Synthesis and in vitro enzymatic and antiviral evaluation of phosphoramidate d4T derivatives as chain terminators
Shiqiong
Yang
a,
Christophe
Pannecouque
b,
Eveline
Lescrinier
a,
Anne
Giraut
a and
Piet
Herdewijn
*a
aLaboratory of Medicinal Chemistry, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Minderbroedersstraat 10, B-3000, Leuven, Belgium. E-mail: piet.herdewijn@rega.kuleuven.ac.be; Fax: +32 16 337340; Tel: +32 16 337387
bDepartment of Microbiology and Immunology, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Minderbroedersstraat 10, B-3000, Leuven, Belgium
First published on 8th November 2011
Abstract
The anti-HIV activity of nucleoside analogues is highly related to their substrate specificity for cellular and viral kinase and, as triphosphate, for HIV-RT. A series of phosphoramidate d4T derivatives have been synthesized and evaluated as substrates for HIV-1 RT, and also tested for their in vitro anti-HIV activity. Compounds 2 and 4 are able to inhibit HIV-1 replication to the same extent as d4T and d4TMP in MT-4 cells as well as in CEM/0 cells and CEM/TK−cells. The data suggests that these phosphoramidates are hydrolysed to d4T before exerting their antiviral activity.
Introduction
The 2′,3′-dideoxynucleoside (ddN) analogues, which are potent inhibitors of human immunodeficiency virus (HIV), act in their metabolic active 5′-triphosphate forms as substrates for reverse transcriptase (RT).1 In essence, the ddN triphosphates (ddNTPs) compete with the natural substrates at the RT level for incorporation in DNA and pyrophosphate (PPi) functions as a leaving group.1,2 In most cases, the bottleneck in the metabolic activation of dideoxynucleosides is the enzymatic phosphorylation of the modified nucleoside by cellular kinases. Therefore, a number of prodrug strategies have been developed to improve anti-HIV efficacy and reduce toxicity by delivering the modified nucleoside monophosphate into the cell.3–11 It is worth noting that phosphoramidates of antiviral nucleosides are especially efficient as a nucleotide delivery system, particularly if an amino acid is used.4 Among them, the phosphoramidate derivatives, such as a number of alkyl and aryl phosphoramidate mono-,5,6 or di-esters7,8 of AZT, d4T, and 3′-fluoro-dideoxythymidine (FLT), have shown promise as potential anti-HIV agents since they show enhanced antiviral activity and lower cytotoxicity when compared to the parent nucleoside. In these modifications the phosphate group is conjugated with hydrophobic amino acid esters, such as alanine, L-phenylalanine and L-tryptophan methyl ester. These pronucleotides are able to be internalized by PBMCs and CEM cells and further transformed to their monophosphates and corresponding triphosphates.10 Additionally, ddN triphosphate analogues were also investigated for producing anti-ddNTP antibodies12 and inhibiting RT-mediated drug resistance.13In our continuous effort to search for amino acid derivatives as PPi mimics in enzymatic DNA synthesis,14–203-phosphono-L-alanine, L-aspartic acid, iminodiacetic acid and iminodipropionic acid proved to be efficient leaving groups when catalyzed by HIV-1 RT, suggesting efficient metal-chelation of these amino acid derivatives inside the polymerase active site. 3-Phosphono-L-Ala-dNMP (N = A, T and G) is efficiently incorporated in DNA resulting in 77–95% conversion to a P + 1 strand at 50 μM after 60 min polymerase reaction by HIV-1 RT.19 Therefore, we have studied the potential of the corresponding phosphoramidate dideoxynucleotide analogues to act as chain terminators. Hence, in the current study, d4T monophosphate was conjugated with 3-phosphono-L-alanine mono- or di-ester and the conjugates 1 and 2 (Fig. 1) were evaluated as chain terminators of HIV-1 RT. Additionally, L-Asp-d4TMP (3) and IDA-d4TMP (4) were synthesized and evaluated as substrates of HIV-1 RT. As well as ester derivatives (1, 2), free acids (3, 4) have been evaluated to investigate the structural requirements for cellular uptake of such derivatives. Among these analogues, 3-phosphono-L-alanine monoester phosphoramidate of d4TMP (2) proved to be efficient in single nucleotide incorporation and resulted in 75% conversion to P + 1 strand at 100 μM after 60 min. Full chain termination can be observed for compound 2 at higher substrate concentration (200 μM to 1 mM). It was found that the steady-kinetics study of compound 2 demonstrated a typical Michaelis–Menten kinetic profile. In addition, to increase the hydrophobicity of the potential prodrugs, which is important for cellular uptake, the aryl phosphoramidate analogue 5 was synthesized. The selection of the fenol leaving group is based on the work of McGuigan and coworkers.4,7,8 Compounds 1–5 were further tested for their anti-HIV activity in the cells.
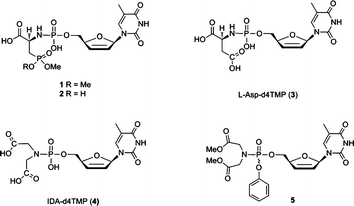 | ||
| Fig. 1 Structure of phosphoramidate derivatives of d4T. | ||
Results and discussion
Synthesis of d4T phosphoramidate derivatives
The synthesis of the amino acid phosphoramidate derivatives of d4T (1–5) was carried out by two synthetic routes. Compounds 1 and 2 were synthesized from d4T monophosphate (d4TMP) and 3-phosphono-L-alanine trimethyl ester (7) by an EDAC-mediated coupling, followed by deprotection in basic conditions. The synthesis of compounds 1 and 2 is shown in Scheme 1. The synthetic strategy starts by removal of the Boc protecting group of 6 with trifluoroacetic acid in CH2Cl2.21,22 The resulting 3-phosphono-L-alanine trimethyl ester (7) can directly be used for further amidation. The monophosphate of d4T (d4TMP) was prepared by treatment of d4T with POCl3 as described before.23,24 In order to avoid purification problems, this reaction was carried out using a small amount (120 mg) of d4T. The EDAC-mediated coupling of d4TMP with the primary amino function of 7 yielded the phosphoramidate methyl ester precursor (8) in 57.1%. Deprotection of both the carboxylic acid ester and phosphonate ester is accomplished in basic condition to give the desired products (1 and 2) in 78.6% overall yield. Column chromatographic purification followed by HPLC was used to purify the final products.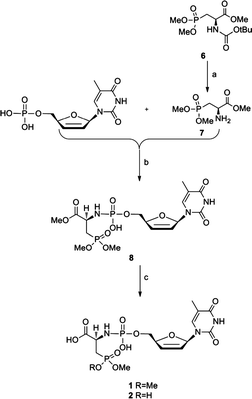 | ||
Scheme 1 Reagents and conditions: (a) CF3COOH, CH2Cl2, rt, 4 h; (b) EDAC, H2O, rt, 3 h; (c) 0.2 M NaOH in MeOH/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, rt, 4–24 h. :2, rt, 4–24 h. | ||
In addition, the hydrolysis of the phosphonate ester function of 8 was also investigated using trimethylsilyl bromide (TMSBr) as reagent in anhydrous CH2Cl2. The reaction failed even at 0 °C, and only degradation products were obtained (demonstrated by the isolation of the nucleobase thymine). This means that we were not able to obtain the fully deprotected congener of compound 8.
The second synthetic route used for the synthesis of the phosphoramidate derivatives of d4T is based on the one pot procedure described by Ludwig and Yoshikawa et al..25,26 The synthesis of L-Asp and IDA phosphoramidate derivatives (3 and 4) of d4T is shown in Scheme 2. 2′,3′-Didehydrodideoxythymidine (d4T) was first phosphorylated and the resulting nucleoside phosphodichloridate was then reacted with the hydrochloride salt of the appropriate amino compound in the presence of tributylamine. After conversion to the triethylammonium salt, the resulted phosphoramidate compounds 9 and 10 were deprotected in basic media and subjected to ion exchange chromatography for purification, affording the final compounds 3 and 4 as triethylammonium salts in good yields (total 37.2–46.7%). As the phosphorylation of d4T requires anhydrous reaction conditions, this one-pot method is convenient when using the commercially available hydrochloride salt of amino acid ester.
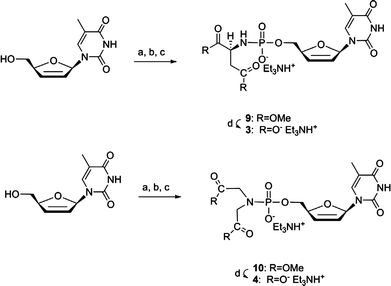 | ||
| Scheme 2 Reagents and conditions: (a) POCl3, PO(OMe)3, 0 °C, 3 h; (b) (L-Asp)Me2·HCl or (IDA)Me2·HCl, n-Bu3N, DMF dry, 0 °C to rt, 3 min; (c) 1 M TEAB, rt, 30 min; (d) 0.4 M NaOH in MeOH/H2O 1:2, rt, 4 h. | ||
Synthesis of phenyl phosphoramidate derivative of d4T
The synthesis of phenylphosphoramidate derivative (5) was achieved following Scheme 3, which is similar to the method described before for the synthesis of arylphosphoramidate derivatives of d4T.27,28 In brief, phenylphosphorodichloride was condensed with iminodiacetate methylester hydrochloride to obtain the monochloridate (11). Treatment of monochloridate (11) in methylene chloride and triethylamine with d4T furnished the desired phenylphosphoramidate derivative 5. Due to the stereochemistry of the phosphorous chiral center,9,11phenyl substituted phosphoramidate derivatives of d4T exist as a mixture of two possible diastereoisomers as evidenced from their 1H,13C and 31P NMR spectra. These isomers could not be separated by regular column chromatography and only fractions enriched with one of the other isomer were obtained.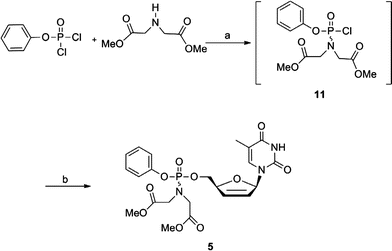 | ||
| Scheme 3 Reagents and conditions: (a) TEA, dry CH2Cl2, −78 °C to rt, 15 h; (b) d4T, rt, 5 d. | ||
Single primer extension by HIV-1 RT with d4T phosphoramidate derivatives (1–4) as substrates
As a key enzyme to replicate the single-stranded RNA genome and produce a double-stranded proviral DNA in HIV life-cycle, RT represents one of the main target enzymes in treating HIV infection. A high mutation rate with HIV-1 RT indicated its important flexibility and high tolerance towards chemically modified nucleotide substrates.29 In previous studies,14–19 it was shown that HIV-1 RT was able to accept amino acid dNMPs as substrate instead of natural dNTPs, during template-directed DNA synthesis. The potential to use this “new leaving group” concept for chain termination in DNA synthesis was evaluated using d4T as example and the amino acid derivatives 1–4. The ability of 3-phosphono-L-alanine di- or mono-methyl ester phosphoramidate analogues (1 and 2) to function as substrates for HIV-1 RT was investigated by the gel-based single nucleotide incorporation assay (Fig. 2). The natural dideoxynucleoside triphosphate (d4TTP) was used as reference.![Single incorporation of primer of P1T1 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1. Panel A: incorporation of compound 1; panel B: incorporation of compound 2; d4TTP (10 μM) used as reference.](/image/article/2012/OB/c1ob06214j/c1ob06214j-f2.gif) | ||
| Fig. 2 Single incorporation of primer of P1T1 (125 nM) by HIV-1 RT with phosphoramidate substrate concentrations and time intervals (min) as indicated, [HIV-1 RT] = 0.025 U μL−1. Panel A: incorporation of compound 1; panel B: incorporation of compound 2; d4TTP (10 μM) used as reference. | ||
During a single nucleotide incorporation assay, compound 1 only resulted in 30% conversion to a P + 1 strand in 30 min at 1 mM. However, the use of compound 2 resulted in 75% incorporation in 60 min at 100 μM. With higher concentrations (200 μM to 1 mM), similar results were obtained and only P+1 product was formed. As expected, a single incorporated d4TMP functions as a chain terminator of HIV-1 RT, Compound 2 is 10-fold more efficient than compound 1 in the single primer-extension assay. This suggests that the 3-phosphono-L-alanine methyl ester can be better accommodated in the active site of the polymerase than the diester.
Under identical HIV-1 RT concentration, L-Asp-d4TMP (3) functioned as a suboptimal substrate: 20–25% P + 1 incorporation product was obtained after 120 min at 500 μM concentration. High concentration of the substrate IDA-d4TMP (4) was required in order to incorporate d4TMP into DNA in a low yield (11–16% P + 1 incorporation product after 120 min at 1 mM concentration).
The efficiency of compound 2 was further investigated by determining the kinetic parameters Km and Kcat.30,31 Steady-state kinetic analysis (Table 1) indicates that the incorporation of compound 2 by HIV-1 RT results in 26-fold increase in the Km value, and only 1.2-fold decrease in Kcat value, compared to those for d4TTP. Although the overall catalytic efficiency, the Kcat/Km ratio, is decreased 31-fold, the Kcat value indicates the catalytic efficiency of HIV-1 RT for the phosphoramidate nucleotide (2) is only a little lower than for d4TTP. It should be mentioned that these kinetics measure the whole process of incorporation (including translocation and DNA release) and that the real incorporation kinetics may be underestimated.32 Whatever, these data indicate efficient nucleophilic displacement of 3-phosphono-L-alanine monomethyl ester when the phosphoramidate is bound in the active site.
| Compound | V max (nM min−1) | K cat (min−1) | K m (μM) | K cat/Km (×10−3 min−1 μM−1) |
|---|---|---|---|---|
| d4TTP | 7.92 ± 0.39 [HIV-1 RT] = 0.0125 U μL−1 = 18.1 nM | 0.44 ± 0.02 | 3.57 ± 0.53 | 123.2 |
| Compound 2 | 13.22 ± 1.08 [HIV-1 RT] = 0.025 U μL−1 = 36.2 nM | 0.37 ± 0.03 | 93.33 ± 16.55 | 3.96 |
Antiviral evaluation of phosphoramidate analogues (1–5) of dideoxynucleotides against HIV-1 and HIV-2
The antiviral activity of compounds 1–5 against HIV-1 and HIV-2 were evaluated in MT-4 cells, CEM/0 and CEM/TK−cells (Table 2). In all of these experiments, d4T and d4TMP were used as references. Compound 1 is 14 and 11-fold less active than compounds 2–4 in MT-4 cells and CEM/0 cells, respectively. Compound 4 is more efficient than compound 3 in CEM/0 cells. Compound 2 shows antiviral activity against HIV-1 and HIV-2 at concentrations of 0.47 μM and 0.19 μM, respectively, in MT-4 cells. Additionally, compound 2 has similar activity as d4T in CEM/0 cells. However, all of these d4T derivatives are much less active in CEM/TK−. These results indicate that the d4T phosphoramidate analogues and d4TMP itself were probably hydrolyzed to d4T in culture medium before penetration into the CEM/TK−cells.| MT-4 cells a | CEM/0b | CEM/TK−c | CEM/0b | |||
|---|---|---|---|---|---|---|
| Compound | IC50 (μM) HIV-1 | IC50 (μM) HIV-2 | CC50 (μM) | IC50 (μM) HIV-2 | IC50 (μM) HIV-2 | CC50 (μM) |
| a HIV-1 (IIIB), HIV-2 (ROD). b CEM/0 cells (ROD). c CEM thymidine kinase deficient cells (ROD). | ||||||
| 1 | 4.26 ± 0.58 | 1.18 ± 0.29 | >259 | 16.52 ± 1.11 | 258.64 ± 0.00 | 259 |
| 2 | 0.47 ± 0.26 | 0.19 ± 0.03 | 145.76 ± 4.73 | 1.44 ± 0.11 | 176.52 ± 28.65 | 266 |
| 3 | 0.36 ± 0.12 | 0.24 ± 0.05 | 169.70 ± 2.86 | 6.55 ± 4.88 | 183.66 ± 28.92 | 298 |
| 4 | 0.31 ± 0.02 | 0.21 ± 0.09 | 172.68 ± 3.72 | 2.13 ± 0.46 | 155.14 ± 19.99 | 298 |
| 5a | 183.66 ± 28.92 | 81.39 ± 13.51 | >239 | >239 | >239 | >239 |
| 5b | 14.90 ± 1.01 | 22.54 ± 0.80 | >239 | >239 | >239 | >239 |
| 5c | 17.11 ± 0.63 | 19.04 ± 1.43 | >239 | >239 | >239 | >239 |
| d4T | 0.30 ± 0.03 | 0.19 ± 0.07 | 145.76 ± 4.73 | 3.02 ± 1.52 | 217.42 ± 11.07 | 268 |
| d4TMP | 0.39 ± 0.10 | 0.33 ± 0.10 | 187.05 ± 2.33 | 1.64 ± 0.48 | 170.11 ± 21.34 | 411 |
In general, compounds 1–4 apparently serve as a source of d4T in cells and none of these phosphoramidate analogues possess marked cytotoxicity. The lower activity of compound 1 may be due to its less efficient hydrolysis to d4T than compound 2. In addition, compound 5 has a limited activity against HIV in MT-4 cells. Three mixtures consisting of varying ratios of the Rp and Sp isomers of the phosphotriester were evaluated for their anti-HIV activity. As we did not assign the absolute phosphorus stereochemistry, the isomer showing δ4.35 ppm (31P NMR) was called the upfield isomer and the other was the downfield isomer. The isomer mixtures denominated 5b (ratio upfield/downfield = 2:1) and 5c (ratio upfield/downfield = 1:1) were almost equipotent, whereas the mixture of isomers 5a (ratio upfield/downfield = 1:2) was 12- and 4-fold less active than 5b against HIV-1 and HIV-2, respectively. The diastereomeric excess was determined by 31P NMR after high pressure liquid chromatography (HPLC). The observed differences in anti-HIV activity result from differences in hydrolytic stability of the different isomers. Most probably the poor anti-HIV activity of compound 5a–c is due to the fact that both carboxylic acids are present as their methyl esters.
Conclusion
In summary, the investigation of the synthesis and evaluation of the phosphoramidate analogues of d4TMP as chain terminators for HIV-1 RT has shown that compound 2 is a good substrate of HIV-1 RT resulting in 75% incorporation in 60 min at 100 μM. Compounds 1, 3 and 4 are suboptimal substrates of HIV-1 RT and higher concentrations are needed to reach 11%–38% incorporation after 120 min. The capability to function as leaving groups in the enzymatic synthesis of DNA catalyzed by HIV-1 RT increase in the order of iminodiacetic acid < L-aspartic acid < 3-phosphono-L-alanine mono methylester, when conjugated with d4TMP.In addition, compound 3 is 3-fold less efficient than compound 4 in CEM/0 cells. Compounds 2 and 4 exhibit similar anti-HIV activity as d4T and d4TMP in MT-4 cells and is 1.5–2-fold more efficient than d4T in CEM/0 cells. However, these compounds are less potent in CEM/TK−cells, as also observed for d4T and d4TMP. This suggests that d4TMP and the phosphoramidate analogues (1–4) are hydrolyzed to d4T in the extracellular medium or in an intracellular compartment. Indeed, following the McGuigan approach, the phosphoramidate diesters are taken up by cells and release the nucleoside monophosphate into the cell.7 The negatively charged phosphoramidate monoesters, as produced by Wagner and coworkers,5 can also be taken up by cells. The outcome of the phosphoramidate prodrug approach seems to be cell type and compound dependent. This, together with the different stability of the prodrugs in the extracellular medium, makes this approach difficult to predict.33 In addition, compound 5 lacks anti-HIV activity. This fully protected phosphoramidate seems to be resistant to enzymatic/chemical degradation to d4T in the given reaction circumstances. Preliminary results suggest a difference in hydrolytic stability of the different diastereomers of the phosphotriester in cell culture. The synthesis of new ester derivatives of the compounds mentioned in this article will be necessary to find a good balance between enzymatic stability, cellular uptake and intracellular processing.
Experimental section
General for chemical synthesis
For all reactions, analytical grade solvents were used. All moisture sensitive reactions were carried out in oven-dried glassware (135 °C). A Bruker Avance II 600 MHz or 500 MHz spectrometer and a Bruker Avance 300 MHz apparatus were used for 1H NMR, 13C NMR and 31P NMR. All chemical shifts are listed in ppm. For sake of clarity of the NMR signal assignment, sugar protons and carbons are numbered with a prime. 31P NMR chemical shifts are referenced to an external 85% H3PO4 standard (δ = 0.000 ppm). UV spectra were recorded on a Varian Cary-300-Bio UV/Vis Spectrophotometer. Mass spectrometry was performed on a quadrupole orthogonal acceleration time-of-flight mass spectrometer (Q-Tof-2, Micromass, Manchester, UK) equipped with a standard electrospray probe (Z-spray, Micromass, Manchester, UK). Samples were dissolved in acetonitrile/water (1:1 v/v) and infused with a flow rate of 5 μL min−1. Electrospray capillary voltage was set to 3000 V and cone voltage to 30 V. Nitrogen gas was used for nebulization and desolvation. Accurate masses were determined by coinfusion of the samples with leucine enkephalin (YGGFL) and recalibration of the spectrum using the peak at m/z 556.2771 as lock mass. Precoated aluminum sheets (MN ALUGRAM SIL G/UV254 20 × 20 cm) were used for TLC; the spots were examined with UV light. Column chromatography was performed on ICN silica gel 63-200, 60 Å.
:5:1 to 4:4:1). Then, the N-deprotected product 7 (0.43 g, 75.5%) was obtained as yellowish syrup and it can directly be used for the subsequent amidation reaction. TLC (CH2Cl2/CH3OH 9:1, v/v): Rf 0.36; 1H NMR (300 MHz, CDCl3): δ 12.28 (br, 2H), 4.12 (m, 1H), 3.74–3.79 (m, 9H), 2.35–2.62 (m, 2H); 31P NMR (121 MHz, CDCl3): δ 29.56; MS for C6H14N1O5P1 (M + H)+ calcd: 212.1 found: 211.9.
Synthesis of d4TMP
To a solution of 3′-deoxy-2′,3′-didehydrothymidine (100 mg, 0.45 mmol) in trimethylphosphate (1.25 mL) was added phosphoryl chloride (0.11 mL, 1.18 mmol) at 0 °C. The reaction mixture was stirred at room temperature for 1.5 h and another 0.11 mL of phosphoryl chloride was added. The reaction was continued to stir for 15 h and quenched by adding 1 M TEAB (pH 7.4) 15 mL. After the solvents were evaporated in vacuo, the residue was purified by silica column chromatography eluting with CHCl3, CHCl3/MeOH 5:1 to i-PrOH/NH3/H2O 7:1:1 to yield the desired d4TMP ammonium salt (55 mg, 36.5%) as white solid. 1H NMR (300 MHz, D2O): δ 7.63 (s, 1H, H-6), 6.96 (m, 1H, H-1′), 6.51 (dd, J1 = 1.6 Hz, J2 = 6.0 Hz, 1H, H-2′), 5.91 (d, J1 = 6.2, J2 = 1.5 Hz, 1H, H-3′), 5.07 (m, 1H, H-4′) 3.92 (m, 2H, H-5′ and H-5′′), 1.90 (q, 3H, -CH3); 13C NMR (75 MHz, D2O): δ 166.34 (C-4), 151.89 (C-2), 137.76 (C-6), 134.16 (C-3′), 124.60 (C-2′), 111.10 (C-5), 89.70 (C-1′), 85.99 (d, 3J(C, P) = 8.3 Hz, C-4′), 65.06 (d, 2J (C, P) = 4.6 Hz, C-5′), 11.08 (CH3); 31P NMR (121 MHz, D2O): δ 1.57 (ammonium salt) or 3.36 (triethylamine salt); MS for C10H13N2O7P1 (M-H)− calcd: 303.0, found: 302.6.
Coupling reaction
2′,3′-Dideoxy-2′,3′-didehydrothymidine-5′-monophosphate ammonium salt (50 mg, 0.15 mmol) and the crude 3-phosphono-L-alanine trimethyl ester (103 mg, 0.49 mmol) were suspended in 1 mL water under Argon. Then, N-ethyl-N′-(3-dimethylamino-propyl)-carbodiimide hydrochloride (EDAC, 94 mg, 0.49 mmol) was added to the suspension, and the reaction was stirred for 2 h at room temperature. The progress of the reaction was monitored by TLC (CHCl3/MeOH/H2O 5:3:0.5). The reaction mixture was evaporated in vacuum at 30 °C and further lyophilized to dryness. The residue was purified by silica column chromatography eluting with CHCl3, CHCl3/MeOH = 5:1, CHCl3/MeOH/H2O = 5:2:0.25 to yield compound 8 (42 mg, 57.1%) as yellowish solid. TLC (i-PrOH/NH3/H2O 7:1:2, v/v): Rf 0.62; 1H NMR (300 MHz, D2O): δ 7.64 (s, 1H, H-6), 6.99 (m, 1H, H-1′), 6.47 (dd, J1 = 6.1, J2 = 1.8 Hz, 1H, H-2′), 5.92 (dd, J1 = 6.7, J2 = 1.8 Hz, 1H, H-3′), 5.08 (m, 1H, H-4′), 3.94 (m, 2H, H-5′ and H-5′′), 3.68–3.80 (m, 9H, 3 OMe), 3.54 (m, 1H, -CHCOO), 2.28–2.36 (m, 2H, -CH2PO3), 1.90 (q, 3H, -CH3); 13C NMR (75 MHz, D2O): δ 174.15 (d, 3J(C, P) = 5.1 Hz, -COOH), 166.33 (C-4), 151.88 (C-2), 137.96 (C-6), 134.19 (C-2′), 124.80 (C-2′), 110.97 (C-5), 89.61 (d, 3J(C, P) = 12.8 Hz, C-4′), 85.68 (C-1′), 64.68 (d, 2J (C, P) = 5.1 Hz, C-5′), 52.59 (d, 2J(C, P) = 3.0 Hz, -CHCOOH), 52.49 (d, 2J (C, P) = 5.7 Hz, POCH3), 49.60 (COOCH3), 28.27 (d, 1J(C, P) = 140.1 Hz, -CH2PO3), 11.10 (CH3); 31P NMR (121 MHz, D2O): δ 31.86 (-CH2PO3), 4.79 (N-PO3); MS for C16H25N3O11P2 (M–H)− calcd: 496.1, found: 495.8.
Deprotection reaction
To the obtained ester intermediate 8 (39 mg, 0.08 mmol) was added 6 mL 0.2 M NaOH in MeOH/H2O 1:2 and the mixture were stirred at room temperature. The progress of the reaction was monitored by TLC (i-PrOH/NH3/H2O 7:1:2) until the disappearance of ester. The hydrolysis is stopped after 4 h and 24 h, respectively, to give the desired products as 1 and 2. The reaction mixture was neutralized by addition of 1 M triethylammonium acetate (TEAA). The solvent was evaporated to dryness in vacuum. The residue was purified by silica column chromatography eluting with CHCl3, CHCl3/MeOH = 5:1, CHCl3/MeOH/H2O = 5:2.5:0.4, yielding compound 1 (18 mg, 46.6%), then i-PrOH/NH3/H2O (7:1:1 to 7:1:1.5) and compound 2 (12 mg, 32.0%) as white solid. An analytical sample was obtained by purification with preparative HPLC (waters 1525-2487 system) with a gradient of CH3CN in 50 mM TEAB buffer (pH = 7.4) on using Prep C18 5 μm column 19 × 150 mm at the flow rate of 3 mL min−1.
:1:2, v/v): Rf 0.53; UV: (H2O) λmax = 266 nm (log ε = 3.97); 1H NMR (300 MHz, D2O, 5 °C): δ 7.70 (s, 1H, H-6), 6.97 (m, 1H, H-1′), 6.48 (ddd, J1 = 6.1 Hz, J2 = 1.8 Hz, J3 = 1.8 Hz, 1H, H-2′), 5.92 (dd, J1 = 6.0 Hz, J2 = 1.8 Hz, J3 = 1.8 Hz, 1H, H-3′), 5.07 (m, 1H, H-4′), 3.97 (m, 2H, H-5′ and H-5′′), 3.70–3.75 (m, 6H, 2 OMe), 3.57 (m, 1H, -CHCOO), 2.23-2.35 (m, 2H, -CH2PO3), 1.92 (q, 3H, -CH3); 13C NMR (75 MHz, D2O, 5 °C): δ 178.14 (d, 3J(C, P) = 9.0 Hz, -COOH), 166.42 (C-4), 151.89 (C-2), 137.92 (C-6), 134.10 (C-3′), 124.86 (C-2′), 111.24 (C-5), 89.61 (C-1′), 85.85 (C-4′), 65.10 (d, 2J (C, P) = 5.3 Hz, C-5′), 52.64 (dd, 2J1(C, P) = 2.0 Hz, 2J2(C, P) = 6.8 Hz, -CHCOOH), 51.87 (d, 2J (C, P) = 3.4 Hz, POCH3), 29.15 (d, 1J(C, P) = 135.6 Hz, -CH2PO3), 11.10 (CH3); 31P NMR (121 MHz, D2O): δ 33.90 (-CH2PO3), 5.66 (N-PO3); HRMS for C15H23N3O11P2 (M-H)− calcd: 482.0735, found: 482.0738.
:1:2, v/v): Rf 0.40; UV: (H2O) λmax = 266 nm (log ε = 3.97); 1H NMR (600 MHz, D2O, 5 °C): δ 7.62 (s, 1H, H-6), 6.90 (m, 1H, H-1′), 6.45 (d, J = 6.0 Hz, 1H, H-2′), 5.89 (m, 1H, H-3′), 5.12 (m, 1H, H-4′), 3.92 (m, 2H, H-5′ and H-5′′), 3.61 (m, 1H, -CHCOOH), 3.45 (q, 3H, OMe), 1.86-1.99 (m, 2H, -CH2PO3), 1.84 (q, 3H, -CH3); 13C NMR (150 MHz, D2O, 5 °C): δ 181.9 (-COOH), 169.63 (C-4), 155.02 (C-2), 141.05 (C-6), 137.28 (C-3′), 127.53 (C-2′), 114.25 (C-5), 92.54 (C-1′), 88.92 (d, 3J(C, P) = 9.1 Hz, C-4′), 67.60 (d, 2J (C, P) = 4.8 Hz, C-5′), 55.73 (d, 2J(C, P) = 18.1 Hz, -CHCOOH), 53.98 (d, 2J (C, P) = 4.8 Hz, POCH3), 33.87 (d, 1J(C, P) = 130.5 Hz, 3J(C, P) = 7.5 Hz, -CH2PO3), 14.21 (CH3); 31P NMR (202 MHz, D2O): δ 22.0 (-CH2PO3), 6.10 (N-PO3); HRMS for C14H21N3O11P2 (M-H)− calcd: 468.0578, found: 468.0600.
General procedure for synthesis of intermediates 9, 10 and compounds 3, 4: 2′,3′-dideoxy-2′,3′-didehydrothymidine-5′-(dimethyl-L-aspartate)phosphoramidate triethylammonium salt (9, intermediate for compound 3)
All solids were dried in a desiccator under high vacuum one day prior to reaction. The nucleoside (50 mg, 0.22 mmol) was placed under argon atmosphere and dissolved in trimethylphosphate (500 μL, 50 μL/10 mg nucleoside). Upon cooling down to 0 °C, phosphorous trichloride (42 μL, 0.44 mmol, 2 eq.) was added slowly via syringe. The resulting solution was stirred for 3 h at 0 °C and the reaction was monitored by TLC (i-PrOH/NH3/H2O 6:3:1, v/v). Upon total conversion of the starting nucleoside, a solution of dimethyl L-aspartic acid hydrochloride (434 mg, 2.2 mmol, 10 eq.) and n-tributylamine (500 μL, 30 eq.) in dried DMF (2.2 mL) was added via syringe. The resulting mixture was stirred vigorously for 3 min and pipetted into a cold TEAB 1 M solution (5 mL). The resulting clear mixture was further stirred at room temperature for 30 min. Upon completion, all volatiles were evaporated under high vacuum. The product was purified by flash chromatography using a gradient of methanol and water in chloroform (5:1:0, 5:2:0.25, 5:3:0.5). The corresponding fractions were evaporated, lyophilized and the final compound (70 mg, colourless solid, yield 73%) was stored at −20 °C.
TLC (i-PrOH/NH3/H2O 6:3:1, v/v) Rf 0.77; 1H NMR (D2O, 300 MHz): δ 7.56 (s, 1H, H-6), 6.89 (s, 1H, H-1′), 6.41 (s, 1H, J = 5.97 Hz, H-2′), 5.86 (s, 1H, J = 5.37 Hz, H-3′), 5.00 (bs, 1H, H-4′), 3.97 (m, 1H, Hα), 3.88 (bs, 2H, H-5′), 3.70, 3.65 (2 s, 2 × 3H, CH3-O), 3.13 (q, 6H, N-CH2-CH3, Et3N+), 2.72–2.70 (m, 2H, Hβ), 1.83 (s, 3H, CH3), 1.21 (t, 9H, N-CH2-CH3, Et3N+) ppm. 13C NMR (D2O, 75 MHz): δ 173.04 (CO Asp 2x), 166.57 (C-4T), 152.16 (C-2T), 138.19 (C-6), 134.05 (C-2′), 125.24 (C-3′), 111.25 (C-5′), 89.75 (C-1′), 85.58 (C-4′, J = 8.71 Hz), 64.71 (C-5′), 52.80 (CH3-O), 50.93 (CH3-O), 43.21 (Et3N+), 38.20 (CHα), 35.48 (CHβ), 11.27 (CH3-T), 8.14 (Et3N+) ppm. 31P NMR (D2O, 121 MHz): δ 5.59 ppm. HRMS (negative ionization): calculated for C16H21N3O10P−: 446.0970, found: 446.0966.
:1 (1 mL) and stirred at room temperature until complete deprotection was achieved. Solvents were evaporated and the residue was submitted to preparative ion exchange chromatography (Source 15Q, Pharmacia) running a gradient of TEAB in water (0 to 30% in 15 min). The final compound was obtained as a white solid (yield 51–64%) and stored at −20 °C.
1H NMR (D2O, 300 MHz): δ 7.58 (s, 1H, H-6), 6.88 (s, 1H, H-1′), 6.39 (s, 1H, J = 6.09 Hz, H-2′), 5.84 (s, 1H, J = 6.06 Hz, H-3′), 5.00 (bs, 1H, H-4′), 3.90 (bs, 2H, H-5′), 3.70–3.67 (m, 1H, Hα), 3.14 (q, 6H, N-CH2-CH3, Et3N+), 2.61–2.58 (m, 2H, Hβ), 1.83 (s, 3H, CH3), 1.21 (t, 9H, N-CH2-CH3, Et3N+) ppm. 13C NMR (D2O, 125 MHz): δ 178.24 (CO Asp), 176.08 (CO Asp), 166.32 (C-4T), 151,85 (C-2T), 137.87 (C-6), 134.05 (C-2′), 124.81 (C-3′), 111.10 (C-5), 89.50 (C-1′), 85.70 (C-4′, J = 9.65 Hz), 64.52 (C-5′), 46.30 (Et3N+), 39.70 (CHα), 34.75 (CH2β), 11.13, 7.86 (Et3N+) ppm. 31P NMR (D2O, 121 MHz): δ 6.34 ppm. HRMS (negative ionization): calculated for C14H17N3O10P−: 418.0657, found: 418.0659.
TLC (i-PrOH/NH3/H2O 6:3:1, v/v) Rf 0.50; 1H NMR (D2O, 300 MHz): δ 7.52 (s, 1H, H-6), 6.85 (s, 1H, H-1′), 6.39 (s, 1H, J = 5.97 Hz, H-2′), 5.83 (s, 1H, J = 5.58 Hz, H-3′), 4.95 (bs, 1H, H-4′), 3.80–3.60 (m, 5H, H-5′ and 2 × CH2), 3.57 (bs, 6H, CH3-O), 3.11 (q, 6H, N-CH2-CH3, Et3N+), 1.82 (s, 3H, CH3) 1.19 (t, 9H, N-CH2-CH3, Et3N+) ppm. 13C NMR (D2O, 126 MHz): δ 173.83 (2 × CO Ac), 166.62 (C-4T), 152.144 (C-2T), 138.44 (C-6), 134.62 (C-2′), 124.73 (C-3′), 111.06 (C-5′), 89.70 (C-1′), 85.85 (C-4′, J = 10.5 HZ), 64.85 (C-5′), 52.13 (CH2 IDA), 48.69 (CH3-O), 46.58 (Et3N+), 11.37, 8.16 (Et3N+) ppm. 31P NMR (D2O, 121 MHz): δ 6.45 ppm. HRMS (negative ionization): calculated for C16H21N3O10P−: 446.0970, found: 446.0976.
1H NMR (D2O, 300 MHz): δ 7.51 (s, 1H, H-6), 6.85 (s, 1H, H-1′), 6.38 (s, 1H, J = 6.18 Hz, H-2′), 5.84 (s, 1H, J = 5.58 Hz, H-3′), 4.97 (bs, 1H, H-4′), 3.91–3.88 (m, 5H, H-5′), 3.67, 3.64 (2 s, 4H, 2 × CH2), 3.11 (q, 6H, N-CH2-CH3, Et3N+), 1.81 (s, 3H, CH3), 1.19 (t, 9H, N-CH2-CH3, Et3N+) ppm. 13C NMR (D2O, 151 MHz): δ 178.91 (2 × CO Ac), 166.42 (C-4), 151.94 (C-2), 137.88 (C-6), 134.00 (C-2′), 124.81 (C-3′), 111.14 (C-5′), 89.62 (C-1′), 85.60 (C-4′, J = 9.50 Hz), 64.92 (C-5′), 52.96 (CH2 IDA), 46.37 (Et3N+), 11.16, 8.10 (Et3N+) ppm. 31P NMR (D2O, 121 MHz): δ 6.97 ppm. HRMS (negative ionization): calculated for C14H17N3O10P−: 418.0657, found: 418.0663.
:2, 2:1 and 1:1, determined by the relative intensity of two peaks of 31P NMR (δ 4.35, 5.11 ppm).
TLC (CHCl3/CH3OH 18:1, v/v): Rf 0.35–0.40; 1H NMR (500 MHz, CDCl3, 5 °C): δ 8.20 (s, 1H, NH), 7.34–7.36 (m, 2H, phenyl), 7.32 (s, 1H, H-6), 7.17–7.21 (m, 3H, phenyl), 7.03 (m, 1H, H-1′), 6.34 (m, 1H, H-2′), 5.88 (m, 1H, H-3′), 5.04 (m, 1H, H-4′), 4.37 (m, 2H, H-5′ and H-5′′), 3.88–3.98 (m, 4H, 2-CH2), 3.67 (s, 6H, 2-OCH3), 1.78 (q, 3H, -CH3); 13C NMR (126 MHz, CDCl3, 5 °C): δ 170.20 (-COOCH3), 163.41 (C-4), 150.52 (C-2), 135.67 (C-6), 133.19 (C-2′), 129.68 (C-phenyl), 126.93 (C-3′), 125.22 (C-phenyl), 120.15 (C-phenyl), 111.27 (C-5), 89.29 (C-1′), 84.48 (d, 3J(C, P) = 8.8 Hz, C-4′), 65.98 (d, 2J (C, P) = 3.1 Hz, C-5′), 52.24 (-OCH3), 47.44 (-CH2), 47.41 (-CH2), 12.35 (CH3); 31P NMR (202 MHz, CDCl3): δ 4.35, 5.11; HRMS for C22H26N3O10P (M-H)− calcd: 522.1288, found: 522.1281.
Enzymatic synthesis
:2 and heating the mixture to 70 °C for 10 min, followed by slow cooling to room temperature over a period of 2 h. The complex of P1T1 was used for the single nucleotide incorporation study of compounds 1–4. A series of 20-μL-batch reactions were performed for the enzyme HIV-1 RT (Ambion, Inc.; 10 U μL−1 stock solutions). The final mixture with HIV-1 RT contained 125 nM primer-template complex, RT buffer (50 mM Tris-HCl, 50 mM KCl, 10 mM MgCl2, 0.5 mM spermidine, 10 mM DTT; pH 8.3), 0.025 U/μL HIV-1 RT, and different concentrations of phosphoramidate substrates. Mixtures were incubated at 37 °C, and aliquots (2.5 μL) were removed and quenched after 10, 20, 30, 60, 120 min. In the control reaction with the natural nucleotides, 10 μM d4TTP was used.
Biological evaluation of phosphoramidate analogues of dideoxynucleotides against HIV-1 and HIV-2
The biological evaluation of phosphoramidate analogues of d4T against HIV-1 and HIV-2 was carried out including d4T and its 5′-O-monophosphate as references.The methodology of the anti-HIV assays was as follows. The anti-HIV activity and cytotoxicity of the compounds were evaluated against wild-type HIV-1 strain IIIB and HIV-2 strain ROD in MT-4 cell cultures using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method.35 Briefly, virus stocks were titrated in MT-4 cells and expressed as the 50% cell culture infective dose (CCID50). MT-4 cells were suspended in culture medium at 1 × 105cells mL−1 and infected with HIV at a multiplicity of infection of 0.02. Immediately after viral infection, 100 μL of the cell suspension was placed in each well of a flat-bottomed microtiter tray containing various concentrations of the test compounds. The test compounds were dissolved in DMSO at 50 mM or higher. After 4 days of incubation at 37 °C, the number of viable cells was determined using the MTT method. Compounds were tested in parallel for cytotoxic effects in uninfected MT-4 cells.
CEM cells were infected as previously described. Briefly, 4 × 105cells mL−1 were infected with HIV-2 at ≈ 100 CCID50 (50% cell culture infective dose) per mL of cell suspension. The thymidine kinase-deficient CEM (CEM/TK−) cell cultures were also infected with HIV-2. Then, 100 μL of the infected cell suspensions was transferred into 96-well microtiter plate wells and mixed with 100 μL of the appropriate dilutions of the test compounds. After 4 days of incubation at 37 °C, the giant cell formation was recorded microscopically in the HIV-infected cell cultures.
Acknowledgements
This work was financed by a grant from K. U. Leuven (GOA) and from ESF. We would like to thank Dr Jef Rozenski for HRMS and Chantal Biernaux for editorial help. We are indebted to Kristien Erven, Cindy Heens and Kris Uyttersprot for evaluating the compounds for their antiviral activity. Luc Baudemprez is acknowledged for running the NMR spectra. Present address of Shiqiong Yang is Department of Chemistry, Fudan University, Shanghai 200433, P.R. China.Notes and references
- B. Oberg, Antiviral Res., 2006, 71, 90–95 CrossRef.
- R. F. Schinazi, J. Mellors, H. Bazmi, S. Diamond, S. Garber, K. Gallagher, R. Geleziunas, R. Klabe, M. Pierce, M. Rayner, J. T. Wu, H. Zhang, J. Hammond, L. Bacheler, D. J. Manion, M. J. Otto, L. Stuyver, G. Trainor, D. C. Liotta and S. Erickson-Viitanen, Antimicrob. Agents Chemother., 2002, 46, 1394–1401 CrossRef CAS.
- D. P. Drontle and C. R. Wagner, Mini Rev. Med. Chem., 2004, 4, 409–419 CAS.
- D. Cahard, C. McGuigan and J. Balzarini, Mini Rev. Med. Chem., 2004, 4, 371–381 CAS.
- S. Chang, G. W. Griesgraber, P. J. Southern and C. R. Wagner, J. Med. Chem., 2001, 44, 223–231 CrossRef CAS.
- T. W. Abraham, T. I. Kalman, E. J. McIntee and C. R. Wagner, J. Med. Chem., 1996, 39, 4569–4575 CrossRef CAS.
- C. McGuigan, D. Cahard, H. M. Sheeka, E. De Clercq and J. Balzarini, J. Med. Chem., 1996, 39, 1748–1753 CrossRef CAS.
- C. McGuigan, H. W. Tsang, D. Cahard, K. Turner, S. Velazquez, A. Salgado, L. Bidois, L. Naesens, E. De Clercq and J. Balzarini, Antiviral Res., 1997, 35, 195–204 CrossRef CAS.
- C. A. Roman, J. Balzarini and C. Meier, J. Med. Chem., 2010, 53, 7675–7681 CrossRef CAS.
- E. J. McIntee, R. P. Remmel, R. F. Schinazi, T. W. Abraham and C. R. Wagner, J. Med. Chem., 1997, 40, 3323–3331 CrossRef CAS.
- G. Wang, N. Boyle, F. Chen, V. Rajappan, P. Fagan, J. L. Brooks, T. Hurd, J. M. Leeds, V. K. Rajwanshi, Y. Jin, M. Prhavc, T. W. Bruice and P. D. Cook, J. Med. Chem., 2004, 47, 6902–6913 CrossRef CAS.
- T. Brossette, A. Valleix, L. Goujon, C. Creminon, J. Grassi, C. Mioskowski and L. Lebeau, Tetrahedron Lett., 1999, 40, 3391–3394 CrossRef CAS.
- J. Deval, K. Alvarez, B. Selmi, M. Bermond, J. Boretto, C. Guerreiro, L. Mulard and B. Canard, J. Biol. Chem., 2004, 280, 3838–3846 CrossRef.
- O. Adelfinskaya and P. Herdewijn, Angew. Chem., Int. Ed., 2007, 46, 4356–4358 CrossRef CAS.
- O. Adelfinskaya, M. Terrazas, M. Froeyen, P. Marliere, K. Nauwelaerts and P. Herdewijn, Nucleic Acids Res., 2007, 35, 5060–5072 CrossRef CAS.
- M. Terrazas, P. Marliere and P. Herdewijn, Chem. Biodiversity, 2008, 5, 31–39 CAS.
- A. Giraut, X. P. Song, M. Froeyen, P. Marliere and P. Herdewijn, Nucleic Acids Res., 2010, 38, 2541–2550 CrossRef CAS.
- I. Zlatev, A. Giraut, F. Morvan, P. Herdewijn and J. J. Vasseur, Bioorg. Med. Chem., 2009, 17, 7008–7014 CrossRef CAS.
- S. Yang, M. Froeyen, E. Lescrinier, P. Marliere and P. Herdewijn, Org. Biomol. Chem., 2011, 9, 111–119 CAS.
- X. P. Song, C. Bouillon, E. Lescrinier and P. Herdewijn, ChemBioChem, 2011, 12, 1868–1880 CrossRef CAS.
- N. Sakai and Y. Ohfune, J. Am. Chem. Soc., 1992, 114, 998–1010 CrossRef CAS.
- D. Sperandio, A. R. Gangloff, J. Litvak, R. Goldsmith, J. M. Hataye, V. R. Wang, E. J. Shelton, K. Elrod, J. W. Janc, J. M. Clark, K. Rice, S. Weinheimer, K. S. Yeung, N. A. Meanwell, D. Hernandez, A. J. Staab, B. L. Venables and J. R. Spencer, Bioorg. Med. Chem. Lett., 2002, 12, 3129–3133 CrossRef CAS.
- J. Beres, W. G. Bentrude, J. Balzarini, E. Declercq and L. Otvos, J. Med. Chem., 1986, 29, 494–499 CrossRef CAS.
- R. R. Chawla, J. J. Freed, F. Kappler and A. Hampton, J. Med. Chem., 1986, 29, 797–802 CrossRef CAS.
- J. Ludwig, Acta Biochim. Biophys. Hung., 1981, 16, 131–133 CAS.
- M. Yoshikawa, T. Kato and T. Takenish, Tetrahedron Lett., 1967, 8, 5065–5068 CrossRef.
- T. K. Venkatachalam, S. Qazi and F. M. Uckun, Bioorg. Med. Chem., 2006, 14, 5161–5177 CrossRef CAS.
- D. Laduree, C. Fossey, Z. Delbederi, E. Sugeac, S. Schmidt, G. Laumond and A. M. Aubertin, J. Enzyme Inhib. Med. Chem., 2005, 20, 533–549 CrossRef CAS.
- A. Herschhorn and A. Hizi, Cell. Mol. Life Sci., 2010, 67, 2717–2747 CrossRef CAS.
- M. S. Boosalis, J. Petruska and M. F. Goodman, J. Biol. Chem., 1987, 262, 14689–14696 CAS.
- M. F. Goodman, S. Creighton, L. B. Bloom and J. Petruska, Crit. Rev. Biochem. Mol. Biol., 1993, 28, 83–126 CrossRef CAS.
- K. A. Johnson, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1041–1048 CrossRef CAS.
- J. Kim, T. F. Chou, G. W. Griesgraber and C. R. Wagner, Mol. Pharmaceutics, 2004, 1, 102–111 CrossRef CAS.
- E. C. R. Smith, L. A. Mcquaid, J. W. Paschal and J. Dehoniesto, J. Org. Chem., 1990, 55, 4472–4474 CrossRef CAS.
- C. Pannecouque, D. Daelemans and E. De Clercq, Nat. Protoc., 2008, 3, 427–434 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |