Complexation and conjugation approaches to evaluate siRNA delivery using cationic, hydrophobic and amphiphilic peptides†
Jung Woo
Park
,
Eun-Kyoung
Bang
,
Eun Mi
Jeon
and
Byeang Hyean
Kim
*
Department of Chemistry, Division of IBB, Pohang University of Science and Technology, Pohang, 790-784, Republic of Korea. E-mail: bhkim@postech.ac.kr; Fax: 82542793399; Tel: 82542792115
First published on 19th September 2011
Abstract
In this study, we used solid phase synthesis to prepare three kinds of peptides and then formulated their peptide–siRNA complexes and peptide–siRNA conjugates. Both the complexation and conjugation systems were nontoxic and allowed the delivery of siRNA into the cytoplasm without the need for any transfection agents and with subsequent inhibition of gene expression.
Introduction
Small interfering RNA (siRNA) is a powerful tool for gene therapy. siRNA strands having lengths of 21–23 nucleotides can recognize and silence complementary sequences of target mRNA in the cell.1 Therefore, siRNA can inhibit the protein expression of a desired target gene. Although siRNA has great therapeutic potential, the application of siRNA remains problematic because of low cellular uptake, poor target specific delivery, and weak nuclease resistance.2 One approach toward overcoming the limitations of gene therapy is the development of efficient siRNA delivery systems.3Among the most interesting siRNA delivery systems are peptide-based gene delivery systems.4 In general, peptide-based delivery materials exhibit no or low toxicity toward cells or tissues because peptides are common in our bodies and rarely induce immune responses. An additional advantage to using peptides is that their functions depend on the properties of their constituent amino acids. For example, cell-penetrating peptides (CPPs), such as Penetratin,5 HIV-1 Tat,6 and oligo-Arg peptide,7 can penetrate the plasma membrane and transfer a variety of cargoes into the cytoplasm or nucleus;8 targeting peptides, which are usually isolated through ex vivo and in vivo screening of phage-displayed peptide libraries, can transport different cargoes to specific cells or tissues.9 Therefore, it might be possible to achieve siRNA-based therapy if we could select a peptide exhibiting a specific function and then complex or conjugate it with an appropriate siRNA for a desired purpose.
In this study, we attempted to deliver vascular endothelial growth factor (VEGF) siRNA10 into cells using two different systems: peptide–siRNA complexes11 and peptide–siRNA conjugates.12 First, we synthesized three peptides, each containing a Cys residue at the C-terminus for peptide–siRNA conjugation: (i) the synthetic short cationic peptide C(PyS)RRRKK (PEP-C); (ii) the hydrophobic peptide C(PyS)PFVYLI (PEP-H), described previously by Davis,13 that constitutes a short active part of the C-terminus of C105Y, which itself penetrates the cell membrane and nucleus of live HuH7 cells; (iii) the amphiphilic peptide C(PyS)RRRKKPFVYLI (PEP-A), which combines the features of PEP-C and PEP-H. Using these three peptides, we tested their complexation with natural VEGF siRNA (VEGF-N): PEP-C–X, PEP-H–X, and PEP-A–X. We then prepared novel three peptide–siRNA conjugates: PEP-C–J, PEP-H–J, and PEP-A–J. Finally, we investigated the delivery properties of the both systems and their efficacy at suppressing the target protein.
Results and Discussion
To insert a thiol group as a linker into siRNA, we prepared specific phosphoramidite 3 in three steps. At first, compound 1 was synthesized by the coupling of two 6-mercapto-1-hexanol units. After one primary OH group was protected with a DMTr unit, the other OH group was converted to the phosphoramidite 3. We synthesized 4 by the incorporation of phosphoramidite 3 at the 5′-terminus of the siRNA sense strand by automated synthesizer. After a reductive cleavage reaction with dithiothreitol (DTT), the disulfide bond of 4 was broken. The reaction mixture was desalted using an NAP-10 column (GE Healthcare, 17-0854-02) and then subsequently purified through reverse phase HPLC. We finally synthesized the 5′-thiol modified VEGF siRNA sense strand 5 (Scheme 1).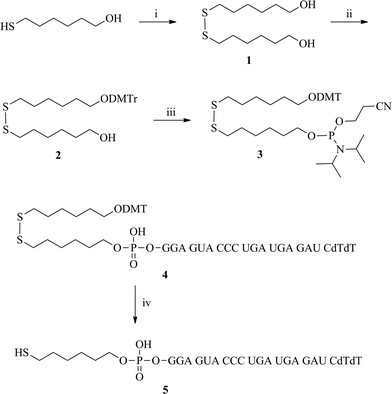 | ||
| Scheme 1 The synthesis of the 5′-thiol-modified VEGF siRNA sense strand: (i) Et3N, I2, MeOH, 0 °C → rt, 4 h; (ii) DMTr-Cl, pyridine, rt, 4 h; (iii) chloro-(2-cyanoethoxy)-N,N-diisopropylaminophosphine, NMP, CH2Cl2, rt, 15 min; (iv) DTT, 50 mM TEAA buffer, pH 8, rt, 2h. | ||
Scheme 2 presents our synthetic strategy for peptides. The peptides PEP-C, PEP-H, and PEP-A were prepared through solid phase syntheses on Rink Amide resin. After activation of their Cys residues with a 2,2′-dipyridyldisulfide, the peptides were reacted with the 5′-thiol-modified VEGF siRNA sense strand 5 under 0.1 M phosphate buffer (pH 7) conditions to synthesize the peptide–siRNA conjugates (Scheme 3). After purifying these peptide–siRNA conjugates through HPLC, we used MALDI-TOF mass spectrometry to determine their masses and checked their chemical properties through the measurement of CD spectra and values of Tm. Secondary structure of our synthetic peptide–siRNA conjugates was A-form RNA (Fig. S1 A in ESI†) and thermal stability of peptide–siRNA was increased when three peptides were conjugated to a VEGF siRNA (Fig. S1 B in ESI†).
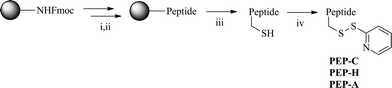 | ||
| Scheme 2 The synthesis of peptides. Reagents and conditions: (i) 30% piperidine in DMF, rt, 30 min; (ii) Fmoc-amino acid, 1-hydroxybenzotriazole, TBTU, DIPEA, DMF, rt, 15 h; (iii) TFA, triethylsilane, CH2Cl2, rt, 2.5 h; (iv) 2,2′-dipyridyldisulfide, DMF/H2O, rt. | ||
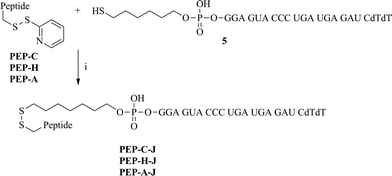 | ||
| Scheme 3 The synthesis of peptide–siRNA conjugates. Reagents and conditions: (i) 0.1 M phosphate buffer (pH 7), 45 °C, 2 h. | ||
In our first attempt to deliver the siRNA into the cells effectively, we tested the effect of using peptides to neutralize the anionic charge of the siRNA. To confirm that our three peptides interacted with siRNA to form complexes, agarose gel retardation assays were performed after mixing siRNA with each of the three peptides at various peptide/siRNA molar ratios. In the case of the cationic peptide, PEP-C (Fig. 1A), when the molar ratio of peptide was increased up to 120, no accumulated bands appeared in the wall; however, the mobility in the gel was decreased and band was broadened. This finding suggests that even if it was difficult to form a complete peptide–siRNA complex, PEP-C could interact with VEGF siRNAvia electrostatic interactions. When the molar ratio of the peptide was between 160–360, upper bands appeared in the wall, representing partially formed peptide–siRNA complexes, but the lower bands did not disappear (Fig. 1E). This finding suggests that it was not possible to form this peptide–siRNA complex, even at excessive molar ratios. In the case of the short hydrophobic peptidePEP-H, increasing the molar ratio of peptide had no effect on the band mobility in the gel (Fig. 1B). From this result, we could suggest that PEP-H cannot interact with anionic siRNAvia electrostatic interaction, because PEP-H has no cationic residues. In contrast, the amphiphilic peptide PEP-A clearly formed a complex with siRNA at and beyond a molar ratio of 32 (Fig. 1C). Interestingly, a mixture containing the individual peptidesPEP-C and PEP-H did not result in the formation of a complex with siRNA (Fig. 1D). From these interesting results, we suggest that for a peptide to form a complex with siRNA, it must feature both cationic and hydrophobic moieties. The cationic part contributes to their electrostatic interaction between siRNA and peptide as mentioned above, and then the hydrophobic part assists their compact complexation via hydrophobic interaction.
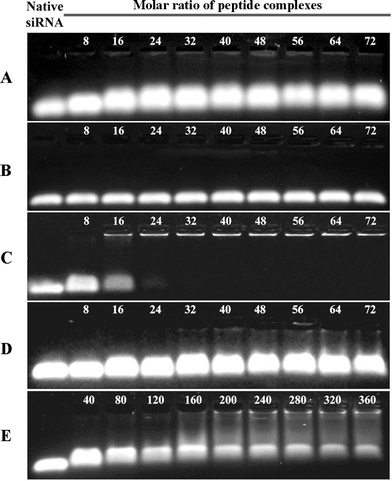 | ||
| Fig. 1 Agarose gel electrophoresis images from representative gel retardation assays. Each assay was performed with various molar ratios of each peptide to 50.0 pmol VEGF siRNA. A) PEP-C was complexed with VEGF siRNA; B) PEP-H was complexed with VEGF siRNA; C) PEP-A was complexed with VEGF siRNA; D) PEP-C and PEP-H were complexed with VEGF siRNA; E) PEP-C was complexed with VEGF siRNA at excess peptide molar ratios. | ||
Prior to performing cellular experiments, WST-1 assays were performed to test the cytotoxicities of our complexation and conjugation systems. All our complexes PEP-C–X, PEP-H–X, and PEP-A–X exhibited no toxicity in the cells, regardless of their molar ratio (Fig. 2A); the synthesized conjugates PEP-C–J, PEP-H–J, and PEP-A–J also exhibited no cytotoxicity (Fig. 2B). Each sample provided a cell viability of greater than 80% of that of Lipofectamine™ 2000. Therefore, neither method (complexation or conjugation) imparted any cytotoxicity on the Huh-7 cell line.
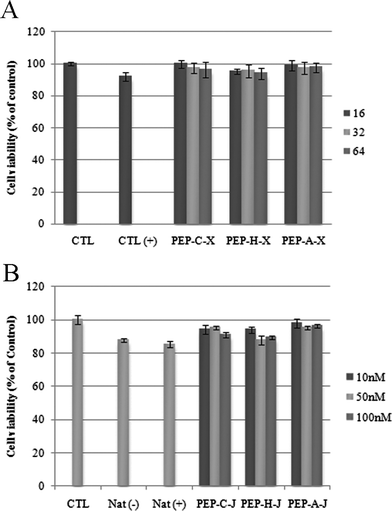 | ||
| Fig. 2 WST-1 assay. A) Peptide–siRNA complexation systems. Concentration of siRNA: 100 nM. Molar ratios of peptide: 16 (dark gray), 32 (bright gray), 64 (gray); B) Peptide–siRNA conjugation systems. Concentrations of conjugates: 10.0 nM (dark gray), 50.0 nM (bright gray), 100 nM (grey). | ||
Next, we used confocal microscopy to analyze the efficacies of the two different siRNA delivery systems (Fig. 3). In the case of a complex system, cellular uptakes are tested at a molar ratio of 64, and we used fluorescein-labeled siRNA (100 nM) in both systems. In the absence of the transfection agent Lipofectamine™ 2000 (Invitrogen), no fluorescence appeared in the cytoplasm (Fig. 3A, 3B, 3I and 3J). In the presence of PEP-C–X and PEP-A–X, intense fluorescence appeared in the cytoplasm (Fig. 3C, 3D, 3G and 3H). In the presence of PEP-H–X, however, the fluorescence intensity was low (Fig. 3E and 3F). These results corresponded well with those of the above gel retardation assays and zeta potential measurementes (Table S1 in ESI†). The PEP-A, which complexed with siRNA at a molar ratio of 64 very well, showed good cellular uptake because the positive charge of PEP-A could completely neutralize the negative charge of siRNA. PEP-H, which did not interact with siRNA at all, however, showed poor cellular uptake. Interestingly, even though PEP-C did not formulate a complete complex with siRNA, it showed good cellular uptake because the positive charge of PEP-C could partly neutralize the negative charge of siRNA. Meanwhile the peptide–siRNA conjugates (Fig. 3 K–P) exhibited higher cellular uptake than did the peptide–siRNA complexes (Fig. 3C–H). Our three peptide–siRNA conjugates PEP-C–J, PEP-H–J, and PEP-A–J all provided intense fluorescence in the cytoplasm.
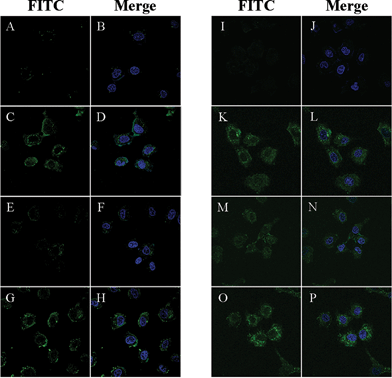 | ||
| Fig. 3 Live cell images obtained using confocal microscopy. Concentration of fluorescein-labeled siRNA: 100nM. Cell line: Huh-7. Green color arose from fluorescein units; blue color arose from the nuclear dye DAPI. No transfection agent was used. A, B) 5′-Fluorescein-labeled siRNA incubated without transfection agent; C, D) PEP-C–X/fluorescein-labeled siRNA complex; E, F) PEP-H–X/fluorescein-labeled siRNA complex; G, H) PEP-A–X/fluorescein-labeled siRNA complex. Molar ratio of all peptides in C–H: 64. I, J) Fluorescein-labeled siRNA incubated without transfection agent; K, L) PEP-C–J; M, N) PEP-H–J; O, P) PEP-A–J. All conjugates systems were annealed with an equivalent 5′-fluorescein-labeled antisense strand of VEGF siRNA. | ||
We conducted enzyme-linked immunosorbent assays (ELISAs)14 to examine the suppression of the VEGF protein in Huh-7 cells. First, we tested the effects of the peptide–siRNA complexes (PEP-C–X, PEP-H–X, PEP-A–X) at molar ratios of 16, 32, and 64 in the presence of 50.0 nM siRNA (Fig. 4A). As the molar ratio is increased, gene down-regulation became more efficient. In the case of PEP-H, which did not form a complex with siRNA, the efficacy of the suppression of VEGF was far lower than that of the other peptides, even when the molar ratio was high. Notably, PEP-C–X and PEP-A–X suppressed the VEGF protein by more than 80% when their molar ratio was 64. Next, we checked the suppression of VEGF protein using the peptide–siRNA conjugates (PEP-C–J, PEP-H–J, PEP-A–J) at various concentrations (5.00, 25.0, and 50.0 nM) in the presence and absence of Lipofectamine™ 2000 (Fig. 4B). Even though the efficacy of gene down-regulation induced by the peptide–siRNA conjugates was lower than that induced in the presence of the transfection agent, they could indeed deliver the siRNA into the cell to suppress the VEGF protein. Relative to the complexation method, the conjugation method allowed the use of smaller amounts of the peptides. Notably, whereas PEP-H–X could not deliver the siRNA into the cells when using the complexation method (because PEP-H–X features no cationic residues and, therefore, did not bind to the siRNA), the use of PEP-H–J and the conjugation method provided an inhibition efficacy of the VEGF protein that was as good as those of the other conjugates.
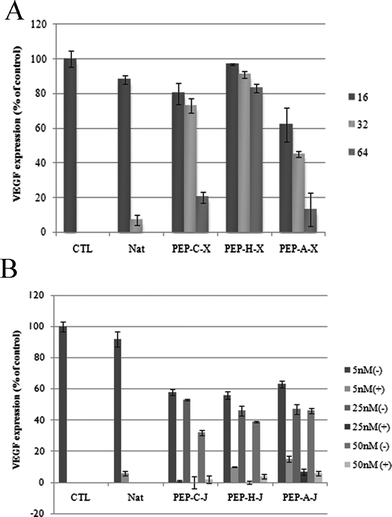 | ||
| Fig. 4 Data from ELISAs. A) Peptide–siRNA complexation systems. Concentration of siRNA: 50.0 nM. Molar ratio of peptide: 16 (dark gray), 32 (bright gray), 64 (gray); B) peptide–siRNA conjugation systems. Concentration of conjugate: 5.00, 25.0, or 50.0 nM. | ||
Conclusions
We used solid phase synthesis to prepare three different characteristic peptides, which we then applied to formulate new peptide–siRNA complexes, PEP-C–X, PEP-H–X, and PEP-A–X, and peptide–siRNA conjugates, PEP-C–J, PEP-H–J, and PEP-A–J. We tested the physical properties of these materials through gel retardation assays. WST-1 assays confirmed that none of our synthetic materials was cytotoxic. Both the complexation and conjugation methods allowed the efficient delivery of siRNA into cells. Notably, even when the peptide sequence was too short or too hydrophobic for its interaction or binding to siRNA, the conjugation method allowed the successful delivery of the siRNA into the cells.Experimental Section
Synthesis of linker
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to yield a white solid (1.43 g, 97.5%)
:1) to yield a white solid (1.43 g, 97.5%)
M.p.: 37.5–37.9 °C; 1H NMR (300 MHz, CDCl3): δ 3.65 (dt, Jd = 5.3 Hz, Jt = 6.3 Hz, 4H), 2.69 (t, J = 7.3 Hz, 4H), 1.72–1.60 (m, 4H), 1.58–1.56 (m, 4H), 1.45–1.38 (m, 8H); 13C NMR (300 MHz, CDCl3): δ 63.03, 39.18, 32.74, 29.27, 28.39, 25.52; IR (neat): 3332, 2929, 2856, 1459, 1053, 728 cm−1; HRMS-FAB (m/z): calcd for C12H26O2S2+ [M + H]+, 266.1374; found, 266.1371.
:1) provided a bright yellow liquid (1.17 g, 54.1%).
1H NMR (300 MHz, CDCl3): δ 7.45–7.42 (m, 2H; -ODMTr), 7.33–7.25 (m, 6H; –ODMTr), 7.21–7.16 (m, 1H; –ODMTr), 6.82 (d, J = 8.8 Hz, 4H; –ODMTr), 3.78 (s, 6H; –ODMTr), 3.62 (t, J = 6.4 Hz, 2H), 3.03 (t, J = 6.5 Hz, 2H), 2.66 (dt, Jd = 6.95 Hz, Jt = 5.88 Hz, 4H), 1.73–1.51 (m, 8H), 1.40–1.35 (m, 8H); 13C NMR (300 MHz, CDCl3): δ 157.8, 144.9, 136.2, 129.5, 127.7, 127.2, 126.0, 112.5, 85.2, 62.8, 62.3, 54.7, 38.6, 38.5, 32.1, 29.4, 28.7, 28.6, 27.9, 27.7, 25.4, 24.9; IR (neat): 3355, 2930, 2857, 1608, 1582, 1509, 1461, 1300, 1250, 1176, 1154, 1072, 1036, 829, 791 cm−1; HRMS-FAB (m/z): calcd for C33H44O4S2+ [M + H]+, 568.2681; found, 568.2685.
:1) provided the compound (546 mg, 57.3%).
1H NMR (300 MHz, CDCl3): δ 7.44–7.41 (m, 2H), 7.34–7.25 (m, 6H), 7.21–7.16 (m, 1H), 6.81 (dt, J = 4.9, 2.9 Hz, 4H), 3.85–3.80 (m, 2H), 3.78 (s, 6H), 3.65–3.55 (m, 4H), 3.03 (t, J = 6.5 Hz, 2H), 2.69–2.60 (m, 6H), 1.70–1.59 (m, 8H), 1.40–1.35 (m, 8H), 1.28–1.16 [m; 1H]; 13C NMR (300 MHz, CDCl3): δ 157.8, 144.9, 136.2, 129.5, 127.7, 127.1, 126.0, 112.4, 85.1, 63.2, 62.9, 62.8, 57.9, 57.6, 54.7, 42.6, 42.4, 38.5, 38.5, 30.6, 30.5, 29.4, 28.7, 28.6, 27.9, 27.7, 25.5, 25.1, 24.2, 24.1, 24.0, 19.9, 19.8; 31P NMR (300 MHz, CDCl3): δ 148.47; HRMS-FAB (m/z): calcd for C42H61N2O5PS2+ [M + H]+, 768.3760; found, 769.2323.
Synthesis of peptides
Peptides were synthesized on a solid support, Rink amide AM resin (100 mg, 0.068 mmol, 200–400 mesh, Novabiochem), generating C-terminally amidated peptides. Stepwise coupling reactions were performed using Fmoc-protected amino acids (Novabiochem), 1-hydroxybenzotriazole (HOBT, 27.6 mg, 0.204 mmol), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU, 65.5 mg, 0.204 mmol), and N,N-diisopropylethylamine (DIPEA, 71.3 μL, 0.408 mmol) as the coupling reagent in 1.00 mL of DMF at room temperature. The reactions were performed in polypropylene syringes (capacity: 5 mL), which were shaken overnight on an IKAVibrax-KS 130 Basic orbital shaker. Anhydrous solvents were used for all reactions. Cleavage and deprotection were achieved through treatment with trifluoroacetic acid (TFA), CH2Cl2, and triethylsilane (60:38:2) for 2 h; the resin was removed through filtration. The peptides were precipitated through the addition of cold Et2O. The precipitates were washed with cold Et2O (12.0 mL) and lyophilized from t-BuOH/H2O (3:1, 1.50 mL). The peptides were purified using reverse-phase high-performance liquid chromatography (RP-HPLC) and a semi-preparative Vydac C-18 column (Cat. 218TP510): buffer A, 0.1% TFA in H2O; buffer B, MeCN. Elution involved a gradient from 5% (0 min) to 98% (25 min) to 5% (30 min) buffer B at a flow rate of 2.50 mL min−1. Peptides were detected at 220 nm and characterized using MALDI-TOF mass spectrometry. Each peptide was then reacted with 2,2′-dipyridyldisulfide (Aldrich, 20 equiv.) in H2O/DMF at rt for 1 h. The solution was lyophilized and purified through RP-HPLC under the conditions mentioned above. All the peptides were characterized using MALDI-TOF mass spectrometry.
Synthesis of oligonucleotides
Oligonucleotides were synthesized on a CPG support (1.00 μmol scale, 1000 Å pore size) using standard phosphoramidite methods on an automated Expedite 8909 synthesizer. All monomers and modifiers such as compound 3 or 5′-fluorescein phosphoramidite (Glen Research, 10-5901-90) were dissolved in MeCN to form 70.0 mM solutions. The coupling time in each cycle was 3 min for natural monomers and 15 min for modifiers. The activator was 0.50 M 5-ethylthiotetrazole in anhydrous MeCN. Cleavage of the synthesized oligonucleotides from the solid support was performed through treatment with a mixture (1.50 mL) of 41% aqueous MeNH2 (Fluka, 65580)/33% ethanolic MeNH2 (Fluka, 65590) (3:1) for 4–5 h at room temperature. The solution was lyophilized and reacted sequentially with 1-methyl-2-pyrrolidone (Fluka, 69120; 120 μL), anhydrous Et3N (60.0 μL), and Et3N·3HF (Aldrich, 344648; 80.0 μL) for 2 h at 65 °C to desilylate of 2′ position. The solution was quenched with 3 M NaOAc (pH 5.4, 26.0 μL) and then 1-butanol (3 times the total volume, 900 μL) was added. After standing at −20 °C overnight, the solution was subjected to centrifugation (3500 rpm, 15 min, 4 °C), washed with EtOH, and dried. The supernatant was discarded and the remaining RNA pellets were redissolved in distilled 1.00 mL of water and desalted using an NAP-10 column (GE Healthcare, 17-0854-02) and then lyophilized. The oligonucleotides were subsequently purified through RP-HPLC using a C-18 column (kromasil, 10 × 250 mm); a gradient from 5% MeCN/0.1 M TEAA buffer (pH 7.0) to 50% MeCN/0.1 M TEAA buffer was run over 30 min at 2.50 mL min−1; detection at 254 nm. The products were characterized using MALDI-TOF mass spectrometry.
The synthesis of 5′-thiol-modified oligonucleotide was the same as other oligonucleoside syntheses except for one procedure. Before the NAP-10 column procedure, the oligonucleotide 4 were treated with dithiothreitol (DTT, 3.10 mg, 20.0 μmol) in 1.00 mL of 50.0 mM triethylammonium acetate (TEAA) buffer (pH 8) for 2 h at room temperature. The resulting solution was desalted using an NAP-10 column and then lyophilized. The 5′-thiol-containing oligonucleotide 5 was subsequently purified through RP-HPLC using a C-18 column under same HPLC condition. The confirmed sense and antisense strands were dissolved in the annealing buffer [100 mM KOAc, 30 mM HEPES–KOH, 2 mM Mg(OAc)2; pH 7.4], heated at 90 °C for 5 min, slowly cooled to 37 °C, and then incubated at 37 °C for 1 h; Natural VEGF siRNA (VEGF-N); antisense strand of VEGF–siRNA (5′-GAUCUCAUCAGGGUACUCCdTdT), m/z 6603.9 (calcd 6603.8); sense strand of VEGF-N (5′-GGAGUACCCUGAUGAGAUCdTdT), m/z 6708.2 (calcd 6709.7); 5′-fluorescein-labeled antisense strand of VEGF–siRNA (Fluorescein-GAU CUC AUC AGG GUA CUC CUdT-3′), m/z 7141.7 (calcd 7141.4); 5′-thiol modified sense strand of VEGF siRNA [5′-SH–(CH2)6–GGAGUACCCUGAUGAGAUCdTdT], m/z 6903.5 (calcd 6904.1).
Synthesis of peptide–siRNA conjugates
5′-Thiol-modified siRNA sense strands (30.0 nmol) were conjugated with PEP-C, PEP-H or PEP-A (90.0 nmol) in 0.1 M phosphate buffer (pH 7) for 2 h at room temperature. The mixtures were purified through an NAP-10 column (GE Healthcare, 17-0854-02) and then lyophilized. The dried products were purified through RP-HPLC using a Xterra MS C-18 column (2.5 μm); buffer A: 0.1 M TEAA buffer solution (pH 7.0); buffer B, MeCN. Elution was a gradient from 5% (0 min) to 50% (25 min) to 5% (30 min) buffer B at a flow rate of 2.5 mL min−1; oligonucleotides were detected at 254 nm. MALDI-TOF mass spectral data is available (see the supporting information†). The confirmed peptide–siRNA sense and antisense strands were anealed by the method above: PEP-C–J [NH2–KKRRRC–S–S–(CH2)6–GGA GUA CCC UGA UGA GAU CUdT-3′], 30.8% yield; PEP-H–J [NH2–ILYVFPC–S–S–(CH2)6–GGA GUA CCC UGA UGA GAU CUdT-3′], 20.7% yield; PEP-A–J [NH2–ILYVFPKKRRRC–S–S–(CH2)6–GGA GUA CCC UGA UGA GAU CUdT-3′], 31.6% yield.Complexation and gel retardation
Peptides were dissolved in water for making the stock solution. Peptide complexes were prepared by mixing appropriate amounts of siRNAs and each peptides based on their peptide/siRNA molar ratios. After mixing, the solutions were incubated at room temperature for 20 min prior to use. VEGF siRNA concentration: 5.00 pmol.WST-1 assay
Peptides were complexed with VEGF siRNA and added to the cells. In the case of the peptide–siRNA conjugates, the conjugates were added directly to the cells. Cells were incubated with the complexes at 37 °C under 5% CO2 for 24 h. After incubation, the medium was removed, the cells were washed three times with DPBS buffer (100 μL), and then WST-1 reagent (1 mg mL−1 of WST-1 dissolved in phenol red free medium, Roche; 100 μL) was added to each well. The mixtures were incubated at 37 °C for 2 h. After incubation, absorbances were measured at a wavelength of 450 nm using a microplate reader (Asys) and converted to the percentage of cell viability (relative to control cells).Cell culture and transfection
Huh-7 cells were cultured in Dulbecco's modified Eagle's medium (HyClone), supplemented with 10% fetal bovine serum (HyClone), streptomycin (100 μg mL−1), and penicillin (100 U mL−1) at 37 °C in a 5% CO2 incubator. Cells were split, using trypsin/EDTA medium, when almost confluent. Huh-7 cells were seeded at a density of 2.50 × 104cells per well – each well contained 10% FBS supplemented DMEM (2.00 mL) – and incubated for 4 h.Huh-7 cells were transfected in the absence of serum with PEP-C–J, PEP-H–J, and PEP-A–J and VEGF siRNAs using peptidePEP-C, PEP-H, PEP-A, and Lipofectamine™ 2000 (Invitrogen), or without any transfection reagent. The cells were left to incubate at 37 °C for 6 h in a CO2 incubator, followed by replacement of DMEM (2.00 mL) containing 10% FBS. After an additional 18 h of incubation, the cell medium was collected and analyzed for the level of VEGF expression using a VEGF ELISA kit (QIA51 VEGF ELISA Kit, Human, Calbiochem) and following the manufacturer's instructions.
Confocal microscopy
Cover glasses (1.13 cm2, Deckglaser) were placed over a six-well plate. Huh-7 cells were seeded at a density of 1.5 × 105cells per well – each well contained 10% FBS supplemented DMEM (2.00 mL) – and incubated for 12 h. In the peptide–siRNA complexes experiment, Huh-7 cells were transfected in the absence of serum with 100 nM fluorescein-labeled VEGF siRNA using Lipofectamine™ 2000 (Invitrogen) and PEP-C, PEP-H, or PEP-A (peptide/siRNA molar ratio: 64) or without any transfection reagent. In the peptide–siRNA conjugates experiment, Huh-7 cells were transfected in the absence of serum with 100 nM FITC-tagged VEGF siRNA using Lipofectamine™ 2000 (Invitrogen) or FITC-tagged PEP-C–J, PEP-H–J, or PEP-A–J without any transfection reagent. The cells were incubated at 37 °C in the presence of the oligonucleotides for 4 h in a CO2 incubator, followed by replacement of DMEM (2.00 mL) containing 10% FBS. For preparation, 4 h after transfection, the cells were washed three times with DPBS, and then the nuclei of the cells were stained with DAPI. After washing three times with PBS, the cover glasses were detached from the bottom of the plate and transferred onto slide glass. Confocal microscopy images were obtained from the live cells by viewing with an Olympus FluoView™ FV1000 confocal microscope (Olympus Optical, Tokyo).Abbreviations
| CD | circular dichroism |
| CEP | chloro-(2-cyanoethoxy)-N,N-diisopropylaminophosphine |
| DAPI | 4′-6-diamidino-2-phenylindole |
| DMF | N,N-dimethylformamide |
| DMAP | N,N-dimethylaminopyridine |
| DMTr-Cl | 4,4′-dimethoxytritylchloride |
| DMEM | Dulbecco's modified Eagle's medium |
| DPBS | Dulbecco's Phosphate buffered saline |
| DTT | dithiothreitol |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | fetal bovine serum |
| Fmoc | 9-fluorenylmethyloxycarbonyl |
| MALDI-TOF | matrix assisted laser desorption ionization-time of flight |
| NMP | 4-methylmorpholine |
| PBS | Phosphate buffered saline |
| PyS | pyridinesulfenyl |
| RP-HPLC | reverse-phase high performance liquid chromatography |
| T m | melting temperature |
Acknowledgements
This work was supported by grants [(no. 2011-0018691), WCU (R31-10105)] and KNRRC program from the National Research Foundation (NRF) and the EPB Center (2011-0001019). We thank Professor Sungjee Kim for assistance with the zeta potential measurements.Notes and references
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- (a) R. M. Schiffelers, A. Ansari, J. Xu, Q. Zhou, Q. Tang, G. Storm, G. Molema, P. Y. Lu, P. V. Scaria and M. C. Woodle, Nucleic Acids Res., 2004, 32, e149 CrossRef; (b) B. S. Hoshika, N. Minakawa and A. Matsuda, Nucleic Acids Res., 2004, 32, 3815–3825 CrossRef; (c) D. D. Paula, M. Vitoria, L. B. Bentley and R. I. Mahato, RNA, 2007, 13, 431–456 CrossRef; (d) J. Soutschek, A. Akinc, B. Bramlage, K. Charisse, R. Constien, M. Donoghue, S. Elbashir, A. Geick, P. Hadwiger, J. Harborth, M. John, V. Kesavan, G. Lavine, R. K. Pandey, T. Racie, K. G. Rajeev, I. Rohl, I. Toudjarska, G. Wang, S. Wuschko, D. Bumcrot, V. Koteliansky, S. Limmer, M. Manoharan and H.-P. Vornlocher, Nature, 2004, 432, 173–178 CrossRef CAS.
- (a) S. M. Park, J. Woo, E. M. Jeon and B. H. Kim, Chem. Commun., 2010, 46, 1523–1525 RSC; (b) H. W. Yang, J. W. Yi, E.-K. Bang, E. M. Jeon and B. H. Kim, Org. Biomol. Chem., 2011, 9, 291–296 RSC; (c) S. P. Patil, J. W. Yi, E.-K. Bang, E. M. Jeon and B. H. Kim, Med. Chem. Commun., 2011, 2, 505–508 RSC.
- P. Järver and Ü. Langel, Drug Discovery Today, 2004, 9, 395–402 CrossRef.
- D. Derossi, A. H. Joliot, G. Chassaing and A. Prochiantz, J. Biol. Chem., 1994, 269, 10444–10450 CAS.
- E. Vivés, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef.
- J. B. Rothbard, S. Garlington, Q. Lin, T. Kirschberg, E. Kreider, P. L. McGrane, P. A. Wender and P. A. Khavari, Nat. Med., 2000, 6, 1253–1257 CrossRef CAS.
- M. Pooga, M. Hällbrink, M. Zorko and Ü. Langel, FASEB J., 1998, 12, 67–77 CAS.
- (a) M. Essler and E. Ruoslaht, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2252–2257 CrossRef CAS; (b) J. A. Hoffman, E. Giraudo, M. Singh, L. Zhang, M. Inoue, K. Porkka, D. Hanahan and E. Ruoslahti, Cancer Cell, 2003, 4, 383–391 CrossRef CAS; (c) R. Pasqualini and E. Ruoslahti, Nature, 1996, 380, 364–366 CrossRef CAS.
- Antisense strand of VEGF-siRNAs (5′-GAUCUCAUCAGGGUACUCCdTdT), m/z 6603.9 (calcd 6606.2); sense strand of VEGF-N (5′-GGAGUACCCUGAUGAGAUCdTdT), m/z 6708.2 (calcd 6709.2).
- (a) S. Veldhoen, S. D. Laufer, A. Trampe and T. Restle, Nucleic Acids Res., 2006, 34, 6561–6573 CrossRef CAS; (b) P. Kumar, H.-S. Ban, S.-S. Kim, H. Wu, T. Pearson, D. L. Greiner, A. Laouar, J. Yao, V. Haridas, K. Habiro, Y.-G. Yang, J.-H. Jeong, K.-Y. Lee, Y.-H. Kim, S. W. Kim, M. Peipp, G. H. Fey, N. Manjunath, L. D. Shultz, S.-K. Lee and P. Shankar, Cell, 2008, 134, 577–586 CrossRef CAS.
- (a) J. J. Turner, S. Jones, M. M. Fabani, G. Ivanoca, A. A. Arzumanoc and M. J. Gait, Blood Cells, Mol., Dis., 2007, 38, 1–7 CrossRef CAS; (b) N. Venkatesan and B. H. Kim, Chem. Rev., 2006, 106, 3712–3761 CrossRef CAS.
- M. Rhee and P. Davis, J. Biol. Chem., 2006, 218, 1233–1240 Search PubMed.
- (a) E. Engvall and P. Perlman, Immunochemistry, 1971, 8, 1–4 CrossRef; (b) B. K. Van Weemen and A. H. Schuurs, FEBS Lett., 1971, 15, 232–236 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CD and Tm data, zeta potential measurements, NMR spectra, HPLC profiles, MALDI-TOF mass spectrometric data. See DOI: 10.1039/c1ob06042b |
| This journal is © The Royal Society of Chemistry 2012 |