Developing asymmetric iron and ruthenium-based cyclone complexes; complex factors influence the asymmetric induction in the transfer hydrogenation of ketones†
Jonathan P.
Hopewell
,
José E. D.
Martins
,
Tarn C.
Johnson
,
Jamie
Godfrey
and
Martin
Wills
*
Department of Chemistry, The University of Warwick, Coventry, UK CV4 7AL. E-mail: m.wills@warwick.ac.uk; Fax: (+44) 24 7652 3260; Tel: (+44) 24 7652 4112
First published on 1st September 2011
Abstract
The preparation of a range of asymmetric iron and ruthenium-cyclone complexes, and their application to the asymmetric reduction of a ketone, are described. The enantioselectivity of ketone reduction is influenced by a single chiral centre in the catalyst, as well as by the planar chirality in the catalyst. This represents the first example of asymmetric ketone reduction using an iron cyclone catalyst.
Introduction
The ruthenium complex 1 (the Shvo catalyst)1–4 reversibly splits to give hydride 2 and the unsaturated species 3.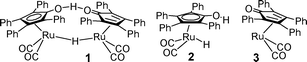
By ‘shuttling’ between 2 and 3, the Shvo catalyst transfers pairs of hydrogen atoms between secondary alcohols and ketones and has been used to good effect in dynamic kinetic resolution (DKR) reactions of alcohols and amines.2,3 There is evidence,4 largely based on kinetic isotope effects, that the hydrogen transfer to ketones and aldehydes, by the Shvo catalyst, takes place via a concerted ‘outer sphere’ mechanism (Fig. 1a). This is analogous to that of ketone reduction by the Noyori catalyst 4 (Fig. 1b).5
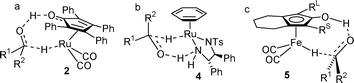 | ||
| Fig. 1 Comparison of mechanisms of hydrogen transfer by a) ruthenium hydride 2, b) Noyori catalyst 4 and c) iron hydride 5. In c, the potential for substituents RL (large) and RS (small) to influence the enantioselectivity is illustrated. | ||
The Shvo catalyst is also an efficient ketone reducing agent when using an excess of an alcohol (usually iPrOH) or formic acid as hydrogen source,1d and can also catalyse hydrogenation reactions.1b,c,4d,6 The closely related iron complex 5 has recently been prepared from the tricarbonyl precursor 67 and employed in catalytic reduction reactions of ketones by Casey and Guan.8 The mechanism appears to be analogous to that of the Shvo catalyst 2 (Fig. 1c). In recent studies, complex 5 has been applied to the oxidation of alcohols using acetone as an acceptor, and a number of its derivatives have been reported and evaluated in this role.9 In our own studies,10 we reported the synthesis and applications of racemic complexes 7a–7g in alcohol oxidations. The complexes were formed by an intramolecular cyclisation from a linear dialkyne precursor 8, followed by diastereoisomer separation.7,11
However, given their proposed mechanism for reduction of ketones, we reasoned that asymmetric derivatives of 7b–7g complexes should be capable of enantioselective ketone reduction reactions. Asymmetric induction would be predicted to be achieved by the steric and/or electronic effects of the groups flanking the central ‘C–O’ bond of the cyclone ligand (Fig. 1c).
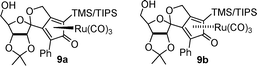
Since the start of our studies in this area, Yamamoto has reported the synthesis of the asymmetric Shvo-type catalyst precursors 9a/9b, which are capable of asymmetric hydrogenation (35 atm H2) of acetophenone in 14–21% ee and up to 100% conversion.12a As far as we are aware this is the first example of an asymmetric Shvo-type catalyst, although Berkessel has very recently published a closely related system based on chiral-at-Fe complexes which catalysed pressure hydrogenation (i.e. with H2 gas) of acetophenone in up to 31% ee.12b Despite the difference in planar chirality, it was found that the same enantiomer of alcohol product (R) was formed by both 9a and 9b. Herein we disclose our own results in the area of asymmetric Shvo-type reduction catalysts using both iron- and ruthenium-based cyclone catalysts in asymmetric transfer hydrogenation (ATH) reactions (Table 1).
|
|
||||
|---|---|---|---|---|
| Complex | Conditions | Time/h | Conv/%b (alcohol) | Ee (R/S) |
| a 10 mol% iron catalyst was used, with 10 mol% Me3NO. b form. = formate byproduct. c ee was 20% (R) at 10 h and decreased with time. d Reactions at 28 °C, 1 M with 7c; 48h, 5% conv, 10% ee (R), 7f; 96h, 2% conv, 29% ee, 7g; 96 h, trace conversion. | ||||
| 7a | FA/TEA 28 °C 1 M | 18 | 60 (<1 form) | n/a |
| 7a | FA/TEA 40 °C 1 M | 18 | 90 (+2 form) | n/a |
| 7a | FA/TEA 60 °C 1 M | 18 | 89 (+10 form.) | n/a |
| 7a | iPrOH 28 °C 0.2 M | 18 | 6.8 | n/a |
| 7a | iPrOH 60 °C 0.2 M | 18 | 52 | n/a |
| 7b | FA/TEA 28 °C 1 M | 48 | 25 | 15 (R) |
| 7b | FA/TEA 40 °C 1 M | 48 | 36 | 10 (R) |
| 7c | FA/TEA 40 °C 1 M | 96 | 10 | 10 (R) |
| 7d | FA/TEA 28 °C 1 M | 48 | 40 | 25 (R) |
| 7d | FA/TEA 28 °C 1 M | 96 | 69 | 23 (R) |
| 7e | FA/TEA 28 °C 1 M | 48 | 46 | 11 (R) |
| 7d | FA/TEA 40 °C 1 M | 96 | 80 (+5 form) | 23 (R) |
| 7e | FA/TEA 40 °C 1 M | 48 | 91 (+5 form.) | 11 (R) |
| 7f | FA/TEA 40 °C 1 Md | 96 | 66 (+10 form.) | 25 (R) |
| 7g | FA/TEA 40 °C 1 Md | 96 | 17 | 5 (R) |
| 12a | FA/TEA 60 °C 1.6 M | 160 | 50 | 12 (R)c |
| 12a | iPrOH 60 °C 0.18 M | 160 | 28 | 11 (R) |
| 12b | FA/TEA 60 °C 1.6 M | 160 | 61 | 3 (R) |
| 12b | iPrOH 60 °C 0.18 M | 160 | 35 | 4 (R) |
| 12c | FA/TEA 60 °C 1.6 M | 150 | 48 | 5 (R) |
| 12c | iPrOH 60 °C 0.18 M | 150 | 58 | 3 (R) |
| 12e | FA/TEA 60 °C 1.6 M | 18 | 31 | 17 (R) |
| 12e | iPrOH 60 °C 0.18 M | 18 | 13 | 11 (R) |
| 13a/b | FA/TEA 60 °C 1.6 M | 150 | 19 | 15 (S) |
| 13a/b | iPrOH 60 °C 0.18 M | 150 | 20 | 5 (S) |
| 16 | FA/TEA 60 °C 1.6 M | 168 | 35 (+5 form.) | n/a |
| 16 | iPrOH 60 °C 0.18 M | 184 | 17 | n/a |
| 17 | FA/TEA 60 °C 1.6 M | 168 | 80 (+7 form.) | n/a |
| 17 | iPrOH 60 °C 0.18 M | 168 | 90 | n/a |
| 18 | FA/TEA 60 °C 1.6 M | 168 | 12.5 | n/a |
| 18 | iPrOH 60 °C 0.18 M | 168 | 21 | n/a |
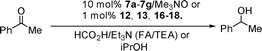
Racemic complex 7a10 was first tested and was found to work efficiently in a formic acid/triethylamine (FA/TEA) system, as commonly used in ATH reactions (Table 1).5 At low temperature only a trace of formate byproduct was observed. Raising the temperature to 60 °C resulted in essentially complete reduction although some formate co-product was formed. The use of 10 mol% catalyst was required, along with 10 mol% of trimethylamine N-oxide to initiate hydride formation.9b,10,13 An inferior result was observed in iPrOH.5 Enantiopure complexes 7b–7g were prepared (Scheme 1) from alcohol (R)-11a (made in 96% ee by reduction of the precursor ketone 10a by ATH using an established catalyst14). Elaboration of (R)-11a following the reported route10 gave in each case (i.e. from 8b–d where R1 = TBDMS, TMS and Ph respectively) two enantiomerically-enriched complexes which were separated by chromatography on silica gel. In our previous studies on the racemic series, we had established the relative configuration of the chiral centres via an X-ray crystallographic solution of 7c; the other iron complexes are assigned by analogy with 7b/c.10 The enantiomeric purity of catalyst 7d was established using a shift reagent and was established to be ca. 92% ee, indicating a small loss of ee relative to the alcohol but a high enough level to be meaningful in these investigations (see supporting information†). Conversions were in all cases measured by integrating the product peaks in the GC against the starting material peaks.
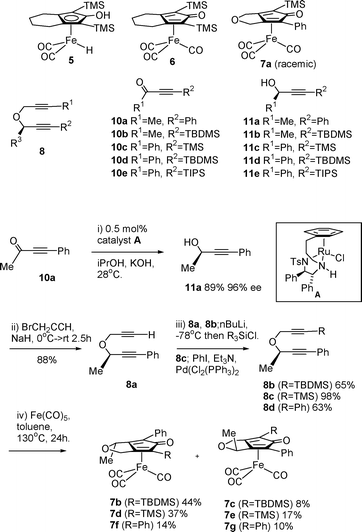 | ||
| Scheme 1 Synthesis of iron catalysts 7b–7g. | ||
The new catalysts were tested in the ATH of acetophenone (Table 1). In the case of 7b/c, the catalyst proved to be of low activity and a reduction product of low ee was formed in the same major enantiomeric form. Each purified, enantiomerically-pure diastereoisomer of 7d/e was found to be a more effective catalyst for ketone reduction in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 FA/TEA, giving almost complete transformation within 48–96 h at 40 °C. Again, both gave reduction products of the same absolute configuration, mirroring the activity of the ruthenium complexes reported by Yamamoto.12 More interestingly, catalysts 7f/g, which bear identical (phenyl) groups flanking the central C
:2 FA/TEA, giving almost complete transformation within 48–96 h at 40 °C. Again, both gave reduction products of the same absolute configuration, mirroring the activity of the ruthenium complexes reported by Yamamoto.12 More interestingly, catalysts 7f/g, which bear identical (phenyl) groups flanking the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O function in the cyclone, also gave the same alcohol enantiomers. The chiral centre on the backbone of the cyclone clearly has a significant effect on the catalyst enantioselectivity, possibly due to an influence on the conformation of the phenyl rings. Although the ees are low, the isomer with the methyl group positioned proximal to the Fe(CO)3 group gave a higher ee in each case (7b,d,fvs.7c,e,g respectively). The effect may arise via an influence on the positions of the CO ligands which subsequently communicates to the reduction transition state.
O function in the cyclone, also gave the same alcohol enantiomers. The chiral centre on the backbone of the cyclone clearly has a significant effect on the catalyst enantioselectivity, possibly due to an influence on the conformation of the phenyl rings. Although the ees are low, the isomer with the methyl group positioned proximal to the Fe(CO)3 group gave a higher ee in each case (7b,d,fvs.7c,e,g respectively). The effect may arise via an influence on the positions of the CO ligands which subsequently communicates to the reduction transition state.
Monitoring the reductions over time indicated that the ees did not change significantly over time, even after several days (e.g.7d after 48 and 96 h), indicating that product racemisation is not taking place. Although the conversions appear to level out over time, it is at present unclear as to whether this may be due to catalyst deactivation or a solvent effect. Formic acid/triethylamine was used in the traditional azeotrope ratio of 5:2 throughout. A ratio of 1:1 FA:TEA gave a conversion of 25% in 96 h and 21% ee when 7d was used as catalyst. A 1:2 FA:TEA mixture was not homogeneous, whilst a 5:1 FA:TEA mixture gave only 11% conversion after 96h, in 26% ee. These results indicate that the FA:TEA ratio effects the rate but not the ee of the reaction, with 5:2 being the best ratio of those tested.
As far as we are aware, this represents the first application of any iron cyclone complex to the asymmetric reduction of ketones by transfer hydrogenation,12 and also reveals an unusual effect of a backbone chiral centre on the observed enantiocontrol.
We also wished to establish whether the analogous ruthenium-based catalysts would exhibit a similar pattern of enantioselectivity. To this end, complexes 12a–12f were prepared and evaluated, using the corresponding diyne precursors.15
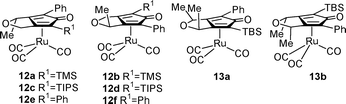
Alcohol (R)-11a (96% ee) was converted to the three derivatives 12a–12f following the precedent for the iron complexes10 but with the use of Ru3(CO)12 in the complexation step.15 Complexes 12a/b, were formed as a mixture of two separable diastereoisomers of product in 27 and 12% isolated yields respectively. Although the relative position of the Me group has not been established, we have assumed that, in analogy with the iron series, that the major enantiomer is that in which the Me on the backbone is proximal to the Ru(CO)3 group. Both 12a and 12b were effective in catalysing the reduction of acetophenone, again in the same absolute sense (Table 1). Unlike the Fe series, there was evidence of racemisation during extended reaction times; for example in the case of 12a, the ee was 20% after 10 h. For the Ru complexes, in situ formation of the hydride using NaOH in THF followed by phosphoric acid was completed prior to the reduction,7a and Me3NO was not required. The ruthenium catalysts were also used at lower loadings (1 mol%) than the iron complexes (10 mol%). In the case of 12c/d, only one isomer was isolated, and in low yield, from the cyclisation, and was assumed to be of the configuration shown in 12c. This complex promoted reduction but in poor enantioselectivity.
Complexes 12e/f have two identical groups flanking the central CO group, analogous to 7c/d. The cyclisation to form 12e/f was achieved in low yield, however only one diastereoisomer, assumed to be 12e, could be isolated. This was capable of reduction of acetophenone in up to 17% ee (R), which, although low, confirms that for the Ru catalysts as well as the iron ones, planar asymmetry is not required for the asymmetric reduction. In order to eliminate the effect of one backbone chiral centre, complexes 13a/b were selected for study – in this case containing two methyl groups in a cis arrangement. This was prepared by reacting alcohol (R)-11b (see below) with mesylated (R)-11a, resulting in inversion of configuration. Cyclisation of the resulting diyne with Ru3(CO)12 gave a mixture from which one isomer of 13 was isolated in pure form. Although the relative positions of the methyl groups to the Ru(CO)3 centre are not known, this catalyst will rely on planar chirality alone in the reduction reaction. The result was that acetophenone was reduced in up to 15% ee, and in this case unexpected (S) configuration (Table 1). Taken together, these results indicate that asymmetric induction by cyclone catalysts arises from a combination of control by backbone chiral centres and planar chirality.
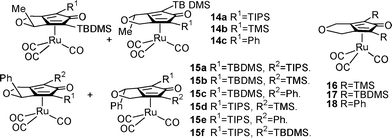
The synthesis of catalysts 14a–c was investigated. The TBS-substituted alcohol (R)-11b was prepared from 10b in quantitative conversion, 88% isolated yield and over 99% ee using the same ATH catalyst as was used for 11a.14 Unfortunately, separation of the diastereomers of these complexes could not be achieved and these were not evaluated in reductions.
In studies directed at improving the d.e.s of the cyclisation through the use of a larger group on the chiral centre, we examined derivatives of 8 where R3 = Ph. Ru/TsDPEN catalysts are less effective at reduction of aryl/propargylic ketones 10c–10e,16 therefore these were reduced using (R)-Alpine borane.17 For variation of group R1, we prepared alcohols (R)-11c–11e however the TMS derivative (R)-11c was prone to loss of the silyl group in the subsequent alcohol alkylation step. Reaction of the dialkyne derivatives of (R)-11d (96% ee) and (R)-11e (97% ee) with Ru3(CO)12 led to the formation of Ru cyclone complexes 15a–f, all of which were formed as inseparable mixtures of diastereoisomers and proved to be poor catalysts (see SI†). For comparison of relative reactivity, symmetrical complexes 16–18 were also prepared and tested in ATH reactions; the TBDMS-substituted catalyst 17 proved to be the most active (Table 1).
In conclusion, a series of enantiomerically enriched iron and ruthenium cyclone complexes have been prepared and applied to the ATH of ketones to alcohols. The iron complexes, in their first reported role as ATH catalysts, are as effective in terms of asymmetric induction as the ruthenium complexes, although higher loadings are required. Other iron-based catalysts have recently been reported to give higher activities and enantioselectivities.18 The results also indicate that the control of enantioselectivity in the reductions, whilst modest, appears to rely on a novel means of asymmetric induction by a remote chiral centre in addition to the planar symmetry of the catalysts.
Experimental section
Procedures for starting material ketones, racemic reductions, synthesis of 16–18, Tables for reductions using 15a–f and NMR spectra may be found in the Electronic Supporting Information†.(R)-4-Phenyl-3-butyne-2-ol 11a14
(R,R)-Tethered Ru(II) catalyst (43 mg, 0.07 mmol)14b was dissolved in iPrOH (135 cm3) at 28 °C. KOH (0.1 M in iPrOH, 3.46 cm3, 0.0346 mmol) was added and the solution was observed to initially turn dark purple and the gradually become lighter in colour. After stirring for 30 min at 28 °C, 4-phenyl-3-butyne-2-one 10a (2.00 g, 13.88 mmol) was added and again the mixture turned dark purple. After 15 h the solvent was removed in vacuo to afford a dark oil which was purified by short path distillation (125 °C, 5.8 mbar) to afford the alcohol as a colourless oil (1.80 g, 12.32 mmol, 89%); [α]28D +32.4 (c 0.966 in CHCl3) (lit.14a [α]23D −35.0 (c 1.0 in CHCl3) 97% ee (S); m/z (ESI) 169 [M + 23]+; (Found (ESI): M + Na 169.0630, C10H10NaO requires 169.0624); νmax 3298, 2978, 2927, 2870, 2225, 1595 and 1490 cm−1; δH (400 MHz, CDCl3) 7.46–7.41 (2H, m, Ar), 7.34–7.28 (3H, m, Ar), 4.77 (1H, m, CH(OH)CH3), 2.00 (1H, d, J 5.13, OH), 1.57 (3H, d, J 7.03, CH3); δC (100 MHz, CDCl3) 131.68 (CH, Ar), 128.42 (CH, Ar), 128.42 (CH, Ar), 122.61 (ipso, Ar), 90.96 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 84.05 (CC), 58.91 (CH(OH)CH3), 24.42 (CH(OH)CH3); The ee was determined using chiral GC of the acetyl derivative of the alcohol synthesised from reacting a sample of the alcohol (<10 mg) with acetic anhydride (<50 μL) and DMAP (<1 mg) in DCM (ca. 1 cm3) overnight; cyclodextrin CB column; 96% ee, 115 °C, H2, 15 psi, 62.72 (S), 64.18 min (R).14b
C), 84.05 (CC), 58.91 (CH(OH)CH3), 24.42 (CH(OH)CH3); The ee was determined using chiral GC of the acetyl derivative of the alcohol synthesised from reacting a sample of the alcohol (<10 mg) with acetic anhydride (<50 μL) and DMAP (<1 mg) in DCM (ca. 1 cm3) overnight; cyclodextrin CB column; 96% ee, 115 °C, H2, 15 psi, 62.72 (S), 64.18 min (R).14b
(R)-Dipropargylic ether 8a (8; R1 = H, R2 = Ph, R3 = Me)19
Sodium hydride (0.190 g, 60% on mineral oil, 4.746 mmol, 1.1 eq.) was added to (R)-4-phenyl-3-butyne-2-ol (0.630 g, 4.315 mmol, 1 eq.) in THF (8 cm3) at 0 °C. After 30 min, propargylic bromide (0.55 cm3, 0.7695 g, 80% in toluene, 5.178 mmol, 1.2 eq.) was added at 0 °C and the reaction was left to warm to room temperature over 2.5 h. The mixture was quenched using saturated NH4Cl(aq) (30 cm3) and extracted with ether (3 × 30 cm3). The combined organic fractions were dried over MgSO4 and the solvent removed in vacuo to afford a yellow oil. Purification by column chromatography (EtOAc/pet. ether (40–60) 1:20) afforded propargylic ether 8a (8; R1 = H, R2 = Ph, R3 = Me) as a yellow oil (0.70 g, 3.804 mmol, 88%); [α]28D +289.04 (c 0.762 in CHCl3); m/z (ESI) 207 (M+ + 23); (Found (ESI): M + Na 207.0788, C13H12NaO requires 207.0780); νmax 3289, 2985, 2932, 2853, 2361, 2223, 2198, 2115, 1958, 1888, 1724, 1671, 1596, 1489, 1439, 1368 and 1325 cm−1; δH (400 MHz, CDCl3) 7.51–7.45 (2H, m, Ar), 7.38–7.32 (3H, m, Ar), 4.68 (1H, q, J 6.5, OCHCH3), 4.44 (1H, dd, J 15.6, 2.0, OCHH), 4.36 (1H, dd, J 15.6, 2.0 OCHH), 2.49 (1H, t, J 2.0, CCH), 1.59 (3H, d, J 6.5, CH3); δC (100 MHz, CDCl3) 131.73 (CH, Ar), 128.43 (CH, Ar), 128.25 (CH, Ar), 122.45 (ipso, Ar), 87.88 (CC), 85.62 (CC), 79.56 ((CCH), 74.42 (CC), 64.62 (OCH), 55.74 (OCH2), 22.00 (CH3).
(R)-Dipropargylic ether 8c (8; R1 = Me3Si, R2 = Ph, R3 = Me)10
To 8a (8; R1 = H, R2 = Ph, R3 = Me) (0.350 g, 1.9022 mmol, 1 eq.) in THF (12.5 cm3) was added nBuLi (1.6 M in hexanes, 1.3 cm3, 2.092 mmol, 1.1 eq.) at −78 °C. After 1 h at −78 °C, trimethylsilylchloride (0.29 cm3, 0.248 g, 2.283 mmol, 1.2 eq.) was added and the mixture stirred at −78 °C for 1 h before being allowed to warm to room temperature overnight. The reaction was quenched using saturated NH4Cl(aq) (15 cm3) and extracted using Et2O (3 × 15 cm3). The combined organics were dried over MgSO4 and the solvent removed in vacuo to afford the product as a dark yellow oil (0.4800 g, 1.86 mmol, 98%) and used without further purification; [α]28D +160.04 (c 0.57 in CHCl3); m/z (ESI) 279 [M + 23]+; (Found (ESI): M + Na 279.1176 C16H20NaOSi requires 279.1176); νmax 2985, 2954, 2927, 2890, 2843, 2168, 1763, 1595 and 1487 cm−1; δH (400 MHz, CDCl3) 7.48–7.45 (2H, m, Ar), 7.34–7.30 (3H, m, Ar), 4.62 (1H, q, J 6.5, OCHCH3), 4.42 (1H, d, J 15.6, OCHH), 4.32 (1H, d, J 15.6, OCHH), 1.57 (3H, d, J 6.5, CH3), 0.20 (9H, s, Si(CH3)3); δC (100 MHz, CDCl3) 131.73 (CH, Ar), 128.39 (CH, Ar), 128.24 (CH, Ar), 122.55 (ipso, Ar), 101.29 (CC), 91.35 (CC), 88.12 (CC), 85.53 (CC), 64.66 (OCH), 55.61 (OCH2), 22.02 (CHCH3), −0.10 (Si(CH3)3).
(R)-Dipropargylic ether 8 (R1 = (iPr)3Si, R2 = Ph, R3 = Me)
To 8a (8: R1 = H, R2 = Ph, R3 = Me) (0.350 g, 1.9022 mmol, 1 eq.) in THF (12.5 cm3) was added nBuLi (1.6 M in hexanes, 1.3 cm3, 2.092 mmol, 1.1 eq.) at −78 °C. After 1 h at −78 °C triisopropylsilylchloride (0.49 cm3, 0.440 g, 2.283 mmol, 1.2 eq.) was added and the mixture stirred at −78 °C for 1 h before being allowed to warm to room temperature overnight. The reaction was quenched using saturated NH4Cl(aq) (15 cm3) and extracted using Et2O (3 × 15 cm3). The combined organics were dried over MgSO4 and the solvent removed in vacuo to afford the product as a dark orange oil in quantitative yield and used without further purification; [α]28D +150.35 (c 0.20 in CHCl3); m/z (ESI) 363 [M + 23]+; (Found (ESI): M + Na 363.2115 C22H32NaOSi requires 363.2115); νmax 2937, 2890 and 2863 cm−1; δH (400 MHz, CDCl3) 7.47–7.44 (2H, m, Ar), 7.34–7.30 (3H, m, Ar), 4.74 (1H, q, J 6.5, (OCHCH3)), 4.41 (2H, s, OCH2), 1.57 (3H, d, J 6.5, CHCH3), 1.12–1.08 (21H, m, Si(CH(CH3)2)3); δC (100 MHz, CDCl3) 131.82 (CH, Ar), 128.39 (CH, Ar), 128.27 (CH, Ar), 122.67 (ipso, Ar), 103.14 (CC), 88.30 (CC), 87.73 (CC), 85.34 (CC), 64.01 (OCH), 56.58 (CH2), 22.03, 18.61, 17.72, 12.31, 11.19.
(R)-Dipropargylic ether 8b (8: R1 = (tBu)3SiMe2, R2 = Ph, R3 = Me)10
Compound 8a (8: R1 = H, R2 = Ph, R3 = Me) (0.195 g, 1.06 mmol) was dissolved in dry THF (15 cm3) and cooled to −78 °C. n-Butyllithium in hexanes (1.6 M, 0.79 cm3, 1.26 mmol) was added dropwise and the mixture was allowed to stir for 1 h after which time tert-butyldimethylsilylchloride (0.207 g, 1.37 mmol) in dry THF (5 cm3) was added. After 17 h the reaction was quenched with H2O (20 cm3), the THF was removed under reduced pressure and the product was extracted into Et2O (3 × 20 cm3). The combined organic phase was dried over Na2SO4, filtered and the solvent was removed under reduced pressure to give the product as a brown oil which was purified by column chromatography on silica with a gradient elution from 100% petroleum ether to 80:20 petroleum ether:ethyl acetate to give the product 8 as a yellow oil (0.206 g, 0.69 mmol, 65%). The measured data is in agreement with that previously reported for the racemic compound.10 [α]24D +150.7 (c 1.0 in CHCl3); δH (300 MHz, CDCl3) 7.42–7.46 (2H, m, Ar), 7.28–7.34 (3H, m, Ar), 4.64 (1H, q, J 6.8, CCH(CH3)O), 4.41 (1H, d, J 15.8, CCH2O), 4.33 (1H, d, J 15.8, CCH2O), 1.55 (3H, d, J 6.8 Hz, (CCH(CH3)O), 0.95 (9H, s, Si(CH3)2C(CH3)3) 0.12 (6H, s, Si(CH3)2C(CH3)3.
(R)-Dipropargylic ether 8d (8: R1 = Ph, R2 = Ph, R3 = Me)
To 8a (8: R1 = H, R2 = Ph, R3 = Me) (0.400 g, 2.1739 mmol, 1 eq.), phenyliodide (0.29 cm3, 0.5322 g, 2.6087 mmol, 1.2 eq.) in triethylamine (7 cm3) at 50 °C was added Pd(Cl2)(PPh3) (0.031 g, 0.043 mmol, 0.02 eq.) After stirring for 5 min, CuI (0.04 g, 0.0217 mmol, 0.01 eq.) was added and the mixture kept at 50 °C for 18 h. The mixture was then hot filtered to remove the amide salts. Removal of the remaining aminein vacuo afforded a yellow oil which was purified by column chromatography to afford the product as a light yellow oil (0.358 g, 1.3769 mmol, 63%); [α]28D +180.00 (c 0.16 in CHCl3); m/z (ESI) 261 [M + 1]+, 283 [M + 23]+; (Found (ESI): M + Na 283.1093 C19H16NaO requires 283.1093); νmax 3052, 2981, 2934, 2843, 2219 and 1716 cm−1; δH (400 MHz, CDCl3) 7.50–7.46 (4H, m, Ar), 7.35–7.30 (6H, m, Ar), 4.73 (1H, q, J 6.5, OCHCH3), 4.65 (1H, d, J 15.6, OCHH), (R1 = Ph, R2 = Ph, R3 = Me).10,19 4.73 (1H, q, J 6.5, OCHCH3), 4.65 (1H, d, J 15.6, OCHH), 4.57 (1H, d, J 15.6, OCHH), 1.61 (3H, d, J 6.5, OCHCH3); δC (100 MHz, CDCl3) 131.81 (CH, Ar), 131.76 (CH, Ar), 128.42 (CH, Ar), 128.40 (CH, Ar), 128.25 (CH, Ar), 128.23 (CH, Ar), 122.61 (ipso, Ar), 122.54 (ipso, Ar), 88.18 (CC), 86.22 (CC), 85.61 (CC), 84.92 (CC), 64.66 (OCH), 56.60 (OCH2), 22.08 (OCHCH3).
(R)-Alcohol 11b20
Tethered-TsDPEN (R,R) Ru(II) catalyst14b (31 mg, 0.005 mmol, 0.5 mol%) was dissolved in anhydrous iPrOH (97 cm3) and warmed to 28 °C. On addition of KOH (in iPrOH, 0.1 M, 2.5 cm3, 0.25 mmol) the colourless solution turned dark purple and was stirred at 28 °C for 30 min before TBDMS-3-butyn-2-one 10b (1.840 g, 10 mmol) was added. After 18 h at 28 °C the solvent was removed in vacuo and the resulting alcohol was purified by column chromatography (EtOAc/hexane 5%) to afford a yellow oil (1.630 g, 8.76 mmol, 88%); m/z (ESI) 207 [M + 23]+; [α]28D +26.29 (c 0.978 in CHCl3); νmax 3315, 2954, 2924, 2887, 2853, 2168, 1470 1358, 1251, 1112, 1072, 1041 and 940 cm−1; δH (300 MHz, CDCl3) 4.52 (1H, dq, J 6.6, 5.3, CH(OH)CH3), 1.77 (1H, d, J 5.3, OH), 1.45 (3H, d, J 6.6, CH(OH)CH3), 0.93 (9H, s, SiC(CH3)3), 0.10 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 108.35 (quat., CC), 86.64 (quat., CC), 58.75 (CH(OH)CH3), 26.00 (CH3), 24.35 (CH3), 16.42 (quat., SiC(CH3)3), −4.71 (CH3, Si(CH3)2). The ee was determined using the acetyl derivative of the alcohol synthesised from reacting a trace amount of the alcohol (<10 mg) with acetic anhydride (<50 μL) and DMAP (<1 mg) in DCM (ca. 1 cm3) overnight. cyclodextrin CB column; 99% ee, 115 °C, H2, 15 psi, 14.19 (S), 14.51 min (R).
(R)-Mesyl derivative of (R)-4-phenyl-3-butyne-2-ol 11a21
To (R)-4-phenyl-3-butyne-2-ol 11a (1.00 g, 1.00 cm3, 6.833 mmol, 1 eq.) and triethylamine (1.90 cm3, 13.46 mmol, 2 eq.) in DCM (10 cm3) at −78 °C was slowly added methanesulfonyl chloride (0.80 cm3, 10.13 mmol, 1.5 eq.) after 1 h the mixture was allowed to warm to room temperature and quenched with NaHCO3(aq) (15 cm3), extracted with DCM (3 × 10 cm3) and dried over Na2SO4. Removal of the solventin vacuo afforded the mesylate as a colourless oil in quantitative yield and was used immediately without further purification; δH (400 MHz, d6-DMSO) 7.50–7.46 (2H, m, Ar), 7.43–7.34 (3H, m, Ar), 5.55 (1H, q, J 6.5, CHCH3), 3.18 (3H, s, SO2CH3), 1.77 (3H, d, J 6.5, CHCH3).(R,S)-Dipropargyl ether precursor of ligand 13a/b
To (R)-4-(t-butyldimethylsilyl)-3-butyne-2-ol 11b (0.3852 g, 2.0710 mmol, 1 eq.) in THF (5 cm3) was added sodium hydride (0.083 g, 2.0710 mmol, 60% in mineral oil, 1 eq.) at 0 °C. After 30 min the mesyl derivative of (R)-4-phenyl-3-butyne-2-ol 11a (0.46 g, 2.0710 mmol, 1 eq.) was added and the reaction was allowed to warm to room temperature over 18 h. After quenching the mixture using saturated aqueous sodium bicarbonate (5 cm3) the mixture was extracted with Et2O (3 × 7 cm3) and dried over NaSO4, removal of the solventin vacuo afforded a yellow oil which was purified by column chromatography (EtOAc/hexane gradient 1:100 to 1:10) yielding the product as a colourless oil (0.1911 g, 0.6125 mmol, 30%); [α]28D +0.19 (c 0.78 in CHCl3); m/z (ESI) 335.2 [M + 23]+; (Found (ESI): M + Na 335.1801 C20H28NaOSi requires 335.1802); νmax 2983, 2952, 2929, 2880 and 2853 cm−1; δH (400 MHz, CDCl3) 7.48–7.42 (2H, m, Ar), 7.33–7.28 (3H, m, Ar), 4.72 (1H, q, J 6.5, CH), 4.51 (1H, q, J 6.5, CH), 1.55 (3H, d, J 6.5, CH3), 1.50 (3H, d, J 6.5, CH3), 0.95 (9H, s, SiC(CH3)3), 0.10 (6H, s, SiCH3); δC (100 MHz, CDCl3) 131.76 (Ar), 131.70 (Ar), 128.29 (Ar), 128.19 (Ar), 128.14 (Ar), 106.30 (CC), 89.35 (CC), 84.73 (CC), 63.47 (CC), 63.45 (CC), 26.04, 22.10, 21.79. −4.70 (Si(CH3)2).
Iron cyclone complexes 7b/c
(R)-Dipropargylic ether 8b (8: R1 = (tBu)3SiMe2, R2 = Ph, R3 = Me, 96% ee) (0.206 g, 0.69 mmol) and Fe(CO)5 (0.27 cm3, 2.05 mmol) were dissolved in dry toluene (3 cm3) and heated at 130 °C for 24 h after which time the solution was allowed to cool to room temperature and the solvent was removed under reduced pressure. The brown residue was filtered through celite using a 9:1 mixture of hexane:ethyl acetate and subsequent purification by column chromatography on silica with a gradient elution from 100% petroleum ether to 40:60 petroleum ether:ethyl acetate gave two diastereomers of product, which were separated. The measured data is in agreement with that previously reported for the racemic compound.10 Minor diastereomer, yellow solid (0.026 g, 0.056 mmol, 8%); [α]26D −47.0 (c 0.05 in CHCl3); δH (300 MHz, CDCl3) 7.99–8.05 (2H, m, Ar), 7.29–7.39 (3H, m, 3H, Ar), 5.56 (1H, q, J 6.8 Hz, CCH(CH3)O), 4.81 (1H, d, J 13.2 Hz, CH2), 4.71 (1H, d, J 13.2 Hz, CH2), 1.53 (3H, d, J 6.8 Hz, CH3), 1.01 (9H, s, SiC(CH3)3) 0.47 (3H, s, Si(CH3)2C(CH3)3), 0.08 (3H, s, Si(CH3)2C(CH3)3). Major diastereomer, brown oil (0.140 g, 0.300 mmol, 44%); [α]28D +20.0 (c 0.05 in CHCl3); δH (300 MHz, CDCl3) 7.47–7.53 (2H, m, Ar), 7.29–7.41 (3H, m, Ar), 5.38 (1H, q, J 6.0 Hz, CCH(CH3)O), 4.79 (1H, d, J 13.2 Hz, CH2), 4.73 (1H, d, J 13.2 Hz, CH2), 1.65 (3H, d, J 6.0 Hz, CH3), 0.97 (9H, s, SiC(CH3)3) 0.51 (3H, s, Si(CH3)2C(CH3)3), 0.06 (3H, s, Si(CH3)2C(CH3)3).
Iron cyclone complexes 7d/e
These complexes (two diastereomers) were synthesised by the same procedure as for 7d/e using (R)-dipropargylic ether 8c (8: R1 = TMS, R2 = Ph, R3 = Me, 96% ee) (0.390 g, 1.53 mmol) and Fe(CO)5 (0.60 cm3, 4.56 mmol) and were purified by column chromatography on silica gel with a gradient elution from 100% petroleum ether to 80:20 petroleum ether:ethyl acetate to give two diastereomers of product which were separated, as brown oils. The measured data is in agreement with that previously reported for the racemic material.10 Minor diastereomer (0.111 g, 0.262 mmol, 17%); [α]28D −166.0 (c 0.05 in CHCl3); δH (300 MHz, CDCl3) 7.99–8.03 (2H, m, Ar), 7.29–7.40 (3H, m, Ar), 5.57 (1H, q, J 6.4 Hz, CCH(CH3)O), 4.81 (1H, d, J 12.8 Hz, CH2), 4.71 (1H, d, J 12.8 Hz, CH2), 1.52 (3H, d, J 6.4 Hz, CH3), 0.33 (9H, s, Si(CH3)3). Major diastereomer (0.240 g, 0.566 mmol, 37%); [α]28D +101.0 (c 0.05 in CHCl3); δH (300 MHz, CDCl3) 7.48–7.52 (2H, m, Ar), 7.30–7.40 (3H, m, Ar), 5.36 (1H, q, J 6.4 Hz, CCH(CH3)O), 4.79 (1H, d, J 13.2 Hz, CH2), 4.71 (1H, d, J 13.2 Hz, CH2), 1.65 (3H, d, J 6.4 Hz, CH3), 0.31 (9H, s, Si(CH3)3).
Iron cyclone complexes 7f/g
These complexes (two diastereomers) were synthesised by the same procedure as for 7b/c using (R)-dipropargylic ether 8d (8: R1 = Ph, R2 = Ph, R3 = Me, 96% ee) (0.165 g, 0.63 mmol) and Fe(CO)5 (0.25 cm3, 1.90 mmol) and were purified by column chromatography on silica with a gradient elution from 100% petroleum ether to 80:20 petroleum ether:ethyl acetate to give two diastereomers of product, which were separated. The measured data is in agreement with that previously reported for the racemic material.10 Minor diastereomer; brown powder (0.026 g, 0.061 mmol, 10%); [α]28D −75.0 (c 0.01 in CHCl3); δH (300 MHz, CDCl3) 8.06–8.11 (2H, m, Ar), 7.86–7.93 (2H, m, Ar), 7.32–7.45 (6H, m, Ar), 5.64 (1H, q, J 6.4 Hz, (CCH(CH3)O), 5.17 (2H, s, CH2), 1.54 (3H, d, J 6.4 Hz, (CCH(CH3)O). A broad resonance exists from 6.5–7.6 ppm in the 1H NMR spectrum that has not been assigned; this may be due to paramagnetic impurities. Major diastereomer; brown powder (0.039 g, 0.091 mmol, 14%); [α]28D +23.0 (c 0.05 in CHCl3); δH (300 MHz, CDCl3) 7.90–7.96 (2H, m, Ar), 7.53–7.59 (2H, m, Ar), 7.32–7.45 (6H, m, Ar), 5.40 (1H, q, J 6.0 Hz, (CCH(CH3)O), 5.25 (1H, d, J 13.2 Hz, CH2), 5.03 (1H, d, J 13.2 Hz, CH2) 1.67 (3H, d, J 6.0 Hz, (CCH(CH3)O). A broad resonance exists from 6.6–7.8 ppm in the 1H NMR spectrum that has not been assigned; this may be due to paramagnetic impurities.
Ruthenium cyclone complex 12a/b
A sealed tube was charged with 8c (8: R1 = Me3Si, R2 = Ph, R3 = Me) (0.3813 g, 1.4894 mmol, 3eq.) and Ru3(CO)12 (0.3173 g, 0.4965 mmol, 1 eq.) in acetonitrile (5 cm3) and the reaction heated to 100 °C over 2 days. The solvent was removed in vacuo and redissolved in DCM then filtered to remove any unreacted Ru3(CO)12 and purified by column chromatography (EtOAc/pet. ether (40–60) 1:10) to afford the major 12a (0.1787, 0.404 mmol, 27%) and minor 12b (0.0768 g, 0.1738 mmol, 12%) diastereoisomers. Configurations assigned by analogy with Fe complexes; Major; m/z (ESI) 471 [M + 1]+, 493 [M + 23]+; [α]28D +79.20 (c 0.11, CHCl3); (Found (ESI): M + H 471.0218 C20H21O5RuSi requires 471.0201); (Found (ESI): M + Na 493.0039 C20H20NaO5RuSi requires 493.0021); νmax 2075, 2006 and 1626 cm−1; δH (400 MHz, CDCl3) 7.43 (2H, d, J 7.0, Ar), 7.35 (2H, t, J 7.5, Ar), 7.30–7.25 (1H, m, Ar), 5.29 (1H, q, J 6.0, OCHCH3), 4.83–4.73 (2H, m, OCH2), 1.59 (3H, d, J 6.0, OCHCH3), 0.32 (9H, s, Si(CH3)3); δC (100 MHz, CDCl3) 193.82 (CO), 179.00 (CO), 130.44 (ipso, Ar), 129.80 (CH, Ar), 128.35 (CH, Ar), 127.64 (CH, Ar), 114.40, 112.85, 80.04, 74.81 (OCHCH3), 67.27, 62.54, 23.73 (OCHCH3), −0.51 (Si(CH3)3); Minor [α]28D −8.30 (c 0.112, CHCl3); m/z (ESI) 471 [M + 1]+, 493 [M + 23]+; (Found (ESI): M + H 471.0200 C20H21O5RuSi requires 471.0201); (Found (ESI): M + Na 493.0019 C20H20NaO5RuSi requires 493.0021); νmax 2080, 2020 and 1989 cm−1; δH (400 MHz, CDCl3) 7.97 (2H, d, J 7.5, Ar), 7.35 (2H, t, J 7.5, Ar),7.29–7.25 (1H, m, Ar), 5.60 (1H, q, J 6.5, OCHCH3), 4.89 (1H, dd, J 12.6, 1.0, OCHH), 4.78 (1H, d, J 12.6, OCHH), 1.43 (3H, d, J 6.5, OCHCH3), 0.33 (9H, s, Si(CH3)3); δC (100 MHz, CDCl3) 193.94 (CO), 180.55 (CO), 132.74 (ipso, Ar), 128.72 (CH, Ar), 127.36 (CH, Ar), 126.54 (CH, Ar), 110.96, 109.65, 76.48 (OCHCH3), 75.74, 70.64, 66.33, 63.86, 27.90, 19.06, −0.40 (Si(CH3)3).
Ruthenium cyclone complex 12c/d
A sealed tube was charged with 8 (R1 = (iPr)3Si, R2 = Ph, R3 = Me) (0.5898 g, 1.798 mmol, 3eq.) and Ru3(CO)12 (0.3830 g, 0.5994 mmol, 1 eq.) in acetonitrile (6 cm3) and the reaction heated to 100 °C over 2 days. The solvent was removed in vacuo and redissolved in DCM and filtered to remove any unreacted Ru3(CO)12 and purified by column chromatography (EtOAc/pet. ether (40–60) 1:10) to afford only one diastereoisomer cleanly (0.1197 g, 0.2213 mmol, 12%); [α]28D +39.11 (c 0.140, CHCl3); m/z (ESI) 555 [M + 1]+, 577 [M + 23]+; (Found (ESI): M + H 555.1148 C22H33O5RuSi requires 555.1142); νmax 2944, 2860, 2077, 2023, 1999 and 1622 cm−1; δH (400 MHz, CDCl3) 7.37–7.32 (2H, m, Ar), 7.29–7.24 (2H, m, Ar), 7.21–7.15 (1H, m, Ar), 5.22 (1H, q, J 6.0, OCHCH3), 4.74 (2H, s, OCH2), 1.53 (3H, d, J 6.0, OCHCH3), 1.32 (3H, sept., J 7.5, Si(CH(CH3)2)3), 1.15–1.08 (18H, m Si(CH(CH3)2)3); δC (100 MHz, CDCl3) 193.83 (CO), 178.95 (CO), 130.38 (ipso, Ar), 129.89 (CH, Ar), 128.37 (CH, Ar), 127.62 (CH, Ar), 116.19, 111.69, 79.37, 74.72 (OCHCH3), 68.36, 62.42, 23.66, 19.12, 12.32.
Ruthenium cyclone complex 12e/f
A sealed tube was charged with 8d (8: R1 = Ph, R2 = Ph, R3 = Me) (0.3580 g, 1.3769 mmol, 3eq.) and Ru3(CO)12 (0.2933 g, 0.4586 mmol, 1 eq.) in acetonitrile (5 cm3) and the reaction was heated to 100 °C over 2 days. The solvent was removed in vacuo and redissolved in DCM and filtered to remove any unreacted Ru3(CO)12 and purified by column chromatography (EtOAc/pet. ether (40–60) 1:20) to afford only one diastereoisomer cleanly (0.1371 g, 0.2898 mmol, 21%); [α]28D −61.96 (c 0.092, CHCl3); m/z (ESI) 475 [M + 1]+, 497 [M + 23]+; (Found (ESI): M + H 475.0123 C23H17O5Ru requires 475.0120); νmax 2074, 1999 and 1622 cm−1; δH (400 MHz, CDCl3) 7.87 (2H, d, J 7.5, Ar), 7.50 (2H, d, J 7.0, Ar), 7.43–7.36 (4H, m, Ar), 7.35–7.28 (2H, m, Ar), 5.34 (1H, dq, J 1.5, 6.0, CHCH3), 5.25 (1H, d, J 12.6, CHH), 5.07 (1H, dd, J 2.0, 12.6, CHH), 1.61 (3H, d, J 6.0, CHCH3); δC (100 MHz, CDCl3) 193.80 (CO), 132.39 (ipso, Ar), 130.08 (ipso, Ar), 128.88 (CH, Ar), 128.47 (CH, Ar), 127.85 (CH, Ar), 127.76 (CH, Ar), 127.00 (CH, Ar), 109.03 (quat.), 107.13 (quat.), 77.98 (quat.), 74.92 (CH), 74.06 (quat.), 67.75 (quat.), 23.74 (CH3).
Ruthenium cyclone complex 13a/b
A sealed tube was charged with the dipropargyl ether precursor of ligand 13a/b (0.083 g, 0.26 mmol, 3eq.) and Ru3(CO)12 (0.056 g, 0.0.887 mmol, 1 eq.) in acetonitrile (1 cm3) and the reaction heated to 100 °C over 2 days. The solvent was removed in vacuo and redissolved in DCM and filtered to remove any unreacted Ru3(CO)12 and purified by column chromatography (EtOAc/hexane 1:20) to afford only one diastereoisomer cleanly (0.040 g, 0.0805 mmol, 31%); [α]28D +38.60 (c 0.020, CHCl3); m/z (ESI) 527 [M]+; (Found (ESI): M + H 527.0822 C24H29O5RuSi requires 527.0828); νmax 2075, 1999 and 1638 cm−1; δH (400 MHz, CDCl3) 7.44–7.40 (2H, m, Ar), 7.37–7.32 (2H, m, Ar), 7.29–7.26 (1H, m, Ar), 5.22 (1H, qd, J 6.0, 1.5, CHCH3) 5.00 (1H, qd, J 6.0, 1.5, CHCH3), 1.58 (3H, d, J 6.0, CHCH3), 1.55 (3H, d, J 6.0, CHCH3), 0.99 (9H, s, SiC(CH3)3), 0.51 (3H, s, SiCH3), 0.18 (3H, s, SiCH3); δC (100 MHz, CDCl3) 193.91 (CO), 180.04 (CO), 130.34 (ipso, Ar), 130.07 (CH, Ar) 128.38 (CH, Ar), 127.70 (CH, Ar), 119.99, 116.40, 78.22, 74.82, 73.51, 60. 83, 31.56, 28.06, 27.23, 23.76, 22.63, 18.81, −2.04, −3.67.
(R)-Alcohol 11d
A flask was charged with (R)-Alpine borane solution (45.08 cm3, 22.54 mmol, 0.5 M in THF) and the solvent was removed in vacuo. The flask was cooled to 0 °C and 3-(t-butyldimethylsilyl)-1-phenyl-prop-2-ynone 10d (5.00 g, 20.49 mmol) was added dropwise. After 4 days acetylaldehyde (5 cm3) was added. After 1 h, NaOH(aq) (10 cm3, 5 N), THF (10 cm3) and hydrogen peroxide (11.5 cm3, 30%wt) added. CAUTION: addition of peroxide is very exothermic. After heating to 40 °C for 4 h, in air, the reaction was extracted using Et2O (3 × 30 cm3) and the combined organic phases washed using brine (30 cm3), dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (EtOAc/hexane 0.5%–5%) afforded a colourless oil (Rf 0.13 (10% EtOAc/hexane), 3.67 g, 14.918 mmol, 73%, 96% ee); [α]28D +4.2 (c 1.054 in CHCl3); m/z 269.2 [M + 23]+; (Found (ESI): M + Na 269.1333. C15H22NaOSi requires 269.1332); νmax 3362 (OH), 2926, 2857, 1671, 1446 and 1003 cm−1; δH (400 MHz, CDCl3) 7.56 (2H, d, J 7.1, Ar), 7.36 (3H, m, Ar), 5.47 (1H, s, Ar), 0.99 (9H, s, SiC(CH3)3), 0.17 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 140.36 (ipso, Ar), 128.49 (CH, Ar), 128.26 (CH, Ar), 126.70 (CH, Ar), 105.69 (quat., CC), 89.81 (quat., CC), 64.90 (CH(OH)), 26.03 (SiC(CH3)3), 16.50 (quat., SiC(CH3)3), −4.71 (Si(CH3)2); δSi (99 MHz, CDCl3) −7.64 (s, Si(CH3)2C(CH3)3). The ee was determined by chiral GC; cyclodextrin CB column; 96% ee, 100 °C, H2, 15 psi, 83.70 min (S), 85.99 min (R).17
(R)-Dipropargylic ether 8 (R1 = H, R2 = Si(tBu)Me2, R3 = Ph)
To a solution of (R)-alcohol 11d (1.1606 g, 6.0976 mmol 1 eq.) in THF (20 cm3) at 0 °C was added NaH (0.2881 g, 60% in mineral oil, 7.2019 mmol, 1.1 eq.). The reaction mixture was stirred for 30 min at 0 °C after which propargylic bromide was added (0.85 cm3, 80%wt in toluene, 1.17 g, 7.86 mmol, 1.2 eq.) and the ice bath removed. After 4 h the reaction was quenched using saturated NaHCO3(aq) solution (20 cm3) and extracted using Et2O (3 × 10 cm3). The combined organic fractions were dried over MgSO4 and the solvent removed in vacuo. Purification column chromatography (EtOAc/hexane 0% to 1%) afforded a colourless oil (1.121 g, 3.948 mmol, 60%); [α]28D +27.3 (c 0.68 in CHCl3); m/z 307.2 [M + 23]+; (Found (ESI): M + Na 307.1483, C18H24NaOSi requires 307.1489); νmax 2952, 2927, 2884, 2855, 1250 and 1059 cm−1; δH (400 MHz, CDCl3) 7.56 (2H, d, J 7.2, Ar), 7.41-7.34 (3H, m, Ar), 5.45 (1H, s, ArCH), 4.41 (1H, dd, 4J 2.3, J 15.7, OCHH), 4.29 (1H, dd, 4J 2.3, J 15.7, OCHH), 2.48 (1H, t, 4J 2.3, CCH), 0.98 (9H, s, SiC(CH3)3), 0.17 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 137.65 (ipso, Ar), 128.69 (CH, Ar), 128.44 (CH, Ar), 126.75 (CH, Ar), 102.50 (quat., CC), 91.77 (quat., CC), 74.82 (quat. CC), 70.44 (ArCH), 55.26 (OCH2), 26.05 (SiC(CH3)3), 16.55 (SiC(CH3)3), −4.70 (Si(CH3)2); δSi (99 MHz, CDCl3) −7.51 (s, Si(CH3)2C(CH3)3).
(R)-Dipropargylic ether 8 (R1 = TMS, R2 = Si(tBu)Me2, R3 = Ph)
A flask was charged with (R)-dipropargylic ether 8 (R1 = H, R2 = Si(tBu)Me2, R3 = Ph) (0.500 g, 1.7606 mmol) in THF (12 cm3) and cooled to −78 °C. Addition of nBuLi (1.21 cm3, 1.94 mmol, 1.6 M in hexane, 1.1 eq.) afforded a dark green solution. After 1 h TMSCl (0.27 cm3, 2.11 mmol, 1.2 eq.) was added and the ice bath removed after 5 min. After 30 min the reaction mixture colour had changed from green to yellow. After 3 h the reaction was quenched using water (10 cm3) and extracted with Et2O (3 × 10 cm3), the organic fractions combined and dried over MgSO4 and the solvent removed under reduced pressure to afford a yellow oil which was purified using column chromatography (EtOAc/hexane 0 to 20%) to afford a pale yellow oil (0.4300 g, 1.208 mmol, 69%); [α]28D +27.3 (c 0.68 in CHCl3); m/z (ESI) 379.2 [M + 23]+; (Found (ESI): M + Na 379.1881. C21H32NaOSi2 requires 379.1884); νmax 2954, 2928, 2898, 2856, 2173, 1249 and 1059 cm−1; δH (400 MHz, CDCl3) 7.56 (2H, d, J 7.5, Ar), 7.40–7.31 (3H, m, Ar), 5.45 (1H, s, ArCH), 4.40 (1H, d, J 15.8, OCHH), 4.28 (1H, d, J 15.8, OCHH), 0.98 (9H, s, SiC(CH3)3), 0.21 (9H, s, Si(CH3)3), 0.17 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 137.77 (ipso, Ar), 128.50 (CH, Ar), 128.41 (CH, Ar), 127.83 (CH, Ar), 102.70 (quat., CC), 101.11 (quat., CC), 91.81 (quat. CC), 91.60 (quat. CC), 70.39 (ArCH), 56.07 (OCH2), 26.08 (SiC(CH3)3), 16.56 (SiC(CH3)3), −0.16 (Si(CH3)3, −4.70 (Si(CH3)2); δSi (99 MHz, CDCl3) −7.57 (s, Si(CH3)2C(CH3)3), −17.73 (s, Si(CH3)3).
(R)-Dipropargylic ether 8 (R1 = TIPS, R2 = Si(tBu)Me2, R3 = Ph
A flask was charged with (R)-dipropargylic ether 8 (R1 = H, R2 = Si(tBu)Me2, R3 = Ph) (0.500 g, 1.7606 mmol) in THF (10 cm3) and cooled to −78 °C. Addition of nBuLi (1.35 cm3, 2.17 mmol, 1.6 M in hexane, 1.2 eq.) afforded a dark green solution. After 30 min TIPSCl (0.50 cm3, 2.36 mmol, 1.3 eq.) was added and the ice bath removed after 30 min. After 5 h the reaction was quenched using water (10 cm3) and extracted with Et2O (3 × 10 cm3), the organic fractions combined and dried over MgSO4 and the solvent removed under reduced pressure to afford a yellow oil which was purified using column chromatography (EtOAc/hexane 0 to 5%) to afford a bright yellow oil (0.219 g, 0.53 mmol, 30%); [α]28D +50.1 (c 0.825 in CHCl3); m/z (ESI) 463.3 [M + 23]+; (Found (ESI): M + Na 463.2821. C27H44NaOSi2 requires 463.2823); νmax 2928, 2889, 2863, 1462, 1249, 1060, 1038,1027 and 1007 cm−1; δH (400 MHz, CDCl3) 7.56–7.53 (2H, m, Ar), 7.40–7.32 (3H, m, Ar), 5.55 (1H, s, ArCH), 4.51 (1H, d, J 16.1, OCHH), 4.31 (1H, d, J 16.1, OCHH), 1.08 (21H, m, Si(CH(CH3)2)3) (0.97 (9H, s, SiC(CH3)3), 0.17 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 137.97 (ipso, Ar), 128.49 (CH, Ar), 128.44 (CH, Ar), 127.87 (CH, Ar), 102.74 (quat., CC), 102.70 (quat., CC), 91.49 (quat. CC), 88.42 (quat., CC), 69.75 (ArCH), 56.22 (OCH2), 26.09 CH3, SiC(CH3)3), 18.61 (Si(CH(CH3)2)3), 16.58 (SiC(CH3)3), 11.18 (CH3, Si(CH(CH3)3)3), −4.68 (Si(CH3)2); δSi (99 MHz, CDCl3) −1.86 (s, Si(CH(CH3)2)3), −7.57 (s, Si(CH3)2C(CH3)3).
(R)-Dipropargylic ether 8 (R1 = Ph, R2 = Si(tBu)Me2, R3 = Ph)
A flask was charged with (R)-dipropargylic ether 8 (R1 = H, R2 = Si(tBu)Me2, R3 = Ph) (0.500 g, 1.7606 mmol), PhI (0.26 cm3, 2.34 mmol, 1.3 eq.) and Et3N (6.3 cm3). To this mixture was added PdCl2(PPh3)2 (27.4 mg, 0.0391 mmol, 0.02 eq.) and after 5 min CuI (3.7 mg, 0.0195 mmol, 0.01 eq.) was added and the reaction heated to 50 °C for 20 h. The reaction mixture was then filtered and the remaining amine removed in vacuo. Purification by column chromatography (EtOAc/hexane 0 to 2%) afforded a light yellow oil (0.3693 g, 1.03 mmol, 58%); [α]28D +58.0 (c 1.02 in CHCl3); m/z (ESI) 361.2 [M + 1]+; 383.1 [M + 23]+; (Found (ESI): M + Na, 383.1802. C24H28NaOSi requires 383.1802); νmax 2952, 2927, 2884, 2855 and 1057 cm−1; δH (400 MHz, CDCl3) 7.56 (2H, d, J 7.6, Ar), 7.50–7.48 (2H, m, Ar), 7.41–7.31 (6H, m, Ar), 5.53 (1H, s, ArCH), 4.63 (1H, d, J 15.7, OCHH), 4.51 (1H, d, J 15.7, OCHH), 1.05 (9H, s, SiC(CH3)3), 0.18 (6H, s, Si(CH3)2); δC (100 MHz, CDCl3) 137.82 (ipso, Ar), 131.81 (CH, Ar), 128.57 (CH, Ar), 128.53 (CH, Ar), 128.45 (CH, Ar), 128.25(CH, Ar), 127.81 (CH, Ar), 122.60 (ipso, Ar), 102.83 (quat., CC), 91.70 (quat., CC), 86.62 (quat., CC), 84.71 (quat., CC), 70.50 (ArCH), 56.10 (OCH2), 26.08 (SiC(CH3)3), 16.56 (SiC(CH3)3), −4.67 (Si(CH3)2); δSi (99 MHz, CDCl3) −7.52 (s, Si(CH3)2C(CH3)3).
(R)-Alcohol 11e
(R)-Alpine borane solution (0.5 M, in THF, 22 mmol, 44 cm3) was introduced to a flask and the solvent removed in vacuo. The flask was cooled to 0 °C and 3-(tri(isopropyl)silyl)-1-phenyl-prop-2-yn-1-one 10e (5.72 g, 20.0 mmol) was added dropwise. The ice bath was removed and after 4 days the reaction was quenched using acetaldehyde (4.5 cm3). After 1 h THF (10 cm3) and NaOH(aq) (5 N, 10 cm3) were added and the reaction mixture put in a water bath and H2O2 (30 wt%, 11.5 cm3) was added carefully. CAUTION: very exothermic. After complete addition of the peroxide the reaction was heated for 4 h in air followed by extraction with Et2O (3 × 30 cm3). The combined organic fractions were then washed with brine (30 cm3) and dried over MgSO4. Removal of the solventin vacuo afforded a colourless oil which on purification by column chromatography (hexane/EtOAc) afforded a colourless oil (2.6031 g, 9.0385 mmol, 45% yield, 97% ee); [α]28D +5.81 (c 1.32, CHCl3); m/z 287.2 [M − 1], 309.2 [M + 23]; (Found (ESI): M − H 287.1816 C18H27OSi requires 287.1826, M + Na 309.1631 C18H26NaOSi requires 309.1645); νmax 3354 (OH), 2941, 2891, 2864 and 2169 cm−1; δH (300 MHz, CDCl3) 7.6–7.56 (2H, m, Ar) 7.42–7.29 (3H, m, Ar), 5.49 (1H, d, J 6.3, ArCH), 2.17 (1H, d, J 6.3, OH), 1.09 (21H, m, Si(CH(CH3)3); δC (75 MHz, CDCl3) 128.54 (ipso, Ar), 128.51 (CH, Ar), 128.30 (CH, Ar), 126.77 (CH, Ar), 106.87 (quat., CC), 98.02 (quat., CC), 65.11 (CH(OH)), 18.59 (Si(CH(CH3)2)3), 11.15 (Si(CH(CH3)2)3); The ee was determined by GC using cyclodextrin CB column; 97% ee, 170 °C, H2, 15 psi, 61.96 min (S), 63.68 min (R).17
(R)-Dipropargylic ether 8 (R1 = H, R2 = Si(iPr)3, R3 = Ph)
To a solution of (R)-alcohol 11e (1.000 g, 3.472 mmol 1 eq.) in THF (11 cm3) at 0 °C was added NaH (0.1527 g, 60% in mineral oil, 3.8194 mmol, 1.1 eq.). The reaction mixture was stirred for 30 min at 0 °C after which propargylic bromide was added (0.45 cm3, 80%wt in toluene, 0.619 g, 4.166 mmol, 1.2 eq.) and the ice bath removed. After 4 h the reaction was quenched using saturated NHCO3(aq) solution (20 cm3) and extracted using Et2O (3 × 10 cm3). The combined organic fractions were dried over MgSO4 and the solvent removed in vacuo. Purification by column chromatography (EtOAc/hexane 0 to 1%) afforded a colourless oil (0.9155 g, 2.8083 mmol, 81%); [α]28D +24.25 (c 1.048, CHCl3); m/z 349.2 [M + 23]+; (Found (ESI): 349.1953 M + Na C21H30NaOSi requires 349.1958); νmax 2941, 2924, 2864, 2169, 2031 and 1461 cm−1; δH (400 MHz, CDCl3) 7.59–7.56 (2H, m, Ar) 7.40–7.31 (3H, m, Ar), 5.48 (1H, s, ArCH), 4.44 (1H, dd, 4J 2.4, J 15.6, OCHH), 4.31 (1H, dd, 4J 2.4, J 15.6, OCHH), 2.47 (1H, t, 4J 2.4, CCH), 1.10 (21H, m, Si(CH(CH3)3); δC (100 MHz, CDCl3) 137.79 (ipso, Ar), 128.47 (CH, Ar), 128.38 (CH, Ar), 127.75 (CH, Ar), 103.62 (quat., CC), 89.95 (quat., CC), 79.35 (CCH), 74.72 (quat., CC), 70.50 (ArCH), 55.19 (OCH2), 18.58 (Si(CH(CH3)2)3), 11.16 (Si(CH(CH3)2)3); δSi (99 MHz, CDCl3) −1.53 (s, Si(CH(CH3)2)).
(R)-Dipropargylic ether 8 (R1 = Me3Si, R2 = Si(iPr)3, R3 = Ph)
To a solution of (R)-dipropargylic ether 8 (R1 = H, R2 = Si(iPr)3, R3 = Ph) (0.190 g, 0.582 mmol) in THF (3 cm3) cooled to −78 °C was added nBuLi (0.4 cm3, 0.6404 mmol, 1.6 M in hexane, 1.1 eq.) affording a dark green solution. After 1 h TMSCl (0.089 cm3, 0.699 mmol, 1.2 eq.) was added and the ice bath removed after 5 min. After 6 h the reaction was quenched using water (3 cm3) and extracted with Et2O (3 × 4 cm3), the organic fractions were combined and dried over MgSO4 and the solvent removed under reduced pressure to afford a yellow oil which was purified using column chromatography (EtOAc/hexane 0 to 1%) to afford a yellow oil (0.1362 g, 0.3424 mmol, 59%); [α]28D +42.2 (c 1.106, CHCl3); m/z 421.2 [M + 23]+; (Found (ESI): 421.2354 M + Na C24H38NaOSi2 requires 421.2353); νmax 2944, 2893, 2865, 1719 and 1250 cm−1; δH (400 MHz, CDCl3) 7.58 (2H, d, J 7.04, Ar), 7.41–7.31 (3H, m, Ar), 5.49 (1H, s, ArCH), 4.44 (1H, d, J 15.8, OCHH), 4.31 (1H, d, J 15.8, OCHH), 1.10 (21H, m, Si(CH(CH3)3), 0.20 (6H, s, Si(CH3)3); δC (100 MHz, CDCl3) 137.92 (ipso, Ar), 128.44 (CH, Ar), 128.38 (CH, Ar), 127.85 (CH, Ar), 103.78 (quat., CC), 101.17 (quat., CC), 91.77 (CCH), 89.85 (quat., CC) 70.43 (ArCH) 56.02 (OCH2) 18.62 (Si(CH(CH3)2)3), 11.19 (CH, Si(CH(CH3)2)3), −0.16 (Si(CH3)3).
(R)-Dipropargylic ether 8 (R1 = Si(tBu)Me2, R2 = Si(iPr)3, R3 = Ph)
To a solution of (R)-dipropargylic ether 8 (R1 = H, R2 = Si(iPr)3, R3 = Ph) (0.400 g, 1.227 mmol) in THF (7 cm3) cooled to −78 °C was added nBuLi (0.84 cm3, 1.3497 mmol, 1.6 M in hexane, 1.1 eq.) affording a dark green solution. After 1 h TBDMSCl (0.222 g, 1.472 mmol, 1.2 eq.) was added and the ice bath removed after 5 min. After 16 h the reaction was quenched using water (10 cm3) and extracted with Et2O (3 × 10 cm3), the organic fractions were combined and dried over MgSO4 and the solvent removed under reduced pressure to afford a crude yellow oil (0.4263 g, 0.9689 mmol, 79%); [α]28D +65.9 (c 0.50, CHCl3); m/z 463.2 [M + 23]+; (Found (ESI): M + Na 463.2819 C27H44NaOSi2 requires 463.2823); νmax 2942, 2927, 2891, 2863, 2173, 1461 and 1063 cm−1; δH (400 MHz, CDCl3) 7.58 (2H, d, J 7.04, Ar) 7.41–7.31 (3H, m, Ar), 5.49 (1H, s, ArCH), 4.44 (1H, d, J 15.8, OCHH) 4.31 (1H, d, J 15.8, OCHH) 1.10 (21H, m, Si(CH(CH3)3) 0.20 (6H, s, Si(CH3)3); δC (100 MHz, CDCl3) 137.92 (ipso, Ar), 128.44 (CH, Ar), 128.38 (CH, Ar), 127.85 (CH, Ar), 103.78 (quat., CC), 101.17 (quat., CC), 91.77 (CH, CCH), 89.85 (quat., CC) 70.43 (CH(OH)) 56.02 (OCH2) 18.62 (Si(CH(CH3)2)3), 11.19 (Si(CH(CH3)2)3), −0.16 (Si(CH3)3).
(R)-Dipropargylic ether 8 (R1 = Ph, R2 = Si(iPr)3, R3 = Ph)
A flask was charged with (R)-dipropargylic ether 8 (R1 = H, R2 = Si(iPr)3, R3 = Ph) (0.500 g, 1.534 mmol), PhI (0.21 cm3, 1.84 mmol, 1.2 eq.) and Et3N (6 cm3). To this mixture was added PdCl2(PPh3)2 (21.5 mg, 0.0306 mmol, 0.02 eq.) and after 5 min CuI (2.9 mg, 0.0153 mmol, 0.01 eq.) was added and the reaction heated to 50 °C for 20 h. The reaction mixture was then filtered and the remaining amine removed in vacuo. Purification by column chromatography (EtOAc/hexane 0 to 0.5%) afforded a light yellow oil (0.5874 g, 1.461 mmol, 95%); [α]28D +45.0 (c 1.088, CHCl3); m/z 403.2 [M + 1]+, 425.2 [M + 23]+; νmax 2942, 2890, 2864, 1762, 1644, 1450 and 1240 cm−1; (Found (ESI): 425.2265 M + Na C27H34NaOSi requires 425.2271); δH (400 MHz, CDCl3) 7.50 (2H, d, J 7.50, Ar), 7.49–7.45 (2H, m, Ar), 7.40–7.31 (6H, m, Ar), 5.56 (1H, s, ArCH), 4.67 (1H, d, J 15.7, OCHH), 4.54 (1H, d, J 15.7, OCHH), 1.12 (21H, m, Si(CH(CH3)3); δC (100 MHz, CDCl3) 138.00 (ipso, Ar), 131.83 (CH, Ar), 128.47 (CH, Ar), 128.45 (CH, Ar), 128.42(CH, Ar), 128.25 (CH, Ar), 127.84 (CH, Ar), 122.65 (ipso, Ar), 103.87 (quat., CC), 90.05 (quat., CC), 86.54 (quat., CC), 84.07 (quat., CC) 70.57 (CH(OH)) 56.04 (OCH2) 18.63 (Si(CH(CH3)2)3), 11.20 (Si(CH(CH3)2)3); δSi (99 MHz, CDCl3) −1.53 (s).
Ruthenium cyclone complex 15b
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = TMS, R2 = Si(tBu)Me2, R3 = Ph), (1.000 g, 2.808 mmol 3 eq.), acetonitrile (10 cm3) and Ru3(CO)12 (0.5986 g, 0.9363 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 2 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (4 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane 0 to 10%) to afford a yellow solid (0.4522 g, 0.7947 mmol, 28%). The metal complex was characterised as a mixture of two diastereoisomers in an approximate ratio of 3:1; m/z 571.0 [M + 1]+; (Found (ESI): 571.0918 M + H C25H33O5RuSi2 requires 571.0911); νmax 2951, 2927, 2894, 2881, 2853, 2077, 2019, 1197 and 1632 cm−1; δH (400 MHz, CDCl3) 7.49–7.44 (1.5H, m, Ar) 7.41–7.36 (3H, m, Ar), 7.20–7.16 (0.5H, m, Ar), 5.85 (1H, s, ArCH), 4.98 (1H, d, J 12.1), 4.85 (0.75H, dd, 2.3, 12.8, OCH2, major) 4.90 (0.25H, d, J 12.5, OCH2, minor), 0.99 (6H, s, SitBu), 0.62 (3H, s, Si(CH3)2), 0.34 (3H, s, Si(CH3)2), 0.30 (6H, s, Si(CH3)3), 0.22 (3H, s, Si(CH3)2), −0.75 (3H, s, Si(CH3)2); δC (100 MHz, CDCl3) 193.80 (quat., CO), 185.80 (quat., CO), 138.75 (ipso, Ar), 138.48 (ipso, Ar), 129.59 (CH, Ar), 129.41 (CH, Ar), 129.14 (CH, Ar), 128.42 (CH, Ar), 128.32 (CH, Ar), 127.67 (CH, Ar), 119.91 (quat.), 119.29 (quat.), 117.12 (quat.), 82.56 (CH, ArCH), 81.33 (CH, ArCH), 67.13 (quat./CH2), 66.92 (quat./CH2), 64.33 (quat./CH2), 64.20 (quat./CH2), 27.67 (CH/CH3), 27.25 (CH/CH3), 19.00 (quat./CH2), 17.74 (quat./CH2), 14.10 (CH/CH3), −0.28 (CH/CH3), −0.45 (CH/CH3), −3.15 (CH/CH3), −4.66 (CH3), −4.78 (CH3); δSi (99 MHz, CDCl3), 4.03 (s, TBS), 3.20 (s, TBS), −3.72 (s, TMS).
Ruthenium cyclone complex 15a
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = TIPS, R2 = Si(tBu)Me2, R3 = Ph) (0.1395 g, 0.3170 mmol 2 eq.), acetonitrile (1 cm3) and Ru3(CO)12 (0.1013 g, 0.1585 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 2 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (2 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane 0 to 10%) to afford a yellow solid (0.1404 g, 0.2151 mmol, 68%). The metal complex was characterised as a mixture of two diastereoisomers in an approximate ratio of 3:1; m/z 655.1 [M + 1]+, 677.1 [M + 23]+; (Found (ESI): 655.1861 M + H C31H45O5RuSi2 requires 655.1851); νmax 2946, 2926, 2862, 2077, 2019, 1998 and1629 cm−1; δH (400 MHz, CDCl3) 7.50–7.47 (1.5H, m, Ar), 7.42–7.37 (3H, m, Ar), 7.22–7.20 (0.5H, m, Ar), 5.85 (1H, s, ArCH), 5.00 (1H, d, J 12.8, OCHH), 4.88 (0.75H, dd, J 2.1, 12.8, OCHH, major), 4.79 (0.25H, d, J 12.5, OCHH, minor), 1.41 (3H, septet, J 7.2, SiCH(CH3)2), 1.18 (18H, pseudo t, J 7.2, SiCH(CH3)2), 0.98 (9H, s, SitBu), 0.24 (3H, s, Si(CH3)2), −0.71 (3H, s, Si(CH3)2); δC (100 MHz, CDCl3) 193.76 (quat., CO), 185.60 (quat., CO), 138.53 (ipso, Ar), 129.62 (CH, Ar), 129.41 (CH, Ar), 129.14 (CH, Ar), 128.95 (CH, Ar), 128.54 (CH, Ar), 128.51 (CH, Ar), 128.32 (CH, Ar), 127.70 (CH, Ar), 121.93 (quat.), 118.11 (quat.), 82.02 (CH), 81.25 (CH), 67.88 (quat./CH2), 64.91 (quat./CH2), 64.01 (quat./CH2), 27.81 (CH/CH3), 27.39 (CH/CH3), 19.67 (CH3), 19.33 (CH3), 19.20 (CH3), 19.15 (CH3), 19.07 (quat.), 15.46 (CH/CH3), 12.37 (CH/CH3), 12.29 (CH/CH3), −3.08 (CH3), −4.40 (CH3); δSi (99 MHz, CDCl3), 4.22 (s, TBS), 3.31 (s, TBS), 2.51 (s, TIPS).
Ruthenium cyclone complex 15c
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = Ph, R2 = Si(tBu)Me2, R3 = Ph), (0.250 g, 0.6944 mmol 2 eq.), acetonitrile (1.5 cm3) and Ru3(CO)12 (0.2219 g, 0.3472 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 3 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (2 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane; 0 to 10%) to afford a yellow solid (0.1799 g, 0.3139 mmol, 45%). The metal complex was characterised as a mixture of two diastereoisomers in an approximate ratio of 4:1; m/z 575.0 [M + 1]+; (Found (ESI): M + H 575.0832 C28H29O5RuSi requires 575.0829); (Found (ESI): M + Na 597.0654 C28H28NaO5RuSi requires 597.0649); νmax 2952, 2928, 2883, 2854, 2064, 2020 and 2009 cm−1; δH (400 MHz, CDCl3) 7.87 (0.3H, d, J 7.3, Ar), 7.83 (1.6H, d, J 7.3, Ar), 7.54–7.51 (1.5H, m, Ar), 7.44–7.35(5H, m, Ar), 7.32–7.24 (1.5H, m, Ar) 5.90 (1H, s, ArCH), 5.40 (0.85H, dd, J 0.9, 12.9, OCHH, major), 5.34 (0.15H, dd, J 1.4, 12.5, OCHH, minor) 5.23 (0.15H, d, J 12.5, OCHH, minor), 5.15 (0.85H, dd, J 2.4, 12.9, OCHH, major), 1.03(7.65H, s, SitBu, major), 0.66 (1.35H, s, SitBu, minor), 0.35 (2.35H, s, Si(CH3)2, major), 0.33 (0.65H, s, Si(CH3)2, minor), −0.03(0.5H, s, Si(CH3), minor), −0.74 (2.5H, s, Si(CH3), major); δC (100 MHz, CDCl3) 193.78 (quat, CO), 193.72 (quat CO), 180.08 (quat, CO), 138.32 (ipso, Ar), 138.08 (ipso, Ar), 132.20 (ipso, Ar), 132.15 (ipso, Ar), 129.78 (CH, Ar), 129.56 (CH, Ar), 129.19 (CH, Ar), 128.88 (CH, Ar), 128.86 (CH, Ar), 128.54 (CH, Ar), 128.38 (CH, Ar), 127.79 (CH, Ar), 127.76 (CH, Ar), 127.13 (CH, Ar), 127.11 (CH, Ar), 115.10 (quat.), 113.14 (quat.), 82.62 (CH), 81.46 (CH), 75.53 (quat./CH2), 67.63 (quat./CH2), 67.30 (quat./CH2), 62.34 (quat./CH2), 34.64 (quat./CH2), 31.56 (quat./CH2), 27.72 (CH/CH3), 27.26 (CH/CH3), 25.26 (quat./CH2), 22.63 (quat./CH2), 19.00 (quat./CH2), 14.10 (CH3), −2.96 (CH3), −4.64 (CH3), −4.72 (CH3, SiCH3); δSi (99 MHz, CDCl3), 4.22 (s, TBS), 3.31 (s, TBS), 2.51 (s, TIPS).
Ruthenium cyclone complex 15e
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = Ph, R2 = Si(iPr)3, R3 = Ph) (0.3906 g, 0.9716 mmol, 2 eq.), acetonitrile (3 cm3) and Ru3(CO)12 (0.3104 g, 0.4858 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 2 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (2 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane 0 to 5%) to afford a yellow solid (Rf 0.2, 0.1994 g, 0.3242 mmol, 33%). Found to be a 3:2 mixture of diastereoisomers; m/z 617.1 [M + 1]+; (Found (ESI): M + H 617.1307 C31H35O5RuSi requires 617.1300); (Found (ESI): M + Na 639.1128 C31H34NaO5RuSi requires 639.1119); νmax 2944, 2864, 2076, 2001 and 1636 cm−1; δH (400 MHz, CDCl3) 7.87–7.81 (2H, m, Ar), 7.53–7.48 (1.3H, m, Ar), 7.41–7.35 (4.5H, m, Ar), 7.31–7.23 (2.2H, m, Ar), 5.97 (0.4H, s, ArCH, minor), 5.92 (0.6H, s, ArCH, major), 5.43 (0.6H, d, J 12.8, OCHH, major), 5.27 (0.4H, d, J 12.4, OCHH, minor), 5.21 (0.4H, d, J 12.4, OCHH, minor), 5.15 (0.6H, dd, J 2 Hz, 12.8, OCHH, major), 1.10 (7.5H, s, SiCH(CH3)2), 1.05 (3.75H, d, J 7.2, SiCH(CH3)2), 1.00 (3.75H, d, J 7.2, SiCH(CH3)2), 0.94 (6H, d, J 6.0, SiCH(CH3)2); δC (100 MHz, CDCl3) 193.89 (quat., CO), 193.80 (quat., CO), 180.18 (quat., CO), 179.75 (quat., CO), 138.44 (ipso, Ar), 137.96 (ipso, Ar), 132.22 (ipso, Ar), 132.20 (ipso, Ar), 129.67 (CH, Ar), 129.53 (CH, Ar), 129.06 (CH, Ar), 128.87 (CH, Ar), 128.39 (CH, Ar), 128.22 (CH, Ar)127.73 (CH, Ar), 127.54(CH, Ar), 127.19 (CH, Ar), 127.16 (CH, Ar), 113.27 (quat.), 113.11 (quat.), 107.34 (quat.), 83.00 (ArCH), 81.85 (ArCH), 75.36 (quat.), 67.13 (quat.), 67.08 (quat.), 61.88 (quat.), 19.76 (CH3/CH), 19.24 (CH3/CH), 19.18 (CH3/CH), 19.11 (CH3/CH), 12.86 (CH3/CH), 12.78 (CH3/CH); δSi (99 MHz, CDCl3), 4.22 (s, TBS), 3.31 (s, TBS), 2.51 (s, TIPS).
Ruthenium cyclone complex 15d
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = TMS, R2 = Si(iPr)3, R3 = Ph) (0.400 g, 1.005 mmol 3 eq.), acetonitrile (3 cm3) and Ru3(CO)12 (0.2141 g, 0.3350 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 3 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (2 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane 0 to 5%) to afford a yellow solid (0.1542 g, 0.2524 mmol, 25%). The metal complex was characterised as a mixture of two diastereoisomers in an approximate ratio of 7:3; m/z 613.1 [M + 1]+; (Found (ESI): MH+ 613.1375 C28H39O5RuSi2 requires 613.1381); νmax 2943, 2891, 2864, 2047 and 1195 cm−1; δH (400 MHz, CDCl3) 7.48–7.43 (1.5H, m, Ar), 7.39–7.33 (3H, m, Ar), 7.19–7.14 (0.5H, m, Ar), 5.92 (0.3H, s, ArCH, minor), 5.86 (0.7H, s, ArCH, major), 5.00 (0.7H, s, J 12.8, OCHH, major), 4.92 (0.3H, d, J 12.5, OCHH, minor), 4.83 (0.7H, dd, J 2.1, J 12.8, OCHH, major), 4.78 (0.3H, d, J 12.5, OCHH, minor), 1.13–1.02 (11H, m, SiiPr), 0.99–0.94 (5H, m, SiiPr), 0.90–0.86 (5H, m, SiiPr), 0.33 (2.7H, s, TMS, minor), 0.29 (6.3H, s, TMS, major); δC (100 MHz, CDCl3) 193.85 (quat., CO), 185.98 (quat., CO), 185.91 (quat., CO), 138.44 (quat., Ar), 138.79(quat., Ar), 138.67 (quat., Ar), 129.49 (CH, Ar), 129.38 (CH, Ar), 128.26 (CH, Ar), 128.13 (CH, Ar), 127.43 (CH, Ar), 119.85 (quat.), 118.49 (quat.), 116.93 (quat.), 114.58 (quat.), 82.90 (ArCH), 81.69 (ArCH), 67.12 (quat.), 66.64 (quat.), 66.58 (quat.), 65.77 (quat.), 64.43 (quat.), 64.05 (quat.), 19.69 (CH3/CH), 19.18 (CH3/CH), 19.12 (CH3/CH), 19.00 (CH3/CH), 17.68 (CH3/CH), 12.80 (CH3/CH), 12.76 (CH3/CH) 12.27(CH3/CH), −0.42 (CH3, TMS), −0.51 (CH3, TMS); δSi (99 MHz, CDCl3), 4.22 (s, TBS), 3.31 (s, TBS), 2.51 (s, TIPS).
Ruthenium cyclone complex 15f
A pressure tube was charged with (R)-dipropargylic ether 8 (R1 = TBDMS, R2 = Si(iPr)3, R3 = Ph) (0.3331 g, 0.7571 mmol 3 eq.), acetonitrile (3 cm3) and Ru3(CO)12 (0.1613 g, 0.2523 mmol, 1 eq.) and purged under a steady stream of N2. The tube was then sealed and heated to 100 °C. After 3 d the reaction mixture was cooled, carefully depressurised and the solvent removed in vacuo. The resulting black semisolid was dissolved in DCM (2 cm3) and filtered through a cotton wool plug and loaded onto a short silica column (EtOAc/hexane 0 to 5%) to afford a sticky red solid (0.104 g, 0.1596 mmol, 21%). The metal complex was characterised as a mixture of two diastereoisomers in an approximate ratio of 3:2; m/z 655.1 [M + 1]+; (Found (ESI): M + H 655.1850 C31H45O5RuSi2 requires 655.1851); νmax 2945, 2927, 2890, 2863, 2079, 2022, 2002 and1620 cm−1; δH (400 MHz, CDCl3) 7.47–7.43 (1 H, m, Ar), 7.39–7.34 (3H, m, Ar), 7.20–7.16 (1H, m, Ar), 5.93 (0.45H, s, ArCH, minor), 5.86 (0.55H, s, ArCH, major), 5.01 (0.55H, dd, J 0.8, 12.8, OCHH, major), 4.93 (0.45H, dd, J 1.2, 12.5, OCHH, minor), 4.85 (0.55H, dd, J 2.2, 12.8, OCHH, major), 4.78 (0.45H, d, J 12.5, OCHH, minor), 1.19–0.6 (30H, m, SiiPr3 and SitBu), 0.43 (1.35H, s, SiCH3, minor), 0.41 (1.65H, s, SiCH3, major), 0.12 (1.35H, s, SiCH3, minor), 0.10 (1.65H, s, SiCH3, major); δC (100 MHz, CDCl3) 193.84 (quat., CO), 185.43 (quat., CO), 185.34 (quat., CO), 138.68 (ipso, Ar), 138.31 (ipso, Ar), 129.53 (CH, Ar), 129.37 (CH, Ar), 129.00 (CH, Ar), 128.56 (CH, Ar), 128.51 (CH, Ar), 128.34 (CH, Ar), 128.14 (CH, Ar), 127.54 (CH, Ar), 127.39 (CH, Ar), 121.64 (quat.), 117.79 (quat.), 82.74 (ArCH), 81.70 (ArCH), 66.97 (quat./CH2), 66.85 (quat./CH2), 66.82 (quat./CH2), 65.65 (quat./CH2), 63.91 (quat./CH2), 61.87 (quat./CH2), 27.42 (CH3), 27.37 (CH3), 19.67 (CH3/CH), 19.20 (CH3/CH), 19.03 (CH3/CH), 18.84 (quat.), 18.78 (quat.), 18.56 (CH3/CH), 12.76 (CH3/CH), 12.63 (CH3/CH) 11.20 (CH3/CH), 11.12 (CH3/CH), 10.95 (CH3/CH), −4.34 (CH3, TBS), −4.70 (CH3, TBS) −4.81; δSi (99 MHz, CDCl3), 4.22 (s, TBS), 3.31 (s, TBS), 2.51 (s, TIPS).
Reduction of acetophenone using iron catalysts and FA/TEA
Complex 7a (7.8 mg, 19.1 μmol), trimethylamine-N-oxide (2.1 mg, 18.9 μmol) and acetophenone (23.0 mg, 0.191 mmol) were dissolved in 5:2 formic acid:triethylamine (0.2 cm3) and heated at 28 °C for 18 h. The reaction was monitored over time by GC (Chrompac cyclodextrin-β-236 M 50 M column, T = 130 °C, inj T = 220 °C, det T = 220 °C, 15 psi He carrier gas). RT: Acetophenone: 13.4 min. 1-Phenylethyl formate: 15.1 (S), 15.5 (R) min. 1-Phenylethanol: 17.4 (R), 18.0 (S) min.1 The above procedure was repeated using other complexes, temperatures and reaction times. The product configurations were assigned by comparison to previously quoted data1 and use of authentic reference samples.
Reduction of acetophenone using iron catalysts and iPrOH
Complex 7a (7.8 mg, 19.1 μmol), trimethylamine-N-oxide (2.1 mg, 18.9 μmol) and acetophenone (23.0 mg, 0.191 mmol) were dissolved in iPrOH (0.96 cm3) and heated at 28 °C for 18 h. The reaction was monitored over time by GC (Chrompac cyclodextrin-β-236 M 50 M column, T = 130 °C, inj T = 220 °C, det T = 220 °C, 15 psi He carrier gas). RT: Acetophenone: 13.4 min. 1-Phenylethanol: 17.4 (R), 18.0 (S) min.1 The above procedure was repeated using other temperatures and concentrations.General ruthenium-hydride synthesis prior to use in reductions12
To Ru(TMS-TMS) (22.6 mg, 0.05 mmol) in THF (1 cm3) was added aqueous sodium hydroxide (0.5 cm3, 1 M, 0.50 mmol) and stirred for 2.5 h. An excess of H3PO4 (0.3 cm3) was then added and the reaction extracted using Et2O (3 × 10 cm3), dried over MgSO4 and the solvent removed all under a nitrogen atmosphere to afford the hydride species as a yellow oil which was used immediately without further purification. Selected data for each hydride this formed is given below.Reduction of acetophenone using ruthenium hydride complexes
:2, 4 cm3) and heated at 60 °C over 30 min. Acetophenone (0.92 cm3, 0.9476 g, 7.90 mmol) was then added and the reaction stirred at 60 °C over 7 days and the reaction was monitored by GC.
Acknowledgements
We thank EPSRC for financial support of JEDM and JPH (EP/F019424) and TCJ (Supergen XIV H-delivery EP/G01244X/1). JG was an MChem project student supported by Warwick University.Notes and references
- (a) B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS; (b) Y. Blum and Y. Shvo, J. Organomet. Chem., 1985, 282, C7–C10 CrossRef CAS; (c) Y. Shvo, D. Czarkie and Y. Rahamim, J. Am. Chem. Soc., 1986, 108, 7400–7402 CrossRef CAS.
- (a) O. Pàmies and J.-E. Bäckvall, Chem. Rev., 2003, 103, 3247–3262 CrossRef CAS; (b) B. A. Persson, A. I. E. Larsson, M. Le Ray and J.-E. Bäckvall, J. Am. Chem. Soc., 1999, 121, 1645–1650 CrossRef CAS; (c) O. Pamies and J.-E. Bäckvall, J. Org. Chem., 2001, 66, 4022–4025 CrossRef CAS.
- (a) L. K. Thalén, D. Zhao, J.-B. Sortais, J. Paetzold, C. Hoben and J.-E. Bäckwall, Angew. Chem., Int. Ed., 2009, 15, 3403–3410 CAS; (b) J. S. M. Samec and J.-E. Bäckvall, Chem.–Eur. J., 2002, 8, 2955–2961 CrossRef CAS; (c) J. Paetzold and J.-E. Bäckwall, J. Am. Chem. Soc., 2005, 127, 17620–17621 CrossRef CAS.
- (a) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 RSC; (b) S. E. Clapham, A Hadzovic and R. H. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS; (c) C. P. Casey, S. W. Singer, D. R. Powell, R. K. Hayashi and M. Kavana, J. Am. Chem. Soc., 2001, 123, 1090–1100 CrossRef CAS; (d) J. B. Johnson and J.-E. Bäckvall, J. Org. Chem., 2003, 68, 7681–7684 CrossRef CAS; (e) Comas-Vives, G. Ujaque and A. Lledós, Organometallics, 2007, 26, 4135–4144 CrossRef CAS; (f) C. P. Casey, S. E. Beetner and J. B. Johnson, J. Am. Chem. Soc., 2008, 130, 2285–2295 CrossRef CAS.
- (a) M. Yamakawa, I. Yamada and R. Noyori, Angew. Chem., Int. Ed., 2001, 40, 2818–2821 CrossRef CAS; (b) C. P. Casey and J. B. Johnson, J. Org. Chem., 2003, 68, 1998–2001 CrossRef CAS; (c) S. Hashiguchi, A. Fujii, K. J. Haack, K. Matsumura, T. Ikariya and R. Noyori, Angew. Chem., Int. Ed. Engl., 1997, 36, 288–290 CrossRef CAS; (d) F. K. Cheung, C. Lin, F. Minissi, A. Lorente Crivillé, M. A. Graham, D. J. Fox and M. Wills, Org. Lett., 2007, 9, 4659–4662 CrossRef CAS; (e) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS.
- C. P. Casey, T. E. Vos, S. W. Singer and H. A. Guzel, J. Org. Chem., 2002, 21, 5038–5046 CAS.
- (a) H.-J. Knölker, E. Baum, H. Goesmann and R. Klauss, Angew. Chem., Int. Ed., 1999, 38, 2064–2066 CrossRef CAS; (b) H. H. Zhang, D. Z. Chen, Y. H. Zhang, G. Q. Zhang and J. B. Liu, Dalton Trans., 2010, 39, 1972–1978 RSC; (c) G. N. Schrauzer, J. Am. Chem. Soc., 1959, 81, 5307–5310 CrossRef CAS.
- (a) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2007, 129, 5816–5817 CrossRef CAS; (b) C. P. Casey and H. Guan, J. Am. Chem. Soc., 2009, 131, 2499–2507 CrossRef CAS; (c) H. Zhang, D. Chen, Y. Zhang, G. Zhang and J. Liu, Dalton Trans., 2010, 39, 1972–1978 RSC.
- (a) M. G. Coleman, A. N. Brown, B. A. Bolton and H. Guan, Adv. Synth. Catal., 2010, 352, 967–970 CAS; (b) S. A. Moyer and T. W. Funk, Tetrahedron Lett., 2010, 51, 5430–5433 CrossRef CAS; (c) M. K. Thorson, K. L. Klinkel, J. Wang and T. J. Williams, Eur. J. Inorg. Chem., 2009, 295–302 CrossRef CAS.
- T. C. Johnson, G. J. Clarkson and M. Wills, Organometallics, 2011, 30, 1859–1868 CrossRef CAS.
- (a) A. J. Pearson and R. J. Shively Jr, Organometallics, 1992, 11, 4096–4104 CrossRef CAS; (b) A. J. Pearson and R. J. Shively Jr, Organometallics, 1994, 13, 578–584 CrossRef CAS.
- (a) Y. Yamamoto, K. Yamashita and M. Nakamura, Organometallics, 2010, 29, 1472–1478 CrossRef CAS; (b) A. Berkessel, S. Reichau, A. van der Höh, N. Leconte and J.-M. Neudörfl, Organometallics, 2011, 30, 3880–3887 CrossRef CAS.
- (a) N. A. Bailey, V. S. Jassal, R. Vefghi and C. White, J. Chem. Soc., Dalton Trans., 1987, 2815–2822 RSC; (b) J. H. Eekhof, H. Hogeveen and R. M. Kellogg, Chem. Commun., 1977, 705 RSC; (c) H.-J. Knölker, E. Baum and J. Heber, Tetrahedron Lett., 1992, 36, 7647–7650.
- (a) K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738–8739 CrossRef CAS; (b) D. J. Morris, A. M. Hayes and M. Wills, J. Org. Chem., 2006, 71, 7035–7044 CrossRef CAS; (c) J. A. Marshall, P Eidam and H. S. Eidam, J. Org. Chem., 2006, 71, 4840–4844 CrossRef CAS.
- Y. Yamamoto, Y. Miyabe and K. Itoh, Eur. J. Inorg. Chem., 2004, 3651–3661 CAS.
- O. Hamed, P. M. Henry and D. P. Becker, Tetrahedron Lett., 2010, 51, 3514–3517 CrossRef CAS.
- M. M. Midland, A. Tramontano, A. Kazubski, R. S. Graham, D. J. S. Tsai and D. B. Cardin, Tetrahedron, 1984, 40, 1371–1380 CrossRef CAS . (R)-Alpine borane is derived from (+)-α-pinene and gives the (R)-reduction product illustrated.
- (a) G. Bauer and K. A. Kirchner, Angew. Chem., Int. Ed., 2011, 50, 5798–5800 CrossRef CAS; (b) A. Naik, T. Maji and O. Reiser, Chem. Commun., 2010, 46, 4475–4477 RSC; (c) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 RSC; (d) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS; (e) C. Sui-Seng, F. Freutel, A. J. Lough and R. H. Morris, Angew. Chem., Int. Ed., 2008, 47, 940–943 CrossRef CAS; (f) A. Mikhailine, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2009, 131, 1394–1395 CrossRef CAS; (g) N. Meyer, A. J. Lough and R. H. Morris, Chem.–Eur. J., 2009, 15, 5605–5610 CrossRef CAS; (h) J.-S. Chen, L.-L. Chen, Y. Xing, G. Chen, W.-Y. Shen, Z.-R. Dong, Y.-Y. Li and J.-X. Gao, Acta. Chim. Sin. (Huaxue Xuebao), 2004, 62, 1745–1750 Search PubMed; (i) S. Zhou, S. Fleischer, K. Junge, S. Das, D. Addis and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8121–8125 CrossRef CAS; (j) A. A. Mikhailine and R. H. Morris, Inorg. Chem., 2010, 49, 11039–11044 CrossRef CAS; (k) P. O. Lagaditis, A. J. Lough and R. H. Morris, Inorg. Chem., 2010, 49, 10057–10066 CrossRef CAS; (l) K. Junge, K. Schroder and M. Beller, Chem. Commun., 2011, 47, 4849–4859 RSC; (m) S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120–5124 CrossRef CAS.
- C. J. Taylor, M. Motevalli and C. J. Richards, Organometallics, 2006, 25, 2899–2902 CrossRef CAS.
- T. Schubert, W. Hummel, M.-R. Kula and M. Müller, Eur. J. Org. Chem., 2001, 4181–4187 CrossRef CAS.
- L. Xu, M. R. Muller, X. Yu and B.-Q. Zhu, Synth. Commun., 2009, 39, 1611–1625 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra. See DOI: 10.1039/c1ob06010d |
| This journal is © The Royal Society of Chemistry 2012 |