Asymmetric cyanation of nitroalkenes catalyzed by a salen–titanium catalyst†
Li
Lin‡
a,
Wen
Yin‡
a,
Xu
Fu
a,
Jinlong
Zhang
a,
Xiaojuan
Ma
a and
Rui
Wang
*ab
aKey Laboratory of Preclinical Study for New Drugs of Gansu Province, State Key Laboratory of Applied Organic Chemistry and Institute of Biochemistry and Molecular Biology, Lanzhou University, Lanzhou, 730000, P. R. China. E-mail: wangrui@lzu.edu.cn; Fax: +86 931 8912567; Tel: +86 931 8912567
bState Key Laboratory of Chiroscience and Department of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hong Kong
First published on 8th September 2011
Abstract
The salen–Ti complex catalyzed cyanation of nitroolefins was accomplished via the silyl nitronate intermediate for the synthesis of chiral β-nitronitriles with e.r. up to 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 and high yields (up to 90%). The catalyst also kept a high turnover frequency at room temperature. The yield and enantioselectivity of the protocol were slightly affected even in a 10 mmol scale.
:8 and high yields (up to 90%). The catalyst also kept a high turnover frequency at room temperature. The yield and enantioselectivity of the protocol were slightly affected even in a 10 mmol scale.
Introduction
The β-peptides, unlike α-peptides, display various advantages such as higher structural diversity, shorter residues to form helix, as well as more stable toward enzymatic degradation.1–2 Thus, β-peptides have received growing attention for their potential pharmaceutical applications.3 Although many methodologies have been developed for the synthesis of β-amino acids, the main shortcomings, also for other general asymmetric catalysis, are that different catalytic systems are necessary for obtaining the different enantioisomers.4 As both nitro and nitrile groups are versatile functional frameworks, a single enantioisomer of chiral β-nitronitrile (2, Scheme 1) could be easily transformed to the two enantioisomers of the corresponding unnatural chiral β-amino acids (3, Scheme 1).5 The simplest method to obtain this versatile synthon was the asymmetric Michael addition of cyanides to nitroolefins.6 Although various methodologies for the Michael addition of nucleophiles to nitroolefins have been developed in recent years,7 asymmetric protocols for conjugate cyanation of nitroolefins have rarely been reported. Only two Cinchona alkaloids derived tetralkylammoniums catalyzed cases have been reported.8 However, these examples both lack high catalytic efficiencies. Reactions always proceeded over days to give ideal results. In addition, to date there is no conclusive study of the mechanism .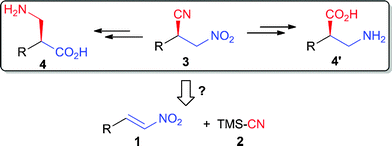 | ||
| Scheme 1 Overview for the synthetic application of β-nitronitriles to β-amino acids. | ||
Organometallic catalysis occupies many advantages for the chiral ligands and metals are easily tunable. In terms of the organometallic catalyzed conjugate cyanations,9 recent reports have just focused on exploiting unsaturated carbonyl substrates such as enones,10 unsaturated imides,11 unsaturated N-acylpyrroles,6,7 and unsaturated diesters.13 Feng and co-workers reported a cinchonidine–titanium complex catalyzed cyanation of activated olefins (5, Fig. 1) with a diphenol as an additive.13 Unfortunately, their protocol was not compatible with using nitrostyrene (1′, Fig. 2) as the substrate. However, we have a different perspective on its structural property. We proposed that the more stable delocalized nature of 1′ and less polarization of its Michael receptor fragment lead to its poorer reactivity towards the conjugate cyanation. In contrast, nitroolefin 1 does not contain an aromatic ring to construct a huge delocalized system. It is much easier to break the conjugated system of alkyl nitroolefin 1 than that of aryl nitroolefin 1′. From this point of view, alkyl nitroolefins (1), which are more polarized and lack aromatic delocalization, would be ideal Michael receptors for the conjugated cyanation.
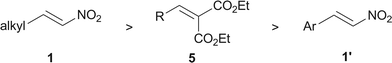 | ||
| Fig. 1 Relative polarization of different conjugated olefins. | ||
Herein, we wish to report a salen–titanium catalyzed asymmetric conjugate cyanation of nitroalkenes. Unlike general reported catalytic systems for the conjugate cyanation, protonic additives were not essential in this work. In situ1H NMR investigation was also accomplished to clarify the hypothesis of this interesting process.
Results and discussion
To confirm our hypothesis, we initially found that quinine in combination with Ti(OiPr)4 indeed can catalyze the conjugate addition of TMSCN to 1a with very poor conversion and enantioselectivity at low temperature (−40 °C). No reaction was observed using only Ti(OiPr)4 to catalyze this conjugate cyanation. We considered that quinine–Ti could not construct a favorable chiral catalytic center. And most importantly, this result suggested that the quinine ligand was not able to affect titanium to display strong enough Lewis acidity to activate TMSCN. Then we intended to use a salen-type chiral ligand to improve the enantioselectivity.Various chiral ligands were screened in the conjugated cyanation of 1a (Scheme 2). As we expected, conjugate cyanation of 1a with TMSCN was smoothly carried out at −15 °C with a moderate enantioselective ratio (e.r.; 82:18) and a high conversion catalyzed by a L1*–Ti complex. Notably, it was critical that the structures of practical ligands (L1*–L5*) must contain two phenolic hydroxy groups which aided in forming strong Lewis acidic Ti-complex. Thus, L6*–L12* in combination with Ti(OiPr)4 did not work to catalyze the model reaction even at room temperature for days. Salen–Ti complexes have been widely used in the cyanation of ketones,14a,baldehydes,14c as well as epoxides.14d However, there was no report on the salen–Ti catalyzed conjugate cyanation of nitroolefins. Based on this encouraging result, a series of conditions, including different solvents and additives, as well as temperature, were then screened to improve the conversion and enantioselectivity of the reaction.
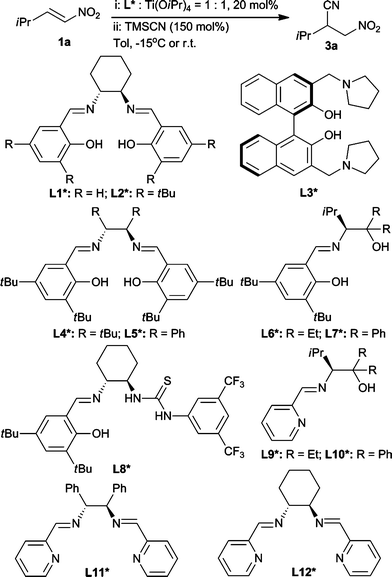 | ||
| Scheme 2 Chiral ligands used in the conjugate cyanation. | ||
Firstly, the reaction was carried out in toluene for the ligand screening. Using 20 mol% of L1* combined with 20 mol% Ti(OiPr)4 as the catalyst, conjugate cyanation of 1a proceeded smoothly to afford 3a with moderate enantioselectivity (82:18 e.r.) at −15 °C (Table 1, entry 1). An improved e.r. value of 88:12 was obtained using L2*-Ti complex (entry 2, Table 1). While using 3,3′-substituted BINOL L3* as ligand, a very low enantioselectivity (61:39 e.r.) was observed (entry 3). For further improving the reaction enantioselectivity, 1,2-diphenyl as well as 1,2-di-tert-butylethylenediamine-derived salens (L4* and L5*) were prepared and examined in the model reaction. Unfortunately, the reaction did not complete after two days at −15 °C catalyzed by neither L4*–Ti nor L5*–Ti. On the other hand, only racemic product was obtained (entry 4–5). Further solvent screening indicated that the enantioselectivity was to some extent affected by different solvents. Catalyzed by L1*–Ti complex, some 10% decreases were observed when using ethers or DCM as the solvent (entry 6–8). Compared with using toluene as the solvent catalyzed by L2*–Ti, similar enantioselectivities were obtained when the reaction proceeded in ethers (entry 9–12). Moderate e.r. values were obtained when the reactions were carried out in chloride hydrocarbon solvents (entry 13–15). A sharp decrease of e.r. value to only 62:38 was observed when the model reaction proceeded in CH3CN (entry 16). Therefore, subsequent studies were all carried out in toluene and catalyzed by L2*–Ti complex.
| Entry | Ligand | Solvent | Time | e.r.c |
|---|---|---|---|---|
|
a The reactions were carried out in 2 ml toluene with 0.1 mmol of nitroolefin, and L*:Ti(OiPr)4:1a:TMSCN = 0.2:0.2:1:1.5.
b Not completed.
c Determined by HPLC analysis.
|
||||
| 1 | L1 * | Tol | overnight | 82:18 |
| 2 | L2 * | Tol | overnight | 88:12 |
| 3 | L3 * | Tol | 48 hb | 61:39 |
| 4 | L4 * | Tol | 24 hb | Racemic |
| 5 | L5 * | Tol | 24 hb | Racemic |
| 6 | L1 * | THF | overnight | 76:24 |
| 7 | L1 * | Et2O | overnight | 77:23 |
| 8 | L1 * | DCM | overnight | 76:24 |
| 9 | L2 * | THF | overnight | 87:13 |
| 10 | L2 * | Et2O | overnight | 86:14 |
| 11 | L2 * | DME | 8 hb | 87:13 |
| 12 | L2 * | MTBE | 6.5h b | 88:12 |
| 13 | L2 * | DCM | overnight | 80:20 |
| 14 | L2 * | DCE | 6 h | 80:20 |
| 15 | L2 * | CHCl3 | 6.5 hb | 85:15 |
| 16 | L2 * | CH3CN | 8 hb | 62:38 |
Different ratios of Ligand to titanium and additives were studied for the enantioselectivity improvement (results in Table 2). No reaction occurred neither in the absence of L2* nor titanium (entry 1–2). The yield and enantioselectivity of the reaction were critically affected by the temperature. An excellent e.r. of 96:4 was observed when the reaction was carried out under −78 °C (entry 3). However, only trace of 3a was obtained after 24 h. Then, it was found that conjugate cyanation of 1a was finished after 19 h with 91:9 e.r. value under −40 °C (entry 4). Reducing the loadings of L2* and Ti(OiPr)4 to 10 mol%, no decrease was observed for the reaction enantioselectivity but with much longer time (34 h) (entry 5). It was pleasing to find that similar enantioselectivity was obtained when the reaction was carried out between −40 °C and −15 °C (entry 6). During the subsequent study, the reaction mixture was precooled to −40 °C before adding TMSCN, and then warmed to −15 °C and stirred until the reaction was complete. The e.r. value of 3a was not affected by either reducing the catalyst loadings (entry 7–8) or changing the ratios of L2* and Ti(OiPr)4 (entry 10–11). But the reaction rate was markedly influenced with the loading of Ti(OiPr)4. Longer reaction time was necessary for the completion of the cyanation when using less Ti(OiPr)4. Unfortunately, 2 mol% of the catalyst failed to promote the conjugate cyanation of 1a (entry 9). Considering the shorter reaction time, 20 mol% of L2* and Ti(OiPr)4 was exploited in the additive screening process. Although it was found that a Lewis base could enhance the turnover frequency of the titanium-catalyst (unpublished results), Lewis bases did not work in improving the reaction enantioselectivity (entry 12–14). In addition, the e.r. value of 3a was also not improved by adding protonic additives which were always explored as promising additives in various cyanations (entry 15–18). The model reaction was restrained by t-BuOK and with very poor enantioselectivity (entry 19). Using molecular sieves led to a slight improvement of the reaction enantioselectivity but with much longer reaction time (entry 20).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | L2 * (mol%) | Ti(OiPr)4 (mol%) | Additiveb | Temperature | Time (h) | e.r.c |
| a The reactions were carried out in 2 ml toluene with 0.1 mmol of 1a and 0.15 mmol of TMSCN. b All of the additive loading was 100 mol% according to nitroolefin 1a except otherwise noted. c Determined by HPLC analysis. d 100 mg of 4 Å MS was added. e No reaction. f Not determined. g Overnight. | ||||||
| 1 | 20% | 0 | None | R.T. | N.R.e | N.D.f |
| 2 | 0 | 20% | — | R.T. | N.R.e | N.D.f |
| 3 | 20% | 20% | — | −78 °C | Trace | 96:4 |
| 4 | 20% | 20% | — | −40 °C | 19h | 91:9 |
| 5 | 10% | 10% | — | −40 °C | 34h | 91:9 |
| 6 | 20% | 20% | — | −15 °C | 8 h | 90:10 |
| 7 | 10% | 10% | — | −15 °C | 13 h | 90:10 |
| 8 | 5% | 5% | — | −15 °C | 18 h | 90:10 |
| 9 | 2% | 2% | — | −15 °C | Trace | N.D.f |
| 10 | 20% | 40% | — | −15 °C | 8 h | 90:10 |
| 11 | 20% | 10% | — | −15 °C | 24 h | 90:10 |
| 12 | 20% | 20% | Py | −15 °C | g | 89:11 |
| 13 | 20% | 20% | DIPEA | −15 °C | g | 88:12 |
| 14 | 20% | 20% | TEA | −15 °C | g | 87:13 |
| 15 | 20% | 20% | i-PrOH | −15 °C | g | 87:13 |
| 16 | 20% | 20% | t-BuOH | −15 °C | g | 88:12 |
| 17 | 20% | 20% | p-cresol | −15 °C | g | 90:10 |
| 18 | 20% | 20% | BHT | −15 °C | g | 90:10 |
| 19 | 20% | 20% | t-BuOK | −15 °C | 16h | 58:42 |
| 20 | 20% | 20% | 4 Å MSd | −15 °C | 48 h | 91:9 |
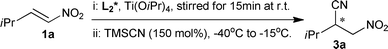
Under the optimal conditions, the substrate scope of this approach was examined and the results are shown in Table 3. Asymmetric cyanation of either linear nitroolefins or cyclic nitroolefin proceeded favorably with moderate to high enantioselectivities. The reaction was not markedly affected by simple alkyl substitution of the nitroolefins. Asymmetric synthesis of β-nitronitriles 3a–c was achieved with high yields and similar enantioselectivities (e.r of 91:9, 92:8 and 91:9, respectively) (entry 1–3). Cyanation of 1d gave 3d with moderate enantioselectivity (86:14 e.r.) due to the negative chelating effect of the methoxy group to the titanium (entry 4). The sterically bulky TBDMS hydroxy-protecting group in 1e–f led to moderate enantioselectivities (e.r. of 80:20 and 85:15 respectively) (entry 5–6). Cyclic nitroolefin 1g was also a suitable substrate for this approach. A relatively longer reaction time (15 h) was needed for the cyanation of 1g. Cyclic β-nitronitrile 3g was obtained with high diastereoselectivity (anti-3g:syn-3g = 16:84) and moderate enantioselectivity (e.r. of 74:26 and 73:27 for anti-3g and syn-3g, respectively) (entry 7).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | Product | Time (h) | Yield (%)b | e.r.c |
|
a The reactions were carried out in 2 ml toluene with 0.1 mmol of nitroolefin, and L2*:Ti(OiPr)4:1:TMSCN = 0.2:0.2:1:1.5.
b Isolated yield.
c Determined by HPLC analysis.
d The S-configuration of 3a was confirmed by comparing with literature optical rotation (ref. 8a).
e The yield referred to both anti-3g and syn-3g. The ratio of anti-3g:syn-3g was 16:84 and determined by 1H NMR of the crude product.
|
|||||
| 1 |
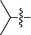
|
3a d | 8 | 73% | 91:9 |
| 2 |
|
3b | 8 | 81% | 92:8 |
| 3 |

|
3c | 8 | 90% | 91:9 |
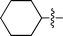
| 4 |
|
3d | 10 | 74% | 86:14 |
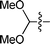
| 5 |
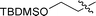
|
3e | 8 | 83% | 80:20 |
| 6 |
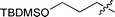
|
3f | 12 | 44% | 85:15 |
| 7 |
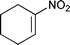
|
3g | 15 | 60%e | 74:26 (anti) |
| 73:27 (syn) |
|||||

In order to synthesize racemic β-nitronitriles for HPLC analysis, cyanation of nitroolefins using racemic salen–Ti complex was carried out at room temperature. It was pleasing to find that the cyanation proceeded very rapidly and completed in minutes. Then enantioselective cyanation of nitroolefins at room temperature was examined with results in Table 4. The asymmetric cyanations was generally completed within ten minutes. The cyanation of 1g needed a longer reaction time (60 min). However, the enantioselectivities and yields did not vary much. The enantioselectivities of 3a–3c were less decreased with e.r. of 84:16, 85:15 and 81:18, respectively (entry 1–3). Much lower yield (26%) of 3f was obtained while the cyanation was carried out at room temperature (entry 6). A similar result was obtained for the cyanation of cyclic nitroolefin 1g (entry 7).
| Entry | R | Product | Time (min) | Yield (%)b | e.r.c |
|---|---|---|---|---|---|
|
a The reactions were carried out in 2 ml toluene with 0.1 mmol of nitroolefin, and L2*:Ti(OiPr)4:1:TMSCN = 0.2:0.2:1:1.5.
b Isolated yield.
c Determined by HPLC analysis.
d The yield referred to both anti-3g and syn-3g. The ratio of anti-3g:syn-3g was 62:38 and determined by 1H NMR of the crude product.
|
|||||
| 1 |
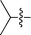
|
3a | 10 | 77% | 84:16 |
| 2 |
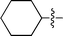
|
3b | 10 | 71% | 85:15 |
| 3 |

|
3c | 10 | 75% | 81:18 |
| 4 |
|
3d | 15 | 76% | 77::23 |
| 5 |
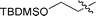
|
3e | 10 | 78% | 77:23 |
| 6 |
|
3f | 15 | 26% | 81:19 |
| 7 |
|
3g | 60 | 63%d | 71:29 (anti) |
| 72:28(syn) |
Cyanation of 1a under standard conditions at low temperature was carried out on a 10 mmol scale affording a high yield of 84% (Scheme 3). It was pleasing to find that the reaction enantioselectivity decreased slightly to e.r. 89:11. Reduction of the nitro group of 3a with Zn/HCl afforded amine 6 favorably.6 Enantioenriched N-Boc protected β-amino acids 8 was obtained by hydrolyzing 6 with H2SO4 (75%) and subsequently protecting 7 with (Boc)2O under basic conditions.6 The total yield of 8 was 35.2% from 3a. The absolute configuration of 8 was confirmed to be S after comparison with the literature optical rotation.15 Thus, the S-configuration of 3a was further confirmed by this result. Based on the configurational relation between L2* and 3a, the enantio-discrimination role of the present L2*–Ti complex could be explained similarly with Feng's report.14c
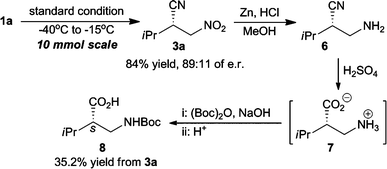 | ||
| Scheme 3 Synthesis of β-amino acids from 3a. | ||
The reported studies on conjugate cyanation indicated that protonic additives such as IPA and phenols were always necessary for the in situ formation of HCN and maintaining high turnovers of the conjugate cyanation catalysts.10–12 However, these protonic additives were not critical in this example. Most importantly, the present salen–Ti complex displayed high turnover frequency even without protonic additives. Based on these observations, we proposed that the asymmetric cyanation of nitroolefin proceeds via an unusual pathway. In situ1H NMR investigation of the asymmetric nitroolefin cyanation was carried out to examine the whole process (Fig. 2). The model cyanation of 1a was carried out in CDCl3 at room temperature. A series of 1H NMR spectra were acquired:
1) S1 referred to the mixture of L2*–Ti complex and 1a. Chemical shifts of the Ha, Hb, and Hc could be easily identified as shown in Fig. 2.
2) S2, S3, and S4 were acquired after adding TMSCN to the above reaction mixture for 2 min, 5 min and 20 min, respectively. Peaks, referring to Ha′, Hb′ as well as Hc′, appeared immediately after addition of TMSCN and disappeared immediately after addition of H2O to quench the reaction. The relative abundances between Ha′ to Ha, Hb′ to Hb, and Hc′ to Hc increased during the reaction.
3) S5 was acquired after quenching the reaction with water. Related Ha′′, Hb′′ and Hc′′ matched well with the spectra of purified 3a.
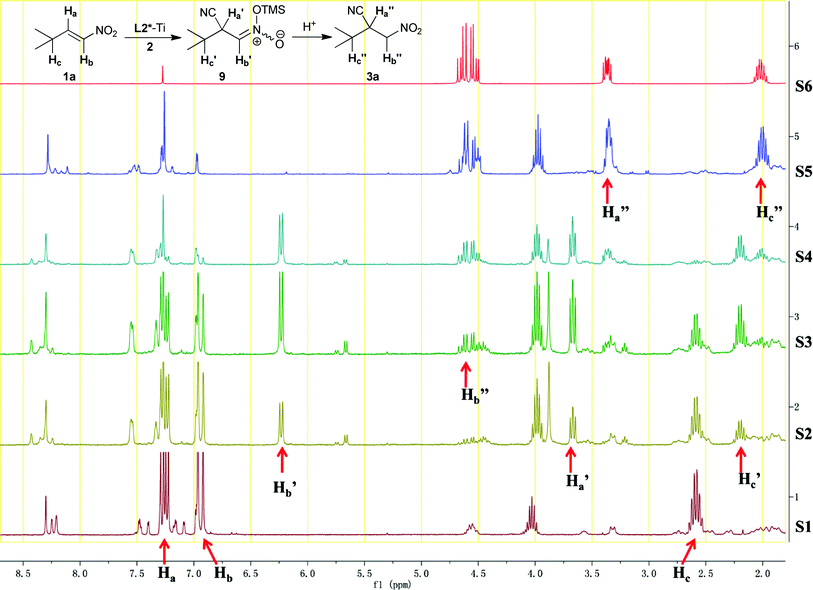 | ||
| Fig. 2
In situ
1H NMR investigation of the asymmetric nitroolefin cyanation. To a NMR tube was added L2*:Ti(OiPr)4:1a = 20 mol%:20 mol%:100mol% in 1 mL CDCl3. S1 was obtained after the reaction mixture standing for 15 min at r.t.. After adding 150 mol% of TMSCN to the above mixture, S2, S3 and S4 were obtained after 2 min, 5 min and 20 min, respectively. S5 was obtained after quenching the reaction by adding 0.5 mL H2O. S6 referred to the 1H NMR of purified 3a. | ||
According to the literature,16 the chemical shift of the silyl nitronate proton was generally between 6.0–6.5 ppm. Thus, Hb′ refers to the silyl nitronate proton (6.23 ppm). For Ha′ was no longer a olefinic proton, its chemical shift was close to that of Ha′′ but at lower field. Chemical shifts of Hc, Hc′ and Hc′′ varied in little, gradually shifting to higher field. Hereby, Ha′, Hb′ and Hc′ were identified as the corresponding protons of the silyl nitronate intermediate. As the phenolic hydroxyl hydrogen of L2* could act as the proton source, the silyl nitronate intermediate was partly protonated and transformed to 3a. The observation of the silyl nitronate intermediate indicated that in situ generated HCN is not needed. In addition, the silyl nitronate intermediate was stable enough under the reaction conditions for a long period of time to be captured by other electrophiles.
Conclusions
In conclusion, the enantioselective cyanation of nitroolefinsviasilyl nitronate intermediate has been achieved by using a salen–Ti complex. The present protocol was used to prepare a series of alkyl nitroolefins with moderate to high enantioselectivities and yields. Although the enantioselectivities of this approach still need to be further improved, the salen–Ti complex maintains a significantly high turnover frequency in the field of conjugate cyanation, especially at room temperature. The present protocol was also successfully applied on a 10 mmol scale. The product could be readily transformed to the corresponding β-amino acids. The silyl nitronate intermediate pathway was confirmed through an in situ1H NMR investigation. Studies on refining the enantioselectivities and applying the silyl nitronate intermediate to useful tandem reactions are underway.Experimental
General methods
All reactions were monitored by thin layer chromatography (TLC), column chromatography purifications were carried out using silica gel. All of the alkyl nitroolefins were prepared according to the literature. 1H NMR and 13C NMR spectra were recorded on a 300 M Bruker® instrument (300 MHz and 75 MHz, respectively) using tetramethylsilane as internal reference. Data for 1H NMR are recorded as follows: chemical shift (δ, ppm), multiplicity (s = singlet, d = doublet, t = triplet, m = multiplet, q = quartet or unresolved, coupling constant(s) in Hz, integration). Data for 13C NMR are reported in terms of chemical shift (δ, ppm). Optical rotations were reported as follows: [α]20D (c: g/100 mL, in solvent). HR-MS was measured with an APEX II 47e mass spectrometer. The e.r. value determination was carried out using chiral HPLC with Daicel Chiracel AD-H/OD-H/OJ column on Waters® with a 996 UV-detector, flow rate = 1.0 mL min−1.General procedure for the asymmetric cyanation of nitroolefins at low temperature
Freshly distilled Ti(OiPr)4 (5.8 μL, 0.02 mmol, 20 mol%) was added to the solution of L2* (10.9 mg, 0.02 mmol, 20 mol%) in toluene (2 mL) under Ar. After stirring for 15 min at room temperature, nitroolefin 1 (0.1 mmol) was added to the above solution which was then cooled to −40 °C. TMSCN (20.0 μL, 0.15 mmol, 150 mol%) in 0.5 mL THF was added to the reaction mixture in dropwise. The reaction was slowly warmed to −15 °C and kept stirring until the reaction was completed (monitored by TLC). Saturated NaHCO3 solution was added to quench the reaction and the mixture was extracted with methylene chloride. The extract was dried with sodium sulfate and concentrated under reduced pressure. After column chromatography on silica gel eluting with 10% ethyl acetate in petroleum, the β-nitronitrile was obtained.:9 e.r. was determined by HPLC analysis (OD-H column, hexane/iPrOH = 90/10); retention times: tmajor = 16.8, tminor = 19.8. [α]21D +3.8 (c 1.05, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.59 (ddd, J = 20.0, 14.0, 7.2 Hz, 2H), 3.37 (ddd, J = 8.4, 6.0, 5.1 Hz, 1H), 2.02 (dtd, J = 13.5, 6.7, 5.1 Hz, 1H), 1.15 (dd, J = 10.1, 6.8 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 116. 9, 73.6, 36.8, 28.5, 20.7, 18.3. Anal. Calcd. for C6H10N2O2: C 50.69%, H 7.09%, N 19.63%. Found C 51.83%, H 7.22%, N 19.69%.
:8 e.r. was determined by HPLC analysis (OD-H column, hexane/iPrOH = 90/10); retention times: tmajor = 17.9, tminor = 21.9. [α]21D −8.9 (c 1.01, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.60 (qd, J = 14.0, 7.2 Hz, 2H), 3.33 (dt, J = 8.4, 5.6 Hz, 1H), 1.97–1.55 (m, 6H), 1.40–1.08 (m, 5H). 13C NMR (75 MHz, CDCl3) δ 117.3, 73.2, 37.4, 36.0, 30.9, 29.0, 25.6, 25.4, 25.3. Anal. Calcd. for C9H14N2O2: C 59.32%, H 7.74%, N 15.37%. Found C 58.93%, H 6.99%, N 14.89%.
:9 e.r. was determined by HPLC analysis (OD-H column, hexane/iPrOH = 95/5); retention times: tmajor = 23.8, tminor = 27.3. [α]21D −19.8 (c 1.01, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.57 (ddd, J = 20.2, 13.9, 7.0 Hz, 2H), 3.40 (ddd, J = 16.5, 7.8, 6.2 Hz, 1H).1.80–1.65 (m, 2H), 1.65–1.47 (m, 2H), 1.46–1.32 (m, 2H), 0.95 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 118.0, 74.6, 29.8, 29.1, 28.6, 21.9, 13.6. Anal. Calcd. for C7H12N2O2: C 53.83%, H 7.74%, N 17.94%. Found C 54.00%, H 7.67%, N 16.62%.
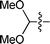
:14 e.r. was determined by HPLC analysis (AD-H column, hexane/iPrOH = 97/3); retention times: tminor = 36.3, tmajor = 38.9. [α]21D +2.9 (c 0.68, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.77–4.61 (m, 3H), 3.70–3.62 (m, 1H), 3.51 (d, J = 1.3 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 115.7, 101.8, 70.8, 56.7, 56.3, 34.5. Anal. Calcd. for C6H10N2O4: C 41.38%, H 5.79%, N 14.35%. Found C 43.83%, H 6.12%, N 14.24%.
:20 e.r. was determined by HPLC analysis (OD-H column, hexane/iPrOH = 95/5); retention times: tmajor = 14.0, tminor = 15.9. [α]21D −13.1 (c 0.61, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.79–4.57 (m, 2H), 3.91–3.77 (m, 2H), 3.73–3.61 (m, 1H), 0.91 (s, 9H), 0.09 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 117.9, 74.5, 59.2, 32.1, 27.1, 25.8, 18.1, −5.6. HRMS (ESI): C11H22N2O3Si +NH4, Calc: 276.1738, Found: 276.1733.
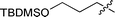
:15 e.r. was determined by HPLC analysis (OJ column, hexane/iPrOH = 98/2); retention times: tminor = 17.4, tmajor = 19.5. [α]21D −9.7 (c 0.72, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.64 (dd, J = 14.0, 7.8 Hz, 1H), 4.52 (dd, J = 14.0, 6.1 Hz, 1H), 3.75–3.63 (m, 2H), 3.61–3.47 (m, 1H), 1.88–1.67 (m, 4H), 0.89 (s, 9H), 0.06 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 118.0, 74.7, 61.8, 29.6, 29.3, 26.6, 25.8, 18.2, −5.5. HRMS (ESI): C12H24N2O3Si +H, Calc: 273.1629, Found: 273.1631.
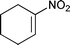
:26 e.r. was determined by HPLC analysis (OJ column, hexane/iPrOH = 85/15); retention times: tmajor = 27.0, tminor = 32.1. [α]28D +28.2 (c 0.78, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.55 (td, J = 10.3, 4.2 Hz, 1H), 3.23 (ddd, J = 11.1, 9.9, 4.1 Hz, 1H), 2.53–2.35 (m, 1H), 2.33–2.19 (m, 1H), 1.95–1.77 (m, 3H), 1.76–1.62 (m, 1H), 1.54–1.30 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 118.4, 84.6, 31.8, 30.5, 27.9, 23.2, 23.1.
:27 e.r. was determined by HPLC analysis (AD-H column, hexane/iPrOH = 95/5); retention times: tmajor = 21.7, tminor = 23.3. [α]28D +37.5 (c 0.64, CHCl3). 1H NMR (300 MHz, CDCl3) δ 4.36 (dt, J = 11.7, 4.0 Hz, 1H), 3.76–3.63 (m, 1H), 2.44 (m, 1H), 2.32–2.17 (m, 1H), 2.17–2.09 (m, 1H), 2.01 (m, 1H), 1.80–1.61 (m, 3H), 1.51–1.32 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 117.3, 82.4, 32.0, 28.1, 26.9, 23.6, 20.9.
General procedure for the asymmetric cyanation of nitroolefins at room temperature
After stirring a solution of freshly distilled Ti(OiPr)4 (5.8 μL, 0.02 mmol, 20 mol%) and L2* (10.9 mg, 0.02 mmol, 20 mol%) in toluene (2 mL) under Ar for 15 min at room temperature, nitroolefin 1 (0.1 mmol) was added. TMSCN (20.0 μL, 0.15 mmol, 150 mol%) was added to the reaction mixture in one-pot. When the reaction was completed (monitored by TLC), the product was obtained following a general work-up procedure.To the above residue was added 5 mL H2SO4 (75%) and heated at reflux for 2 h. The solution was then cooled to 0 °C and carefully adjusted to pH 10 with 40% NaOH. Dioxane (5 mL) was added to the above aqueous solution followed by (Boc)2O (240 mg, 1.1 eq. according to the starting loading of 3a). The solution was warmed to room temperature and stirred for 1 h. The dioxane was removed in vacuo, the aqueous layer was acidified to pH 2 with 1 M NaHSO4 and extracted with ethyl acetate (2 × 15 mL). The organic phase was dried and concentrated in vacuo. The residue was purified by flash column chromatography on SiO2 (33% ethyl acetate/hexane) to afford 8 as a white solid (81.3 mg, 35.2% from 3a). [α]rtD +3° (c 1.0, CHCl3). 1H NMR (300 MHz, CDCl3) δ 10.6 (br,1H), 6.70 & 5.03 (br, 1H), 3.44–3.40 (m, 1H), 3.28–3.09 (m, 1H), 2.52–2.38 (m, 1H), 2.05–1.90 (m, 1H), 1.48–1.44 (m, 9H), 1.02–0.96 (m, 6H). 13C NMR (75 MHz, CDCl3): δ 179.8 & 178.3, 158.1 & 155.9, 81.0&79.6, 52.8 & 52.1, 40.7&39.5, 28.7, 28.3, 28.2, 20.4, 20.3, 19.8. ESI-MS (M+H)+: 232.1.15
Procedure for the in situ1H NMR investigation
To an NMR tube was added L2* (5.5 mg, 0.01 mmol, 20 mol%), Ti(OiPr)4 (2.9 μL, 0.01 mmol, 20 mol%), and 1a (0.05 mmol) in 1 mL CDCl3. 1H NMR S1 was obtained after the reaction mixture had been left for 15 min at room temperature. After addition of 150 mol% of TMSCN to the above mixture, S2, S3 and S4 were obtained after standing for 2 min, 5 min and 20 min, respectively. S5 was obtained after quenching the reaction with 0.5 mL H2O.Acknowledgements
We gratefully acknowledge financial support from NSFC (nos. 21002043, 20932003, and 90813012) and the National S&T Major Project of China (2009ZX09503-017).Notes and references
- Selected reviews: (a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC; (b) D. Seebach and J. Gardiner, Acc. Chem. Res., 2008, 41, 1366 CrossRef CAS.
- (a) G. V. M. Sharma, N. Chandramouli, S. J. Basha, P. Nagendar, K. V. S. Ramakrishna and A. V. S. Sarma, Chem.–Asian J., 2011, 6, 84 CrossRef CAS; (b) S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2010, 132, 13879 CrossRef CAS; (c) F. Fulop, T. A. Martinek and G. K. Toth, Chem. Soc. Rev., 2006, 35, 323 RSC.
- (a) R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219 CrossRef CAS; (b) G. Grasso, A. Pietropaolo, G. Spoto, G. Pappalardo, G. R. Tundo, C. Ciaccio, M. Coletta and E. Rizzarelli, Chem.–Eur. J., 2011, 17, 2752 CrossRef CAS.
- Selected examples: (a) A. R. Minter, A. A. Fuller and A. K. Mapp, J. Am. Chem. Soc., 2003, 125, 6846 CrossRef CAS; (b) M. P. Sibi and K. Patil, Angew. Chem., Int. Ed., 2004, 43, 1235 CrossRef CAS; (c) A. Perdih and M. S. Dolenc, Curr. Org. Chem., 2007, 11, 801 CrossRef CAS; (d) J. M. M. Verkade, L. J. C. van Hemert, P. J. L. M. Quaedflieg and F. P. J. T. Rutjes, Chem. Soc. Rev., 2008, 37, 29 RSC; (e) B. E. Sleebs, T. T. Van Nguyen and A. B. Hughes, Org. Prep. Proced. Int., 2009, 41, 429 CrossRef CAS; (f) B. Weiner, W. Szymanski, D. B. Janssen, A. J. Minnaard and B. L. Feringa, Chem. Soc. Rev., 2010, 39, 1656 RSC; (g) J. L. Acena, A. Simon-Fuentes and S. Fustero, Curr. Org. Chem., 2010, 14, 928 CrossRef CAS.
- (a) R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017 CrossRef CAS; (b) N. Sewald, Angew. Chem., Int. Ed., 2003, 42, 5794 CrossRef CAS; (c) M. X. Wang, G. Deng, D. X. Wang and Q. Y. Zheng, J. Org. Chem., 2005, 70, 2439 CrossRef CAS; (d) B. Shen and J. N. Johnston, Org. Lett., 2008, 10, 4397 CrossRef CAS; (e) C. X. Xu and J. X. Xu, Amino Acids, 2011, 41, 195 CrossRef CAS.
- J. C. Anderson, A. J. Blake, M. Mills and P. D. Ratcliffe, Org. Lett., 2008, 10, 4141 CrossRef CAS.
- (a) Otto M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (b) A. Duursma, A. J. Minnaard and B. L. Feringa, J. Am. Chem. Soc., 2003, 125, 3700 CrossRef CAS; (c) R. Ballini, G. Bosica, D. Fiorini, A. Palmieri and M. Petrini, Chem. Rev., 2005, 105, 933 CrossRef CAS; (d) G. R. Boyce and J. S. Johnson, Angew. Chem., Int. Ed., 2010, 49, 8930 CrossRef CAS; (e) D. Enders, M. H. Bonten and G. Raabe, Angew. Chem., Int. Ed., 2007, 46, 2314 CrossRef CAS.
- (a) D. Enders, R. Syrig, G. Raabe, R. Fernández, C. Gasch, J. M. Lassaletta and J. M. Llera, Synthesis, 1996, 48 CrossRef CAS; (b) L. Bernardi, F. Fini, M. Fochi and A. Ricci, Synlett., 2008, 12, 1857 Search PubMed; (c) P. Bernal, R. Fernández and J. M. Lassaletta, Chem.–Eur. J., 2010, 16, 7714 CrossRef CAS.
- M. North, D. L. Usanov and C. Young, Chem. Rev., 2008, 108, 5146 CrossRef CAS.
- (a) M. S. Taylor, D. N. Zalatan, A. M. Lerchner and E. N. Jacobsen, J. Am. Chem. Soc., 2005, 127, 1313 CrossRef CAS; (b) Y. Tanaka, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2008, 130, 6072 CrossRef CAS; (c) T. Arai, Y. Suemitsu and Y. Ikematsu, Org. Lett., 2008, 11, 333 CrossRef; (d) Y. Tanaka, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 8862 CrossRef CAS; (e) N. Kurono, N. Nii, Y. Sakaguchi, M. Uemura and T. Ohkuma, Angew. Chem., Int. Ed., 2011, 50, 5541 CrossRef CAS.
- (a) G. M. Sammis and E. N. Jacobsen, J. Am. Chem. Soc., 2003, 125, 4442 CrossRef CAS; (b) G. M. Sammis, H. Danjo and E. N. Jacobsen, J. Am. Chem. Soc., 2004, 126, 9928 CrossRef CAS; (c) C. Mazet and E. N. Jacobsen, Angew. Chem., Int. Ed., 2008, 47, 1762 CrossRef CAS; (d) N. Madhavan and M. Weck, Adv. Synth. Catal., 2008, 350, 419 CrossRef CAS.
- (a) T. Mita, K. Sasaki, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 127, 514 CrossRef; (b) N. Yamagiwa, H. Qin, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2005, 127, 13419 CrossRef CAS.
- J. Wang, W. Li, Y. Liu, Y. Chu, L. Lin, X. Liu and X. Feng, Org. Lett., 2010, 12, 1280 CrossRef CAS.
- Selected examples: (a) F.-X. Chen, B. Qin, X. M. Feng, G. L. Zhang and Y. Z. Jiang, Tetrahedron, 2004, 60, 10449 CrossRef CAS; (b) D. A. Nicewicz, C. M. Yates and J. S. Johnson, J. Org. Chem., 2004, 69, 6548 CrossRef CAS; (c) S.-K. Chen, D. Peng, H. Zhou, L.-W. Wang, F.-X. Chen and X.-M. Feng, Eur. J. Org. Chem., 2007, 38, 639 CrossRef; (d) M. H. Yang, C. J. Zhu, F. Yuan, Y. J. Huang and Y. Pan, Org. Lett., 2005, 7, 1927 CrossRef CAS.
- R. Moumné, B. Denise, K. Guitot, H. Rudler, S. Lavielle and P. Karoyan, Eur. J. Org. Chem., 2007, 38, 1912 CrossRef.
- (a) K. R. Knudsen, T. Risgaard, N. Nishiwaki, K. V. Gothelf and K. A. Jørgensen, J. Am. Chem. Soc., 2001, 123, 5843 CrossRef CAS; (b) T. Ooi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 9022 CrossRef CAS; (c) T. Ooi, K. Doda and K. Maruoka, J. Am. Chem. Soc., 2003, 125, 2054 CrossRef CAS.
- HPLC spectra indicate that opposite enantiomer of 3b was obtained in this work compared with the one obtained in ref. 8c (see ESI†). After further HPLC experimentation and carefully re-checking the product rotation values, we became aware some erroneous data in ref. 8c. The optical rotation value of 3b obtained in ref. 8c should be positive but not negative. Similar instance was also found for compound 3c.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR data and spectra of the starting materials 1, HPLC and NMR spectra of the products. See DOI: 10.1039/c1ob05899a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |