3,4′-Linked bis(piperidines) related to the haliclonacyclamine class of marine alkaloids: synthesis using crossed-aldol chemistry and preliminary biological evaluations†
Martin G.
Banwell
*a,
Mark J.
Coster
a,
Natasha L.
Hungerford
a,
Mary J.
Garson
b,
Stephen
Su
c,
Andrew C.
Kotze
d and
Murray H. G.
Munro
e
aResearch School of Chemistry, Institute of Advanced Studies, The Australian National University, Canberra, ACT 0200, Australia. E-mail: mgb@rsc.anu.edu.au
bSchool of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, QLD 4072, Australia
cCytopia Pty Ltd, 576 Swan St, Richmond, VIC 3121, Australia
dCSIRO Livestock Industries, Queensland Bioscience Precinct, Brisbane, QLD 4067, Australia
eDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, New Zealand
First published on 27th September 2011
Abstract
Compounds 2–5, incorporating various elements of the 3,4′-bis(piperidine) core associated with the sponge-derived alkaloid haliclonacyclamine A (HA, 1), have been prepared through, inter alia, aldol-type reactions of N-substituted piperidin-4-ones and certain derivatives. Screening of these compounds in various assays, including an ecological one, reveals that compound 5 exhibits allelochemical properties similar to those associated with HA itself.
Introduction
The natural product haliclonacyclamine A (HA, 1) was first isolated from the sponge Haliclona sp. 628 that inhabits the coral reefs adjacent to Heron Island on the Great Barrier Reef. The structure, including absolute configuration, of this novel tetracyclic alkylpiperidine alkaloid was established using a combination of NMR and X-ray crystallographic techniques.1 Structurally-related alkaloids (halicyclamine A, haliclonacyclamines B–F, tetradehydrohaliclonacyclamine A, halichondramide, tetradehydrohalicyclamine A) varying, inter alia, in the positions and degrees of unsaturation of the hydrocarbon chains linking the 3,4′-bis(piperidine) cores of these systems have been reported.2 Other structural variants include those incorporating hydroxy groups in the lower chain (arenosclerins A–E, 22-hydroxyhalicyclamine A, 22-hydroxyhaliclonacyclamine B),2a,2e,3 differently sized “spacer groups” (halicyclamine B, neopetrosamine, xestoproxamines A–B)4,5 and, intriguingly, a methyl-branched spacer group (xestoproxamine C).5 This extraordinary suite of compounds is probably generated in vivo in a manner similar to that proposed by Baldwin and Whitehead for the biogenesis of the manzamine alkaloids6 and therefore involving, as the pivotal steps, intramolecular Diels–Alder cycloaddition and retro-Mannich-type fragmentation reactions.1c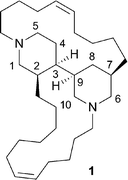
The haliclonacyclamines exhibit strong antimicrobial activity and potent cytotoxicity against various cancer cell lines.1a,1b,2b,2c Furthermore, haliclonacyclamines A and B have recently been shown to exhibit anti-mycobacterial activity against Mycobacterium smegmatis and M. Bovis Bacille de Calmette et Guérin (BCG) under both aerobic and hypoxic conditions.3 A study on the mechanism of anti-mycobacterial action of the related halicyclamine A has been reported.3b Semi-synthetically-derived tetrahydrohaliclonacyclamine A (≡ perhydrohaliclonacyclamine2c) is cytotoxic,1a,1b binds to a variety of ion channels/GPCRs as well as acting as a muscarinic M1antagonist, a mode of action that may be related to its cytotoxicity.7 However, the most intriguing biological activities associated with the haliclonacyclamines are their in situ effects. Thus, an ecological study revealed that the natural mixture of haliclonacyclamines A–D is an effective toxin towards coral tissue and deters feeding by reef fish,8 while pure haliclonacyclamine A exhibits anti-fouling properties because it inhibits the metamorphosis of the larvae of a wide range of taxa, including the ascidian Herdmania momus (syn curvata).9
Despite their fascinating molecular architectures and biological properties, thus far there has been only a handful of reports concerned with the total synthesis of the haliclonacyclamine framework.7 As such the structural features of these natural products responsible for their activities remains uncertain, although the tetra-alkylated 3,4′-bis(piperidine) core is almost certain to be a crucial motif. Herein, therefore, we describe simple synthetic protocols based on, inter alia, aldol-type chemistry that have allowed for the rapid assembly of compounds 2–5 related to the haliclonacyclamine core. We also report on the outcomes of the preliminary biological evaluation of these same compounds which has revealed that the most elaborate of them (compound 5) retains much of the activity associated with the parent haliclonacyclamine framework.
Results and discussion
Synthetic studies
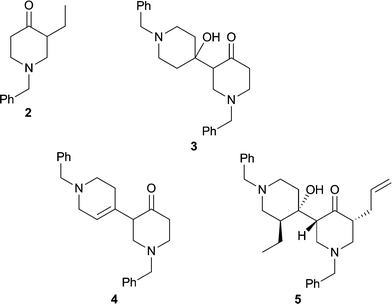
The commercially available 1-benzyl-4-piperidinone (6) served as the starting material for the synthetic aspects of the present study and the capacity of various derived enolates to participate in C-alkylation reactions, including aldol-type processes, was explored as a means of assembling the targeted substructures 2–5 of HA (1). The benzyl group was chosen to protect the secondary amine residue of 4-piperidinone because of its ease of removal with triphosgene.10 Our first experiments (Scheme 1) rapidly established that the previously unreported but readily prepared N,N-dimethylhydrazone (DMH) derivative, 7 (100%), of ketone 6 was smoothly C-ethylated upon sequential treatment with BuLi then ethyl iodide.11 After workup the required ketone 2 was obtained in 81% yield and thereby representing a useful improvement upon a previously reported12 route to this compound. Two distinct reaction conditions were examined in an effort to effect the self-aldol-type reaction of compound 6 which, if successful, would lead to the 3,4′-bis(piperidine) core of HA. When the tin(II)-mediated aldol process was applied to compound 6 at −78 °C then the desired aldol product 3 was obtained in ca. 60% yield. In contrast, when the same substrate (6) was subjected to treatment with 2 molar equivalents of BF3·Et2O in refluxing 1,2-dichloroethane then the unsaturated 3,4′-bis(piperidine) 4 was formed, presumably via intermediate 3, in 64% yield. Compound 4 was accompanied by ca. 16% of the corresponding conjugated enone from which it could be separated by conventional chromatographic methods.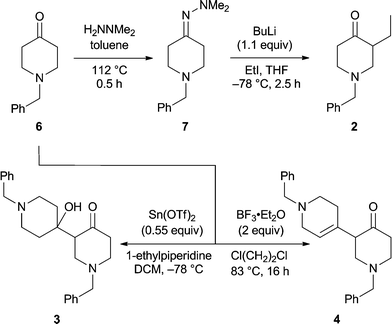 | ||
| Scheme 1 | ||
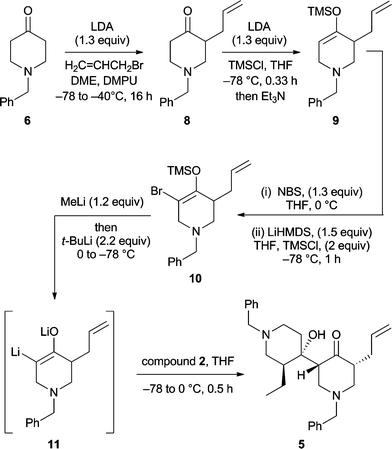
A necessary requirement for the development of a total synthesis of HA was the identification of protocols that allow for the crossed-aldol reaction of two differentially substituted 4-pyridinones. Since neither of the procedures depicted in Scheme 1 proved effective in this regard, alternatives were sought. Ultimately, the use of an α-keto dianion intermediate13 provided the desired crossed-aldol product 5 (Scheme 2). Thus, the enolate derived by treating compound 6 with lithium diisopropylamide (LDA) in the presence of DMPU could be trapped with allyl bromide to give compound 814 in 45% yield. Addition of a THF solution of the latter to a mixture of LDA/trimethylsilyl chloride (TMS-Cl)15 maintained at −78 °C resulted in the selective formation of the kinetically favoured silyl enol ether 9 (quantitative yield) which when treated with N-bromosuccinimide (NBS) gave the expected α-allyl-α′-bromoketone as a ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of cis- and trans-isomers. This sensitive ketone was immediately treated with lithium hexamethyldisilazide (LiHMDS) and the resulting enolate trapped with TMSCl at −78 °C to give the brominated silyl enol ether 10 in 49% yield.
:1 mixture of cis- and trans-isomers. This sensitive ketone was immediately treated with lithium hexamethyldisilazide (LiHMDS) and the resulting enolate trapped with TMSCl at −78 °C to give the brominated silyl enol ether 10 in 49% yield.
 | ||
| Scheme 2 | ||
In the final and pivotal step of the reaction sequence, compound 10 was treated sequentially with methyllithium then t-butyllithium and the α-keto dianion 11 so formed was trapped with 1-benzyl-3-ethyl-4-piperidinone (2) and thereby forming β-hydroxyketone 5 (24%) which could be separated from various co-produced diastereoisomers (44% combined yield) using flash chromatographic techniques. The structure of compound 5 was established by NMR spectroscopic methods, with nOe techniques being used to determine the illustrated stereochemistry (Fig. 1).
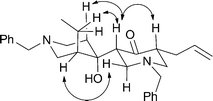 | ||
| Fig. 1 Significant nOe interactions used in assigning the structure of compound 5. | ||
Each of the reaction sequences presented above serves to highlight the capacity of aldol reactions to generate 3,4′-bis(piperidines) including tetra-alkylated variants. As such, this type of chemistry, when used in conjunction with ring-closing metathesis (RCM) protocols, could provide a means for developing total syntheses of the haliclonacyclamines themselves. Work directed towards such ends is now underway in our laboratories.
Biological evaluation of compounds 2–5
Our previous ecological studies have established that HA (1) inhibits the settlement and metamorphosis of sponge, polychaete, gastropod and bryozoan larvae.9 The synthetic compounds 2–5 were screened against small populations of the larvae of the solitary ascidian Herdmania momus, commonly found on the Great Barrier Reef, to test if the various treatments induced or inhibited settlement, and to establish whether larvae subsequently progressed normally through metamorphosis under these conditions. Since there is natural variation in larval settlement rates, the assay runs included experimental controls to assess spontaneous effects in filtered seawater (FSW) or in the presence of the known settlement inducer 40 mM KCl-elevated seawater.9In an initial assay, compounds 2 and 4 were screened at 25 μg and at 50 μg per well. At 25 μg, settlement rates shown by treatment larvae were similar to those in FSW and larvae progressed normally (data not shown). At 50 μg per well [Fig. 2(a)], all sets of treated larvae showed initial settlement rates that were elevated above those of FSW, but these were not as pronounced as for KCl-treated larvae; at 3 h, between 70–80% of treatment larvae had settled compared to <70% of the control larvae (FSW). At 12 h, postlarvae exposed to compound 2 showed normal development (characterised by a rounded morphology and the formation of ampullae) while those exposed to compound 4 were retarded and lacked any sign of ampullar development. After 24 h there was a further pronounced difference in the appearance of the two sets of treated postlarvae, while at 36 h those exposed to compound 2 were behind the control larvae in development although still developing normally. In contrast, larvae exposed to compound 4 were necrotic after 36 h.
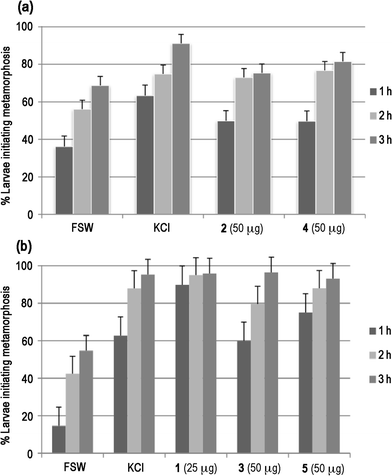 | ||
| Fig. 2 The effects of haliclonacyclamine mimic compounds on settlement of larvae of the Great Barrier Reef ascidian Herdmania momus. (a) Data for compounds 2 and 4 (top); (b) Data for compounds 3 and 5 compared to the natural sponge metabolite haliclonacyclamine A (HA, 1) (bottom). | ||
A second assay run compared compounds 3 and 5 at 25, 50 and 100 μg per well, and compound 4 at 100 μg per well against the inhibitory effects of HA (1) at 25 μg per well. At 50 μg per well for the two mimics, all sets of treated larvae again showed initial settlement rates that were elevated above those of FSW [Fig. 2(b)]; at 3 h, between 70–80% of treatment larvae had settled compared to <60% of the control larvae (FSW). However, settlement rates for compound 3 were not as pronounced as for HA (1) or compound 5, or for KCl-treated larvae. The settlement rates for analogue 5 were comparable to those of KCl-treated larvae, but did not show the rapid response associated with the HA treatment. At 3 h, between 70–80% of these treated larvae had initiated metamorphosis whereas close to 90% of HA-treated larvae initiated metamorphosis within 1 h.9 At 3 h, postlarvae exposed to HA (1) showed abnormal development and within 8 h all of these larvae were dead. Those exposed to compounds 3 and 5 were progressing through normal metamorphosis at the 3 h stage, but were mildly retarded compared to control larvae; evidence of abnormal development was apparent after 8 h, and all of these treated larvae were dead after 12 h. For both compounds 3 and 5, these abnormal developmental effects were less apparent in the 25 μg treatment; larvae exposed to compound 3 continued through metamorphosis for 24 h, but showed some abnormalities, while those exposed to compound 5 progressed normally (but behind KCl) for the first 8 h, but were dead at 12 h. At 100 μg, larvae exposed to both compounds 3 and 5 were significantly retarded compared to controls, and all larvae were dead at 12 h.
In summary, then, these assays revealed that compound 2 had no marked effect on larval settlement and metamorphosis, while compounds 3–5 each inhibited development, but at a higher concentration than the natural metabolite haliclonacyclamine A. The most potent synthetic analogue was compound 5. In ascidians, haliclonacyclamine A inhibits formation of ampullae and other postlarval structures at a key development point in early metamorphosis that corresponds to an increase in gene activity.9a These toxic effects benefit the sponge in two ways: firstly, a potential competitor is removed; secondly, the dead postlarvae are a potential source of food for either the sponge or associated microorganisms.
For comparison with their ecological effects, compounds 2–5 were also subjected to cytotoxicity, antimicrobial, and antiparasitic screening. Table 1 shows that when tested at a concentration of 20 μM, compounds 2–5 inhibited cell growth by >10% in 3, 6, 6, and 10 of the 13 different human cancer cell lines targeted. These data reveal that compounds 2 and 5 were, respectively, the least effective and most effective cytostatic compounds, results that parallel the ecological data. Haliclonacyclamine A was not screened directly against the thirteen cell-line panel, but earlier screening against the NCI 60 cell line panel had previously revealed its significant cytotoxic effects against certain cancer cell lines16 and a detailed study of its effects on cultured human cervical cancer (HeLa) cell lines has recently been presented. Thus, HeLa cells exposed to HA at a concentration of 22 μM appeared abnormal within 2 h, a marked change in cell permeability was observed within 4 h, and there was >90% cell death by 24 h.9b
| Cell linec | Compound | |||
|---|---|---|---|---|
| 2 | 3 | 4 | 5 | |
| a Percent inhibition values were calculated by comparison with vehicle-treated cells. The numbers shown are calculated. Since cell growth has some inherent variability, the values shown are approximate measurements of inhibition of cellular proliferation, and those identified as <0% simply represent no inhibition of cell growth. b DMSO was used as solvent in these assays. c See Experimental Section for details. | ||||
| A375 | −2 | 10 | 18 | 22 |
| A431 | 3 | 9 | 6 | 23 |
| HCT-15 | −2 | 1 | 13 | 77 |
| A549 | −4 | 5 | 5 | 4 |
| PA-1 | −4 | −1 | 4 | 12 |
| HT-1376 | 9 | 11 | 11 | 20 |
| MES-SA-Dx5 | 16 | 20 | 9 | 19 |
| MCF-7 | −9 | −5 | −1 | −2 |
| RAMOS | 9 | 10 | 9 | 20 |
| KHOS-NP | 2 | 4 | −1 | 5 |
| BT-20 | 3 | 9 | 11 | 17 |
| MAT-LyLu | 20 | 20 | 19 | 20 |
| DAUDI | 18 | 24 | 25 | 61 |
Screening against a panel of microorganisms (Table 2) revealed that although none of the three synthetic compounds 3, 4, or 5 tested was as potent as the natural sponge metabolite, they each possessed moderate activity against Bacillus subtilis and the marine fungi Trichophyton mentagrophytes and Cladosporium resinae. Natural 1 and synthetic 2–5 were also subjected to preliminary screening against the free-living larval stages of the parasitic nematodes Haemonchus contortus and Trichostrongylus colubriformis.17 All five compounds showed modest anthelmintic activity and inhibited larval development by 100% when tested at 100 μg mL−1, while at the lower concentration of 10 μg mL−1, significant numbers of larvae were able to develop as normal for each of the compounds (data not shown). Unfortunately, there was insufficient material for additional screening.
Conclusions
Although marine ecological studies frequently target structure–activity relationships among naturally-derived candidate allelochemicals,18 only a few studies have examined the effects of synthetic mimics. Lindel et al. assayed fourteen synthetic analogues, in addition to seven bromopyrrole sponge metabolites, for their inhibitory effects on feeding by the omnivorous reef fish Thalassoma bifasciatum. They found that a pyrrole group was essential for feeding inhibition activity, while an additional imidazole group further enhanced activity.19 A subsequent study explored chemical modifications of the pyrrole ring and highlighted the importance of the bromination pattern.20 Although these and other studies21 emphasise the key role of individual structural elements in determining ecological effects, the spatial arrangement of functional groups also needs to be considered.19As demonstrated by our recent study on the tambjamines,22 access to quantities of synthetically-derived metabolites in larger amounts than can be supplied from natural sources significantly extends the capacity for screening against biological targets. Synthetic chemistry also provides rapid access to natural product libraries for compound screening.23 Our synthetic study provided a suite of mimics of the haliclonacyclamine A skeleton that were then screened against a range of biological targets. The low activity noted for the simple 1-benzylpiperidinone (2) in the various assays highlights the likely importance of a 3,4′-bis(piperidine) core for effective ecological activity. The synthetic protocol employed provided a concise route to mimics with this scaffold. Three synthetic compounds that embodied the heterocyclic core of the haliclonacyclamines were tested, and all showed useful activity in the larval assays, as well as some antimicrobial activity. Assessment of the antiparasitic activity of the mimics was limited by compound availability. The most potent synthetic compound 5 is a very interesting mimic of the structurally complex and biologically potent haliclonacyclamine A (1), as it is the only synthetic compound that replicates a key structural feature of the alkaloid, namely the alkyl substitution at C-2 and at C-7 of 1 that is a natural structural consequence of a biosynthetic pathway involving a nicotinic acid precursor.1c The size and shape of alkyl substituents at these positions may well have a profound influence on ecological activity. Accordingly, the screening of more advanced synthetic intermediates would appear to be worthwhile.
The present work reinforces the notions that “function-oriented synthesis (FOS)”24 and “molecular editing through diverted total synthesis”25 are important concepts in identifying and then preparing simplified analogues of biologically active natural products that possess useful properties.
Experimental
(i) Synthetic studies
Compound
2. A magnetically stirred solution of hydrazone 7 (2.31 g, 10.0 mmol) in dry THF (20 mL) maintained at 0 °C was treated with n-BuLi (8.5 mL of a 1.29 M solution in hexanes, 11 mmol, 1.1 equiv). After 0.16 h the reaction mixture was cooled to −78 °C and iodoethane (1.6 mL, 20 mmol, 2.0 equiv) was added. After a further 2.5 h (at −78 °C) the reaction mixture was quenched by the slow addition of HCl (30 mL of a 1 M aqueous solution). The resulting mixture was stirred at 18 °C for 1 h, cooled to 0 °C, basified [with NaOH (ca. 15 mL of a 2 M aqueous solution)] to pH 9 then diluted with water (50 mL) and diethyl ether (50 mL). The separated aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic phases washed with brine (1 × 20 mL) before being dried (Na2SO4), filtered and then concentrated under reduced pressure to afford a clear, colourless oil. Subjection of this material to flash chromatography (silica, 1:4 → 3:7 v/v ethyl acetate/hexane gradient elution) provided, after concentration of the appropriate fractions (Rf 0.5 in ethyl acetate), compound 212 (1.76 g, 81%) as a clear, colourless oil. 1H NMR (300 MHz) δ 7.36–7.21 (5H, complex m), 3.63 (1H, d, J = 13.2 Hz), 3.52 (1H, d, J = 13.2 Hz), 3.00 (1H, ddd, J = 11.0, 5.3 and 2.2 Hz), 2.92 (1H, m), 2.55–2.31 (4H, complex m), 2.21 (1H, dd, J = 11.0 and 9.5 Hz), 1.81 (1H, m), 1.27 (1H, m), 0.86 (3H, t, J = 7.5 Hz); 13C NMR (75 MHz) δ 210.5 (C), 138.0 (C), 128.6 (CH), 128.1 (CH), 127.0 (CH), 61.8 (CH2), 58.4 (CH2), 53.5 (CH2), 51.3 (CH), 40.9 (CH2), 20.7 (CH2), 11.8 (CH3); IR (NaCl, film) νmax 3028, 2962, 2875, 2800, 2761, 1715, 1602, 1495, 1454, 1356, 1326, 1193, 1135, 1065, 1028, 1002, 867 cm−1; MS (EI)m/z 217 (M+˙, 18%), 216 (13), 202 (19), 126 (9), 91 (100); HRMS calcd for C14H19NO (M+˙) 217.1467. Found 217.1467.
Compound 3. A magnetically stirred suspension of tin(II) triflate (459 mg, 1.10 mmol, 0.55 equiv) in dry CH2Cl2 (2 mL) maintained at 0 °C was treated with 1-ethylpiperidine (179 μL, 1.30 mmol, 0.65 equiv). The resulting mixture was cooled to −78 °C and a solution of 1-benzyl-4-piperidinone (6) (379 mg, 2.00 mmol, 1.0 equiv) in CH2Cl2 (3 mL) was added via cannula. After 1 h at −78 °C the reaction mixture was slowly warmed to −30 °C and held at this temperature for 2 h. The reaction mixture was then treated with sodium potassium tartrate (5 mL of a 1 M aqueous solution) and CHCl3 (5 mL) and the ensuing mixture stirred vigorously at 18 °C for 1 h. The phases were separated and the aqueous one was extracted with CHCl3 (3 × 5 mL). The combined organic phases were washed with brine (1 × 5 mL) before being dried (Na2SO4), filtered and concentrated under reduced pressure. The resulting clear, colourless oil was subjected to flash chromatography (silica, ethyl acetate elution) and concentration of the appropriate fractions (Rf 0.1) gave compound33 (230 mg, 61%) as a clear, colourless oil and containing ca. 5% of the conjugated isomer of compound 4. 1H NMR (500 MHz) δ 7.36–7.21 (10H, complex m), 5.70 (1H, broad s), 3.64 (1H, d, J = 12.7 Hz), 3.53–3.45 (3H, complex m), 3.22 (1H, dt, J = 12.2 and 2.9 Hz), 3.05 (1H, ddd, J = 12.8, 6.0 and 2.9 Hz), 2.83 (1H, ddd, J = 17.6, 10.7 and 6.8 Hz), 2.64–2.50 (4H, complex m), 2.45 (1H, m), 2.40–2.28 (2H, complex m), 2.24 (1H, broad s), 1.60 (1H, ddd, J = 13.2, 12.2 and 4.4 Hz), 1.47 (1H, m), 1.44 (1H, m), 1.35 (1H, td, J = 11.7 and 4.4 Hz); 13C NMR (125 MHz) δ 210.0 (C), 138.6 (C), 136.9 (C), 129.2 (CH), 129.1 (CH), 128.6, (CH), 128.2 (CH), 127.9 (CH), 126.9 (CH), 70.3 (C), 63.1 (CH2), 62.1 (CH2), 58.0 (CH), 53.7 (CH2), 53.2 (CH2), 49.2 (CH2), 49.0 (CH2), 41.8 (CH2), 36.5 (CH2), 36.1 (CH2); IR (NaCl, film) νmax 3270, 3028, 2941, 2815, 1707, 1453, 1345, 1120, 1027, 998, 909, 812 cm−1; MS (EI)m/z 378 (M+˙, 5%), 360 (5), 287 (8), 269 (8), 191 (73), 91 (100); HRMS calcd for C24H30N2O2 (M+˙) 378.2307. Found 378.2300.
When the procedure described above was followed except for allowing the reaction mixture to stand to 18 °C for 2 h then a ca. 1:4 mixture of compound 4 and it conjugated isomer (238 mg, 66% combined yield) was obtained.
Compound
4. A magnetically stirred solution of 1-benzyl-4-piperidinone (6) (8.5 mL, 45.9 mmol, ex. Aldrich Chemical Co.) in dry 1,2-dichloroethane (15 mL) maintained at 40 °C was treated with BF3·OEt2 (11.6 mL, 91.5 mmol, 2 mole equiv). The resulting mixture was heated at reflux for 16 h (during the course of which a viscous brown gum formed) then cooled to 18 °C and poured into NaHCO3 (200 mL of a saturated aqueous solution). The residual gum was treated with MeOH (30 mL) and the mixture heated at reflux for 0.5 h and the ensuing methanolic solution then cooled to 18 °C before being poured into the NaHCO3 (200 mL of a saturated aqueous solution). Water (50 mL) and CHCl3 (100 mL) were added and the phases were separated. The aqueous phase was extracted with CHCl3 (3 × 50 mL), the combined organic portions were washed with NaHCO3 (1 × 50 mL of a saturated aqueous solution), water (1 × 50 mL) and brine (1 × 50 mL) before being dried (Na2SO4), filtered and then concentrated under reduced pressure to afford a brown oil. Subjection of this material to flash chromatography (silica, 3:2 → 4:1 v/v ethyl acetate/hexane gradient elution) provided two fractions, A and B.
Concentration of fraction A (Rf 0.25 in 3:2 v/v ethyl acetate/hexane) afforded compound44 (5.30 g, 64%) as a clear, yellow oil. 1H NMR (500 MHz) δ 7.35–7.24 (10H, complex m), 5.56 (1H, broad s), 3.60 (2H, ABq), 3.58 (2H, ABq), 3.12 (1H, broad dd, J = 9.3 and 5.2 Hz), 3.01 (2H, dd, J = 5.2 and 2.2 Hz), 2.98 (1H, m), 2.94 (1H, m), 2.78 (1H, m), 2.62 (1H, dd, J = 11.3 and 9.2 Hz), 2.58–2.42 (4H, complex m), 2.08 (2H, m); 13C NMR (125 MHz) δ 208.6 (C), 138.1 (C), 138.0 (C), 132.6 (CH), 129.2 (CH), 128.9, (CH), 128.3 (CH), 128.2 (CH), 127.3 (CH), 127.0 (CH), 123.1 (C), 62.8 (CH2), 62.2 (CH2), 57.1 (CH2), 56.7 (CH), 53.6 (CH2), 52.9 (CH2), 49.6 (CH2), 40.9 (CH2), 28.5 (CH2); IR (NaCl, film) νmax 3026, 2908, 2800, 2759, 1715, 1493, 1453, 1364, 1349, 1186, 1123, 1027 cm−1; MS (EI)m/z 360 (M+˙, 19%), 325 (5), 269 (13), 241 (40), 199 (36), 185 (10), 172 (15), 134 (11), 91 (100); HRMS calcd for C24H28N2O (M+˙) 360.2202. Found 360.2201.
Concentration of fraction B (Rf 0.15 in 1:99 v/v ammonia/ethyl acetate) afforded 1,1′-dibenzyl-2′,3′,5,5′,6,6′-hexahydro-1H,1′H[3,4′-bipyridinylidene]-4(2H)-one (the conjugated isomer of compound 4) (1.36 g, 16%) as a clear, yellow oil. 1H NMR (300 MHz) δ 7.36–7.23 (10H, complex m), 3.65 (2H, s), 3.51 (2H, s), 3.39 (2H, broad s), 2.80 (2H, m), 2.78 (2H, m), 2.55 (2H, app t, J = 6.3 Hz), 2.49 (4H, m), 2.22 (2H, app t, J = 5.7 Hz); 13C NMR (75 MHz) δ 201.6 (C), 147.2 (C), 138.1 (C), 138.0 (C), 129.1 (CH), 128.8 (CH), 128.4 (CH), 128.2 (CH), 127.7 (C), 127.3 (CH), 127.0 (CH), 62.8 (CH2), 62.0 (CH2), 55.7 (CH2), 54.7 (CH2), 53.9 (CH2), 51.1 (CH2), 41.5 (CH2), 30.7 (CH2), 30.1 (CH2); IR (NaCl, film) νmax 3027, 2904, 2800, 2757, 1682, 1602, 1494, 1453, 1364, 1347, 1285, 1191, 1126, 1027, 1004 cm−1; MS (EI)m/z 360 (M+˙, 51%), 332 (6), 269 (45), 241 (37), 198 (42), 134 (52), 91 (100); HRMS calcd for C24H28N2O (M+˙) 360.2202. Found 360.2199.
Compound
8. A magnetically stirred solution of diisopropylamine (3.9 mL, 28 mmol, 1.4 equiv) in dry DME (100 mL) maintained at 0 °C was treated with n-BuLi (10.8 mL of a 2.4 M solution in hexanes, 26 mmol, 1.3 mole equiv). After stirring for 0.08 h the reaction mixture was cooled to −78 °C and 1-benzyl-4-piperidinone (6) (3.79 g, 20.0 mmol, ex. Aldrich Chemical Co.) was added dropwise. Dry DMPU (4.8 mL, 40 mmol, 2 equiv) was then added to aid solubility. After 0.16 h at −78 °C the reaction mixture was treated with allyl bromide (2.6 mL, 30 mmol, 1.5 mole equiv) then slowly warmed to −40 °C and after 0.5 h at this temperature quenched with NaHCO3 solution (100 mL of a saturated aqueous solution). The resulting mixture was warmed to 18 °C then diluted with water (100 mL) and diethyl ether (50 mL). The separated aqueous phase was extracted with CH2Cl2 (3 × 50 mL) and the combined organic phases washed with brine (1 × 50 mL) before being dried (Na2SO4), filtered and then concentrated under reduced pressure to give a clear, colourless oil. Subjection of this material to flash chromatography (silica, 1:1 v/v ethyl acetate/hexane elution) provided, after concentration of the appropriate fractions (Rf 0.6), compound 814 (2.05 g, 45%) as a clear, colourless oil. 1H NMR (300 MHz) δ 7.36–7.25 (5H, complex m), 5.73 (1H, m), 5.05–4.96 (2H, complex m), 3.68 (1H, d, J = 13.1 Hz), 3.54 (1H, d, J = 13.1 Hz), 3.10–2.96 (2H, complex m), 2.66–2.35 (5H, complex m), 2.24 (1H, dd, J = 11.2 and 9.7 Hz), 2.05 (1H, m); 13C NMR (75 MHz) δ 210.1 (C), 138.2 (C), 135.7 (CH), 128.9 (CH), 128.4 (CH), 127.3 (CH), 116.7 (CH2), 62.0 (CH2), 58.6 (CH2), 53.6 (CH2), 49.4 (CH), 41.1 (CH2), 32.0 (CH2); IR (NaCl, film) νmax 3064, 3028, 2911, 2801, 1715, 1640, 1453, 1354, 1188, 1133, 1028, 999, 915 cm−1; MS (EI)m/z 229 (M+˙, 26%), 228 (16), 214 (8), 200 (27), 187 (35), 172 (6), 152 (10), 146 927), 91 (100); HRMS calcd for C15H19NO (M+˙) 229.1467. Found 229.1466.
Compound 9. A magnetically stirred solution of diisopropylamine (1.1 mL, 7.8 mmol, 1.6 equiv) in dry THF (100 mL) maintained at 0 °C was treated n-BuLi (3.1 mL of a 2.4 M solution in hexanes, 7.4 mmol, 1.5 mole equiv). After stirring for 0.08 h the reaction mixture was cooled to −78 °C and TMS-Cl (1.3 mL, 10 mmol, 2 mole equiv) was added dropwise. A solution of ketone 8 (3.79 g, 20.0 mmol) in THF (8 mL) was then added via cannula over 0.8 h and the resulting mixture stirred at −78 °C for 0.33 h. Triethylamine (0.5 mL) was then added to the mixture, followed by NaHCO3 (10 mL of a saturated aqueous) and the resulting mixture warmed to 18 °C before being diluted with water (10 mL) and hexane (10 mL). The separated aqueous phase was extracted with hexane (2 × 10 mL) and the combined organic phases were then dried (Na2SO4), filtered and concentrated under reduced pressure to afford compound99 (1.51 g, quant.) as a clear, colourless but unstable oil. 1H NMR (300 MHz) δ 7.36–7.22 (5H, complex m), 5.69 (1H, m), 4.92–4.85 (2H, complex m), 4.76 (1H, broad t, J = ca. 3 Hz), 3.64 (1H, d, J = 13.0 Hz), 3.46 (1H, d, J = 13.0 Hz), 3.02 (1H, ddd, J = 14.8, 3.7 and 1.2 Hz), 2.91 (1H, ddd, J = 14.8, 3.5 and 1.1 Hz), 2.53–2.34 (3H, complex m), 2.16 (2H, m), 0.21 (9H, s).
Compound
10. Step (i): A magnetically stirred solution of compound 9 (1.51 g, 5.00 mmol) in dry THF (25 mL) maintained at 0 °C was treated with NBS (1.16 g, 6.50 mmol, 1.3 mole equiv). After stirring for a further 0.5 h at 0 °C the reaction mixture was treated successively with NaHCO3 (10 mL of a saturated aqueous solution), sodium thiosulfate (10 mL of a 1 M aqueous solution), water (50 mL) and hexane (50 mL). The separated aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic ones were dried (Na2SO4), filtered and then concentrated under reduced pressure to afford a yellow oil. Subjection of this material to flash chromatography (silica, 0.2:5:95 → 0.2:15:85 v/v/v triethylamine/ethyl acetate/hexane gradient elution) provided, after concentration of the appropriate fractions (Rf 0.4 and 0.3 in 0.2:15:85 v/v/v triethylamine/ethyl acetate/hexane), the expected mixture of cis- and trans-α-bromoketones (672 mg, 44%) as a clear, yellow oil. Since this material was found to decompose readily upon standing, it was used immediately in the next step of the sequence.
Step (ii): A magnetically stirred solution of hexamethyldisilazane (338 mL, 1.6 mmol, 1.6 mole equiv) in dry THF (5 mL) maintained at 0 °C was treated with n-BuLi (625 mL of a 2.4 M solution in hexanes, 1.50 mmol, 1.5 equiv). After stirring for 0.08 h the ensuing mixture was cooled to −78 °C and TMS-Cl (254 mL, 2.0 mmol, 2 mole equiv) added dropwise. A solution of the previously prepared mixture of α-bromoketones (308 mg, 1.00 mmol) in THF (3 mL) was added dropwise, via cannula, and the ensuing mixture stirred at −78 °C for 1 h then treated with triethylamine (0.2 mL) and NaHCO3 (5 mL of a saturated aqueous solution) before being warmed to 18 °C and diluted with water (5 mL) then hexane (5 mL). The separated aqueous phase was extracted with hexane (2 × 5 mL) and the combined organic phases were then dried (Na2SO4), filtered and concentrated under reduced pressure to afford a clear, colourless oil. Subjection of this material to flash chromatography (silica, 0.2:5:95 → 0.2:15:85 v/v/v triethylamine/ethyl acetate/hexane gradient elution) provided, after concentration of the appropriate fractions (Rf 0.6 in 0.2:15:85 v/v/v triethylamine/ethyl acetate/hexane), compound1010 (185 mg, 49%) as a clear, colourless but highly unstable oil. 1H NMR (300 MHz) δ 7.33–7.25 (5H, complex m), 5.63 (1H, m), 4.94–4.85 (2H, complex m), 3.66 (1H, d, J = 13.0 Hz), 3.49 (1H, d, J = 13.0 Hz), 3.34 (1H, broad d, J = ca. 14 Hz), 3.14 (1H, broad d, J = ca. 14 Hz), 2.62–2.48 (2H, complex m), 2.42 (1H, m), 2.29–2.16 (2H, complex m), 0.28 (9H, s).
Compound
5. A magnetically stirred solution of compound 10 (180 mg, 0.47 mmol) in dry THF (2.5 mL) maintained at −78 °C was treated with MeLi (1.58 mL of a 0.36 M solution in diethyl ether, 0.57 mmol, 1.2 equiv) and the resulting mixture warmed to 0 °C and stirred at this temperature for 0.75 h. The solution thus obtained was cooled to −78 °C, t-BuLi (720 μL of a 1.45 M solution in pentane, 1.04 mmol, 2.2 equiv) was added dropwise and the resulting yellow solution was warmed to 0 °C and stirred at this temperature for 0.5 h. The mixture was then recooled to −78 °C and ketone 2 (154 mg, 0.71 mmol, 1.8 equiv) in THF (1.5 mL) was added dropwise via cannula and the reaction mixture then stirred at −78 °C for 0.16 h before being warmed to 0 °C. After 0.25 h at 0 °C, pH 7 buffer solution (2 mL) was added, followed by water (5 mL) and hexane (5 mL). The layers were separated and the aqueous phase was extracted with CH2Cl2 (3 × 5 mL). The combined organic portions were dried (Na2SO4), filtered and then concentrated under reduced pressure to afford a yellow oil. Subjection of this material to flash chromatography (silica, 1:4 → 99:1 v/v ethyl acetate/hexane gradient elution) provided three fractions, A, B and C.
Concentration of fraction A (Rf 0.5 in ethyl acetate) provided a ca. 1:1 mixture of compounds 2 and 8 (57.1 mg) as a clear, colourless oil.
Concentration of fraction B (Rf 0.3 in ethyl acetate) provided a clear, yellow oil (93.5 mg) tentatively identified as a mixture of the various diastereoisomers of compound 5 (44%). The various components of this mixture could not be separated from one another by conventional chromatographic methods.
Concentration of fraction C (Rf 0.2 in ethyl acetate) provided compound55 (51 mg, 24%) as a clear, yellow oil. 1H NMR (600 MHz) δ 7.35–7.22 (10H, complex m), 5.70 (1H, m), 5.02–4.95 (2H, complex m), 3.93 (1H, broad s), 3.63 (2H, ABq), 3.57 (1H, d, J = 13.1 Hz), 3.42 (1H, d, J = 13.1 Hz), 3.27 (1H, ddd, J = 11.1, 5.3 and 2.8 Hz), 3.17 (1H, ddd, J = 11.3, 6.0 and 2.8 Hz), 2.96 (1H, dd, J = 11.6 and 5.3 Hz), 2.65 (1H, m), 2.60 (1H, broad d, J = ca. 10 Hz), 2.51 (1H, partially obsc. m), 2.49 (1H, partially obsc. m), 2.35 (1H, broad t, J = 11.1 Hz), 2.31 (1H, app. t, J = ca. 11 Hz), 2.14 (1H, broad t, J = ca. 10 Hz), 2.11 (1H, t, J = 11.3 Hz), 1.92 (1H, m), 1.86 (1H, m), 1.67 (1H, m), 1.51 (1H, broad d, J = 13.2 Hz), 1.34 (1H, m), 1.20 (1H, m), 0.78 (3H, t, J = 7.5 Hz); 13C NMR (75 MHz) δ 212.9 (C), 138.6 (C), 137.8 (C), 135.6 (CH), 129.1 (CH), 128.7 (CH), 128.5 (CH), 128.1 (CH), 127.4 (CH), 126.9 (CH), 116.7 (CH2), 73.0 (C), 63.1 (CH2), 61.8 (CH2), 59.4 (CH2), 55.6 (CH), 55.5 (CH2), 53.0 (CH2), 49.7 (CH), 48.9 (CH2), 42.9 (CH), 35.6 (CH2), 31.1 (CH2), 20.5 (CH2), 12.3 (CH3); IR (NaCl, film) νmax 3478, 3027, 2931, 2806, 1713, 1638, 1494, 1453, 1359, 1138, 1028, 914, 737 cm−1; MS (EI)m/z 447 [(M + H)+, 19%], 328 (7), 230 (100), 218 (29), 119 (53); HRMS calcd for C29H38N2O2 (M + H)+ 447.3012. Found 447.3013.
(ii) Biological studies
Acknowledgements
We thank the Australian Research Council and the Institute of Advanced Studies, Australian National University for generous financial support, and the Director and Staff of Heron Island Research Station for access to scientific and boating facilities. Professor Bernard Degnan and Mr Simon Walker (The University of Queensland) are thanked for their advice regarding larval assays with Herdmania momus.Notes and references
- (a) R. D. Charan, M. J. Garson, I. M. Brereton, A. C. Willis and J. N. A. Hooper, Tetrahedron, 1996, 52, 9111 CrossRef CAS; (b) R. J. Clark, K. L. Field, R. D. Charan, M. J. Garson, I. M. Brereton and A. C. Willis, Tetrahedron, 1998, 54, 8811 CrossRef CAS; (c) I. W. Mudianta, M. J. Garson and P. V. Bernhardt, Aust. J. Chem., 2009, 62, 667 CrossRef CAS.
- (a) Y. R. Torres, R. G. S. Berlinck, A. Magalhães, A. B. Schefer, A. G. Ferreira, E. Hadju and G. Muricy, J. Nat. Prod., 2000, 63, 1098 CrossRef CAS; (b) J. H. H. L. de Oliveira, A. Nascimento, M. H. Kossuga, B. C. Cavalcanti, C. O. Pessoa, M. O. Moraes, M. L. Macedo, A. G. Ferreira, E. Hadju, U. S. Pinheiro and R. G. S. Berlinck, J. Nat. Prod., 2007, 70, 538 CrossRef CAS; (c) I. W. Mudianta, P. L. Katavic, L. K. Lambert, P. Y. Hayes, M. G. Banwell, M. H. G. Munro, P. V. Bernhardt and M. J. Garson, Tetrahedron, 2010, 66, 2752 CrossRef CAS; (d) L. Chill, T. Yosief and Y. Kashman, J. Nat. Prod., 2002, 65, 1738 CrossRef CAS; (e) S. Matsunaga, Y. Miyata, R. W. M. van Soest and N. Fusetani, J. Nat. Prod., 2004, 67, 1758 CrossRef CAS.
- (a) M. Arai, S. Ishida, A. Setiawan and M. Kobayashi, Chem. Pharm. Bull., 2009, 57, 1136 CrossRef CAS; (b) M. Arai, L. Liu, T. Fujimoto, A. Setiawan and M. Kobayashi, Mar. Drugs, 2011, 9, 984 CrossRef CAS.
- (a) B. Harrison, S. Talapatra, E. Lobkovsky, J. Clardy and P. Crews, Tetrahedron Lett., 1996, 37, 9151 CrossRef CAS; (b) X. Wei, K. Nieves and A. D. Rodríguez, Bioorg. Med. Chem. Lett., 2010, 20, 5905 CrossRef CAS.
- B. I. Morinaka and T. F. Molinski, J. Nat. Prod., 2011, 74, 430 CrossRef CAS.
- J. E. Baldwin and R. C. Whitehead, Tetrahedron Lett., 1992, 33, 2059 CrossRef CAS.
- (a) B. J. Smith and G. A. Sulikowski, Angew. Chem., Int. Ed., 2010, 49, 1599 CrossRef CAS; (b) B. J. Smith, T. Qu, M. Mulder, M. J. Noetzel, C. W. Lindsley and G. A. Sulikowski, Tetrahedron, 2010, 66, 4805 CrossRef CAS; (c) I. Sinigaglia, T. M. Nguyen, J.-C. Wypych, B. Delpech and C. Marazano, Chem.–Eur. J., 2010, 16, 3594 CrossRef CAS; (d) G. A. Molander and F. Cadoret, Tetrahedron Lett., 2011, 52, 2199 CrossRef CAS.
- M. J. Garson, R. J. Clark, R. I. Webb, K. L. Field, R. D. Charan and E. J. McCaffrey, Mem. Qld. Mus., 1999, 44, 205 Search PubMed.
- (a) K. M. Green, B. D. Russell, R. J. Clark, M. K. Jones, M. J. Garson, G. A. Skilleter and B. M. Degnan, Mar. Biol., 2002, 140, 355 CrossRef CAS; (b) K. E. Roper, H. Beamish, M. J. Garson, G. A. Skilleter and B. M. Degnan, Mar. Biotechnol., 2009, 11, 188 CrossRef CAS.
- M. G. Banwell, M. J. Coster, M. J. Harvey and J. Moraes, J. Org. Chem., 2003, 68, 613 CrossRef CAS.
- D. B. Collum, D. Kahne, S. A. Gut, R. T. DePue, F. Mohamadi, R. A. Wanat, J. Clardy and G. Van Duyne, J. Am. Chem. Soc., 1984, 106, 4865 CrossRef CAS.
- (a) R. L. Augustine, A. J. Gustavsen, S. F. Wanat, I. C. Pattison, K. S. Houghton and G. Koletar, J. Org. Chem., 1973, 38, 3004 CrossRef CAS; (b) J. Bonjoch, A. Linares, M. Guardià and J. Bosch, Heterocycles, 1986, 26, 2165 Search PubMed.
- C. J. Kowalski, M. L. O'Dowd, M. C. Burke and K. W. Fields, J. Am. Chem. Soc., 1980, 102, 5411 CrossRef CAS.
- M. A. Iorio, G. Damia and A. F. Casy, J. Med. Chem., 1973, 16, 592 CrossRef CAS.
- E. J. Corey and A. W. Gross, Tetrahedron Lett., 1984, 25, 495 CrossRef CAS.
- Unpublished results.
- W. Rungprom, W. Chavasiri, U. Kokpol, A. Kotze and M. J. Garson, Mar. Drugs, 2004, 2, 101 CrossRef CAS.
- M. J. Garson, ‘Marine Natural Products as Antifeedants’ in Comprehensive Natural Products Chemistry II, ed. L. N. Mander, H.-W. Liu, Elsevier, Oxford, 2010, vol. 4, pp. 503–537 Search PubMed.
- T. Lindel, H. Hoffmann, M. Hochgürtel and J. R. Pawlik, J. Chem. Ecol., 2000, 26, 1477 CrossRef CAS.
- M. Assmann, E. Lichte, J. R. Pawlik and M. Köck, Mar. Ecol.: Prog. Ser., 2000, 207, 255 CrossRef CAS.
- (a) H. C. Vervoort, J. R. Pawlik and W. Fenical, Mar. Ecol.: Prog. Ser., 1998, 164, 221 CrossRef CAS; (b) J. A. Diers, J. J. Bowling, S. O. Duke, S. Wahyuono, M. Kelly and M. T. Hamann, Mar. Biotechnol., 2006, 8, 366 CrossRef CAS.
- D. M. Pinkerton, M. G. Banwell, M. J. Garson, N. Kumar, M. Odorico de Moraes, B. C. Cavalcanti, F. W. A. Barros and C. Pessoa, Chem. Biodiversity, 2010, 7, 1311 CAS.
- For a recent example, see: H. Holla, M. Labaied, N. Pham, I. D. Jenkins, K. Stuart and R. J. Quinn, Bioorg. Med. Chem. Lett., 2011, 21, 4793 CrossRef CAS.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS.
- R. M. Wilson and S. J. Danishefsky, J. Org. Chem., 2006, 71, 8329 CrossRef CAS.
- W. C. Still, M. Kahn and A. Mitra, J. Org. Chem., 1978, 43, 2923 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, 5th edn, Butterworth-Heinemann, Amsterdam, 2003 Search PubMed.
- B. M. Degnan, D. Souter, S. M. Degnan and S. C. Long, Dev. Genes Evol., 1997, 206, 370 CrossRef CAS.
- A. W. Bauer, W. M. Kirby, J. C. Sherris and M. Turck, Am. J. Clin. Pathol, 1966, 45, 493 CAS.
- E. Lacey, J. M. Redwin, J. H. Gill, V. M. Demargheriti and P. J. Waller, in Resistance of Parasites to Antiparasitic Drugs, ed. J. C. Boray, P. J. Martin, R. T. Roush, MSD AGVET, Rahway NJ, USA 1990, pp. 177–184 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and/or 13C NMR spectra of compounds 2–5 and 7–10. See DOI: 10.1039/c1ob06418e |
| This journal is © The Royal Society of Chemistry 2012 |