlaugsson, Dalton Trans., 2008, 3801 RSC; (b) S. E. Plush and T. Gunnlaugsson, Org. Lett., 2007, 9, 1919 CrossRef CAS; (c) M. Cantuel, C. Lincheneau, T. Buffeteau, L. Jonusauskaite, T. Gunnlaugsson, G. Jonusauskas and N. D. McClenaghan, Chem. Commun., 2010, 46, 2486 RSC; (d) J. P. Leonard, C. M. G. Dos Santos, S. E. Plush, T. McCabe and T. Gunnlaugsson, Chem. Commun., 2007, 129 RSC; (e) J. P. Leonard, P Jensen, T. McCabe, J. E. O'Brien, R. D. Peacock, P. E. Kruger and T. Gunnlaugsson, J. Am. Chem. Soc., 2007, 129, 10986 CrossRef CAS; (f) T. Gunnlaugsson, J. P. Leonard, K. Senechal and A. J. Harte, J. Am. Chem. Soc., 2003, 125, 12062 CrossRef CAS; (g) T. Gunnlaugsson and J. P. Leonard, Dalton Trans., 2005, 3204 RSC.
- K. Sénéchal-David, S. J. A. Pope, S. Quinn, S. Faulkner and T. Gunnlaugsson, Inorg. Chem., 2006, 45, 10040 CrossRef.
- B. McMahon, P. Mauer, C. P. McCoy, T. C. Lee and T. Gunnlaugsson, J. Am. Chem. Soc., 2009, 131, 17542 CrossRef CAS.
- R. Pal and D. Parker, Org. Biomol. Chem., 2008, 6, 1020 CAS.
- F. Kielar, C. P. Montgomery, E. J. New, D. Parker, R. A. Poole, S. L. Richardson and P. A. Stenson, Org. Biomol. Chem., 2007, 5, 2975 CAS.
- J. Yu, D. Parker, R. Pal, R. A. Poole and M. J. Cann, J. Am. Chem. Soc., 2006, 128, 2294 CrossRef CAS.
- R. J. Aarons, J. K. Notta, M. M. Meloni, J. Feng, R. Vidyasagar, J. Narvainen, S. Allan, N. Spencer, R. A. Kauppinen, J. S. Snaith and S. Faulkner, Chem. Commun., 2006, 909 RSC.
- S. J. A. Pope, B. J. Coe, S. Faulkner, E. V. Bichenkova, X. Yu and K. T. Douglas, J. Am. Chem. Soc., 2004, 126, 9490 CrossRef CAS.
- W. S. Perry, S. J. A. Pope, C. Allain, B. J. Coe, A. M. Kenwright and S. Faulkner, Dalton Trans., 2010, 39, 10974 RSC.
- P. J. Breen, E. K. Hild and W. D. Horrocks, Biochemistry, 1985, 24, 4991 CrossRef CAS.
- P. J. Breen, K. A. Johnson and W. D. Horrocks, Biochemistry, 1985, 24, 4997 CrossRef CAS.
- M. J. Rhee, D. R. Sudnick, V. K. Arkle and W. D. Horrocks, Biochemistry, 1981, 20, 3328 CrossRef CAS.
- C. W. am Ende, H. Y. Meng, M. Ye, A. K. Pandey and N. J. Zondlo, ChemBioChem, 2010, 11, 1738 CrossRef CAS.
- J. P. Macmanus, C. W. Hogue, B. J. Marsden, M. Sikorska and A. G. Szabo, J. Biol. Chem., 1990, 265, 10358 CAS.
- C. W. V. Hogue, J. P. Macmanus, D. Banville and A. G. Szabo, J. Biol. Chem., 1992, 267, 13340 CAS.
- M. Nitz, K. J. Franz, R. L. Maglathlin and B. Imperiali, ChemBioChem, 2003, 4, 272 CrossRef CAS.
- M. Nitz, M. Sherawat, K. J. Franz, E. Peisach, K. N. Allen and B. Imperiali, Angew. Chem., Int. Ed., 2004, 43, 3682 CrossRef CAS.
- L. J. Martin, M. J. Hahnke, M. Nitz, J. Wohnert, N. R. Silvaggi, K. N. Allen, H. Schwalbe and B. Imperiali, J. Am. Chem. Soc., 2007, 129, 7106 CrossRef CAS.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114 CrossRef CAS.
- E. Pazos, D. Torrecilla, M. Vazquez Lopez, L. Castedo, J. L. Mascarenas, A. Vidal and M. E. Vazquez, J. Am. Chem. Soc., 2008, 130, 9652 CrossRef CAS.
- R. T. Kovacic, J. T. Welch and S. J. Franklin, J. Am. Chem. Soc., 2003, 125, 6656 CrossRef CAS.
- J. T. Welch, W. R. Kearney and S. J. Franklin, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3725 CrossRef CAS; S. W. Wong-Deyrup, Y. Kim and S. J. Franklin, JBIC, J. Biol. Inorg. Chem., 2005, 11, 17 CrossRef.
- T. Ito, M. Inoue, K. Akamatsu, E. Kusaka, K. Tanabe and S. Nishimoto, Bioorg. Med. Chem. Lett., 2011, 21, 3515 CrossRef CAS.
- C. S. Bonnet, P. H. Fries, S. Crouzy, O. Seneque, F. Cisnetti, D. Boturyn, P. Dumy and P. Delangle, Chem.–Eur. J., 2009, 15, 7083 CrossRef CAS.
- F. Cisnetti, C. Gateau, C. Lebrun and P. Delangle, Chem.–Eur. J., 2009, 15, 7456 CrossRef CAS.
- F. Cisnetti, C. Lebrun and P. Delangle, Dalton Trans., 2010, 39, 3560 RSC.
- A. M. Reynolds, B. R. Sculimbrene and B. Imperiali, Bioconjugate Chem., 2008, 19, 588 CrossRef CAS.
- I. D. Clark, I. Hill, M. Sikorskawalker, J. P. Macmanus and A. G. Szabo, FEBS Lett., 1993, 333, 96 CrossRef CAS.
- C. S. Bonnet, M. Devocelle and T. Gunnlaugsson, Chem. Commun., 2008, 4552 RSC.
- (a) 1,8-naphthalimides have also been used as antenna for Eu(III) by: D. R. Kauffman, C. M. Shade, H. Uh, S. Petoud and A. Star, Nat. Chem., 2009, 1, 500 CrossRef CAS; (b) M. de Sousa, M. Kluciar, S. Abad, M. A. Miranda, B. de Castro and U. Pischel, Photochem. Photobiol. Sci., 2004, 3, 639 RSC.
- (a) G. J. Ryan, S. Quinn and T. Gunnlaugsson, Inorg. Chem., 2008, 47, 401 CrossRef CAS; (b) R. B. P. Elmes and T. Gunnlaugsson, Tetrahedron Lett., 2010, 51, 4082 CrossRef CAS.
- (a) E. B. Veale and T. Gunnlaugsson, J. Org. Chem., 2010, 75, 5513 CrossRef CAS; (b) E. B. Veale, D. O. Frimannsson, M. Lawler and T. Gunnlaugsson, Org. Lett., 2009, 11, 4040 CrossRef CAS.
- R. Sheppard, J. Pept. Sci., 2003, 9, 545 CrossRef CAS.
- S. F. A. Kettle, in Physical Inorganic Chemistry: A coordination chemistry approach, Oxford University Press, 1996 Search PubMed.
- J.-C. G. Bünzli, S. Comby, A.-S. Chauvin and C. D. B. Vandevyver, J. Rare Earths, 2007, 25, 257 CrossRef.
- V. Wintgens, P. Valat, J. Kossanyi, L. Biczok, A. Demeter and T. Berces, J. Chem. Soc., Faraday Trans., 1994, 90, 411 RSC.
- I. Vaya, C. Jimenez and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 9363 CrossRef CAS.
- F. Tanaka and T. Ishibashi, J. Chem. Soc., Faraday Trans., 1996, 92, 1105 RSC.
- L. L. Liu and K. J. Franz, J. Am. Chem. Soc., 2005, 127, 9662 CrossRef CAS.
- L. S. Natrajan, A. J. Blake, C. Wilson, J. A. Weinstein and P. L. Arnold, Dalton Trans., 2004, 3748 RSC.
- C. Bonnet, A. Gadelle, J. Pecaut, P. H. Fries and P. Delangle, Chem. Commun., 2005, 625 RSC.
- L. Le Clainche, G. Plancque, B. Amekraz, C. Moulin, C. Pradines-Lecomte, G. Peltier and C. Vita, J. Biol. Inorg. Chem., 2003, 8, 334 CAS.
- J. Haiech, J. Derancourt, J. F. Pechere and J. G. Demaille, Biochemistry, 1979, 18, 2752 CrossRef CAS.
- N. Sreerama and R. W. Woody, in Circular Dichroism: Principles and Applications, ed. N. Berova, K. Nakanishi and R. W. Woody, Wiley, New York, 2000 Search PubMed.
- S. Y. Shetty and R. M. Sathe, Talanta, 1976, 23, 46 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbuhler, Talanta, 1985, 32, 1133 CrossRef CAS.
- H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbuhler, Talanta, 1985, 32, 95 CrossRef CAS.
- M. L. Liu, X. A. Mao, C. H. Ye, H. Huang, J. K. Nicholson and J. C. Lindon, J. Magn. Reson., 1998, 132, 125 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ob06567j |
| This journal is © The Royal Society of Chemistry 2012 |