Synthesis of novel 2,8-disubstituted indolo[3,2-b]carbazoles†
Sven
Van Snick
and
Wim
Dehaen
*
Division of Molecular Design and Synthesis, Department of Chemistry, K. U. Leuven, Celestijnenlaan 200F, B-3001, Leuven, Belgium
First published on 31st August 2011
Abstract
A new synthetic pathway towards 2,8-difunctionalised indolo[3,2-b]carbazoles was investigated. The presented method offers a short and high yielding route towards 2,8-dibromo-5,11-dihexyl-6,12-diphenyl-indolo[3,2-b]carbazole. It is demonstrated that the latter compound is a versatile building block, enabling the synthesis of a number of previously unreported 5,11-dialkyl-6,12-diphenyl-indolo[3,2-b]carbazoles in moderate to good yields, using Suzuki and Sonogashira cross-coupling reaction. Furthermore it is shown that 2,8-dibromo-5,11-dihexyl-6,12-diphenyl-indolo[3,2-b]carbazole can be easily formylated, giving rise to the 2,8-diformyl-5,11-dihexyl-6,12-diphenyl-indolo-[3,2-b]carbazole. The latter compound was successfully subjected to condensation reactions.
Introduction
Indolocarbazoles represent an important class of N-heterocycles characterized by an array of applications depending on the considered structural isomer. Among the five possible isomers, 5,11-dihydroindolo[3,2-b]carbazoles (which we will subsequently call indolo[3,2-b]carbazoles or ICZs for short) have attracted a large interest because of their structural and electronic properties (high charge carrier mobility), combined with good stability towards atmospheric conditions, which makes them ideal components for use in organic electronics. Hence, indolo[3,2-b]carbazoles have been successfully implemented in OFETs,1 OLEDs,2 DSSCs3 and even PSCs.4Although the field of applications is broad, the search for new functional materials for ICZ-based electronics has reached an impasse, which can mainly be attributed to the lack of high yielding synthetic pathways towards various functionalised ICZs and the sizeable presence of alternatives such as thiophene-based oligomers and polymers, which benefit from the well established knowledge concerning their reactivity.5,6
One of the first syntheses of indolo[3,2-b]carbazole, by cyclodehydrogenation of N,N′-diphenyl-p-phenylenediamine, was reported in 1961 by Grotta et al.7 Ever since, many other syntheses have been reported in the literature, most of which depend on multi-step sequences characterized by low overall yields.8 In contrast, a notable result was obtained by Pindur et al. who isolated the parent indolo[3,2-b]carbazole in 80% yield by condensation of 3,3′-bis(indolyl)methane with triethyl orthoformate.9 For a detailed overview on both synthesis and characterisation of indolo[3,2-b]carbazoles excellent reviews can be found in the literature.10
Even though many pathways towards indolo[3,2-b]carbazole exist, few allow for the synthesis of 2,8-difunctionalized ICZs. Usually these ICZs are prepared in low to moderate yield by double Fischer cyclisation of phenylhydrazones, obtained through condensation between 1,4-cyclohexanedione and the appropriate substituted hydrazine.11
Herein we report an alternative fast and robust method for synthesizing various 2,8-difunctionalized ICZs in high yield and with improved solubility, by the use of cheap commercially available starting materials.
Results and discussion
The presented method is a continuation of our work previously done on 6-monosubstituted indolo[3,2-b]carbazoles and 6,12-diaryl-indolo[3,2-b]carbazoles.12,13 It was demonstrated that the latter ICZs could be obtained in moderate to good yields using a two-step method involving the condensation of an aromatic aldehyde and indole under acid catalysis (HI) in CH3CN, yielding a mixture of indolo[3,2-b]carbazole and 5,6,11,12-tetrahydro-indolo[3,2-b]carbazole. The reaction proceeded overnight and heating (80 °C) was imperative to ensure higher amounts of the oxidized derivative.However, a second oxidation step with I2 in CH3CN was still necessary to convert the remaining non-oxidized derivative into indolo[3,2-b]carbazole. This method was further optimized to a one-pot procedure and expanded to various aromatic aldehydes, yielding the expected ICZs in good yields.13 (Fig. 1)
![Synthesis of indolo[3,2-b]carbazole. (i) CH3CN, HI (57%), 80 °C, 14 h. (ii) CH3CN, I2, 80 °C, 14 h. R = Aryl.](/image/article/2012/OB/c1ob06298k/c1ob06298k-f1.gif) | ||
| Fig. 1 Synthesis of indolo[3,2-b]carbazole. (i) CH3CN, HI (57%), 80 °C, 14 h. (ii) CH3CN, I2, 80 °C, 14 h. R = Aryl. | ||
While this method produced the desired compounds, the low solubility in organic solvents hampered further functionalisation of the ICZ-backbone.
As a continuation of this work, we planned to address the problem of low solubility by alkylating the two nitrogens present in the ICZ backbone. It has previously been shown that this is indeed a facile and flexible strategy to obtain highly soluble ICZs.1 Compound 1 was easily converted to the 5,11-dihexyl-6,12-diphenyl-indolo[3,2-b]carbazole 3, by treating the former with a 50% NaOH solution and subsequent addition of the 1-bromohexane alkylating reagent at room temperature. The alkylated compound was obtained in 78% yield and indeed shows greatly improved solubility in CH2Cl2 as compared to the non-alkylated derivative. (Scheme 1)

In order to create a functionalisable substrate we envisioned to brominate 3. We have previously demonstrated that bromination of the 6-unsubstituted ICZ derivatives using 3 eq. NBS yielded the 2,6,8-tribrominated ICZ in 20% yield.12 However, upon treating 3 under similar conditions we did not observe any trace of the expected 2,8-dibromo-5,11-dihexyl-6,12-diphenyl-indolo[3,2-b]carbazole 4. Starting compound 3 was recovered nearly completely.
We then considered to functionalise the 5,6,11,12-tetrahydroindolo[3,2-b]carbazole 2 prior to oxidation. Because the latter corresponds to two indole nuclei connected via a methylene bridge, the reactivity of 2 should be similar to that of 2,3-disubstituted indoles and bromination of indoles is well described in literature.14,15 Hence, we explored a way to obtain ICZ 2 as the major reaction product in the initial condensation reaction.
From our previous research it was apparent that elevated temperatures during the condensation favoured the oxidized ICZ 1. Therefore we envisioned to condense indole with benzaldehyde under acid catalysis, but this time at room temperature in order to obtain only non-oxidized ICZ 2.
A convincing result was obtained when MS-analysis and NMR demonstrated the presence of only 5,6,11,12-tetrahydroindolo[3,2-b]carbazole 2, as a mixture of both the cis- and trans-ICZ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2), in the reaction mixture and no trace of the oxidized analogue. This product was easily isolated by filtration (97% yield) and subsequently alkylated to improve its solubility. We found that upon alkylation of 2, the length of the chosen alkyl chain was determinative for the reaction products formed. When methyl iodide was used, ICZ 5a was formed in 90% yield after 48 h stirring at room temperature. 1H NMR analysis of the isolated compound indicated that not only alkylation but also oxidation proceeded, evidenced by the disappearance of the characteristic singlets corresponding to the benzylic protons of both the cis- and trans-5,11-dimethyl-6,12-diphenyl-5,6,11,12-tetrahydroindolo[3,2-b]carbazole 2. The oxidation here apparently is affected by DMSO and/or advantageous oxygen. A similar result was obtained when alkylating 2 with n-propyl bromide. After 48 h, 5b was obtained in 86% yield. Conversely, upon alkylating 2 with longer alkyl chains such as hexyl, we did not observe any oxidation products being formed after 24 h. It was found that the yield of alkylated ICZ 6a did not increase when the reaction proceeded longer than three hours and thus ICZ 6a could be isolated in 67% yield as a mixture of the cis- and trans-isomers (1:3). Alkylation with dodecyl bromide was also performed using these reaction conditions, with a comparable result, however in this case crystallization resulted in the isolation of only the trans-isomer in 50% yield. (Scheme 2, Table 1)
:2), in the reaction mixture and no trace of the oxidized analogue. This product was easily isolated by filtration (97% yield) and subsequently alkylated to improve its solubility. We found that upon alkylation of 2, the length of the chosen alkyl chain was determinative for the reaction products formed. When methyl iodide was used, ICZ 5a was formed in 90% yield after 48 h stirring at room temperature. 1H NMR analysis of the isolated compound indicated that not only alkylation but also oxidation proceeded, evidenced by the disappearance of the characteristic singlets corresponding to the benzylic protons of both the cis- and trans-5,11-dimethyl-6,12-diphenyl-5,6,11,12-tetrahydroindolo[3,2-b]carbazole 2. The oxidation here apparently is affected by DMSO and/or advantageous oxygen. A similar result was obtained when alkylating 2 with n-propyl bromide. After 48 h, 5b was obtained in 86% yield. Conversely, upon alkylating 2 with longer alkyl chains such as hexyl, we did not observe any oxidation products being formed after 24 h. It was found that the yield of alkylated ICZ 6a did not increase when the reaction proceeded longer than three hours and thus ICZ 6a could be isolated in 67% yield as a mixture of the cis- and trans-isomers (1:3). Alkylation with dodecyl bromide was also performed using these reaction conditions, with a comparable result, however in this case crystallization resulted in the isolation of only the trans-isomer in 50% yield. (Scheme 2, Table 1)

With the alkylated, non-oxidized ICZ 6a available we set out to brominate this compound using NBS in acetic acid. 1H NMR analysis of the compound isolated from this reaction after aqueous work-up confirmed the oxidation of 6a, but also the presence of two doublets at 7.37 ppm and 7.10 ppm, indicating the 2,8-dibromination of the ICZ-backbone. Further characterization of 6a by ESI-MS showed the expected mass corresponding to the dibrominated ICZ 4. In view of the failed bromination of the ICZ 3, we can safely assume that bromination takes place before aromatisation.
Since aryl halides are very versatile substrates in various transition metal catalyzed cross coupling reactions,16 we envisioned to expand the conjugation of the ICZs by use of Suzuki, Stille and Sonogashira coupling reactions using the newly obtained 5,11-dihexyl-2,8-dibromo-6,12-diphenyl-indolo[3,2-b]carbazole 4. (Scheme 3)
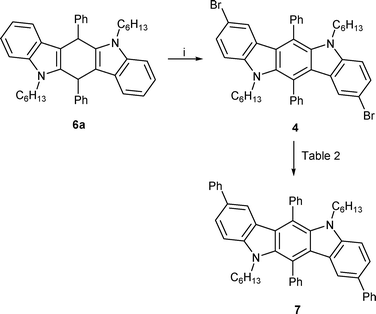 | ||
| Scheme 3 (i) AcOH, 4 eq. NBS, 3 h. | ||
The Suzuki coupling proved to be troublesome at first, but during optimisation procedure it became quickly apparent that the coupling was sensitive to the chosen base. Ultimately, upon treatment of 4 with phenylboronic acid under optimized reaction conditions we were able to isolate the expected compound in 92% yield. (Table 2)
An attempt was also made to react 4 under Sonogashira reaction conditions, using a 1:1 ratio of THF/iPr2NH, CuI and various acetylene substrates, such as TMS-acetylene, phenylacetylene and 2-methyl-3-butyn-2-ol. However, TLC analysis during these reactions showed no reactivity of the ICZ. The Stille coupling with 2-(tributylstannyl)thiophene and the Mizoroki–Heck reaction with styrene also yielded no expected products and in all cases only starting material 4 was recovered.
Compound 4 also allows to synthesize the diformylated derivative 8. By treatment of the former with BuLi at −78 °C in THF, followed by the addition of DMF the diformylated ICZ 8 was obtained in 81% yield. The presence of two aldehyde moieties allowed us to further expand the conjugation of ICZ 8 by conducting condensation reactions, yielding two previously unreported 2,8-difunctionalized ICZs.
Firstly, the Horner–Wadsworth–Emmons modification to the Wittig reaction was performed by condensing 8 with diethylbenzyl phosphonate in the presence of KHMDS, resulting in the formation of 9 in 74% yield. (Scheme 4)
 | ||
Scheme 4 (i) THF, −78 °C, n-BuLi, DMF. (ii) THF, KOtBu, 0 °C, BnP(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OEt2. O)OEt2. | ||
Secondly, in order to overcome the difficulties encountered when trying to introduce an acetylene functional group by Sonogashira reaction and with the diformylated ICZ 8 available, we were able to transform the latter to 11 according to the Corey–Fuchs protocol.17 To accomplish this, 8 was firstly converted to the bisdibromovinyl-ICZ 10, followed by treatment with n-BuLi at −78 °C leading to 11 in 65% yield. (Scheme 5)
 | ||
| Scheme 5 (i) CH2Cl2, CBr4, PPh3, 1 h. (ii) THF, n-BuLi, 2h. (iii) THF, iPr2NH, CuI, Pd(PPh3)4, C6H6Br, Reflux. | ||
The obtained dialkyne 11 was then treated with bromobenzene under standard Sonogashira reaction conditions, yielding 12 without problems in 57% yield.
Conclusions
An efficient method has been developed for the synthesis of 2,8-difunctionalised ICZs in good overall yields by use of cheap starting materials and straightforward reactions. The otherwise not easily accessible 2,8-dibromo-5,11-dihexyl-indolo[3,2-b]carbazole can be obtained in high yield using a one pot bromination/aromatisation of the dialkylated indolocarbazole derivative. The versatility of this dibrominated building block has been demonstrated by implementing several standard reaction protocols. Amongst these, the double Suzuki coupling and the introduction of two aldehyde functional groups and subsequent conversion to styryl and phenylacetylene derivatives offer a solid way for expanding conjugation of the ICZ aromatic system. All of the 5,11-dialkylated ICZs show good solubility in common organic solvents, allowing for easy manipulation in follow-up reactions. Therefore, the 2,8-disubstituted ICZs also represent viable building blocks for ICZ-containing polymers, which will be the subject of our subsequent research.Acknowledgements
The authors thank IMEC for a fellowship, and the F.W.O.-Vlaanderen, The “Ministerie voor Wetenschapsbeleid” (Grant IAP-IV-27), and the University of Leuven for continuing financial support.Notes and references
- Y. Wu, Y. Li, S. Gardner and B. S. Ong, J. Am. Chem. Soc., 2005, 127, 614–618 CrossRef CAS; Y Li, Y. Wu and B. S. Ong, Macromolecules, 2006, 39, 6521–6527 CrossRef; P.-L. T. Boudreault, S. Wakim, N. Blouin, M. Simard, C. Tessier, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 129, 9125–9136 CrossRef; Y. Guo, H. Zhao, G. Yu, C.-a. Di, W. Liu, S. Jiang, S. Yan, C. Wang, H. Zhang, X. Sun, X. Tao and Y. Liu, Adv. Mater., 2008, 20, 4835–4839 CrossRef; P.-L. T. Boudreault, A. A. Virkar, Z. Bao and M. Leclerc, Org. Electron., 2010, 11, 1649–1659 CrossRef.
- H.-P. Zhao, X.-T. Tao, P. Wang, Y. Ren, J.-X. Yang, Y.-X. Yan, C.-X. Yuan, H.-J. Liu, D.-C. Zou and M.-H. Jiang, Org. Electron., 2007, 8, 673–682 CrossRef CAS; H.-P. Zhao, X.-T. Tao, F.-Z. Wang, Y. Ren, X.-Q. Sun, J.-X. Yang, Y.-X. Yan, D.-C. Zou, X. Zhao and M.-H. Jiang, Chem. Phys. Lett., 2007, 439, 132–137 CrossRef; H.-P. Zhao, F.-Z. Wang, C.-X. Yuan, X.-T. Tao, J.-L. Sun, D.-C. Zou and M.-H. Jiang, Org. Electron., 2009, 10, 925–931 CrossRef; S. Lengvinaite, J. V. Grazulevicius, S. Grigalevicius, R. Gu, W. Dehaen, V. Jankauskas, B. Zhang and Z. Xie, Dyes Pigm., 2010, 85, 183–188 CrossRef.
- X.-H. Zhang, Z.-S. Wang, Y. Cui, N. Koumura, A. Furube and K. Hara, J. Phys. Chem. C, 2009, 113, 13409–13415 CAS.
- E. Zhou, S. Yamakawa, Y. Zhang, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2009, 19, 7730–7737 RSC; Y. Xia, X. Su, Z. He, X. Ren, H. Wu, Y. Cao and D. Fan, Macromol. Rapid Commun., 2010, 31, 1287–1292 CrossRef CAS; E. Zhou, J. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890–4895 CrossRef.
- Y. Liu, Y. Liu and X. Zhan, Macromol. Chem. Phys., 2011, 212, 428–443 CrossRef CAS; S. Hotta and T. Yamao, J. Mater. Chem., 2011, 21, 1295–1304 RSC.
- R. D. McCullouggh, Adv. Mater., 1998, 10, 93–116 CrossRef CAS; J. Roncali, P. Blanchard and P. Frère, J. Mater. Chem., 2005, 15, 1589–1610 RSC.
- H. M. Grotta, C. J. Riggle and A. E. Bearse, J. Org. Chem., 1961, 26, 1509–1511 CrossRef CAS.
- H. Ishii, E. Sakurada, K. Murakami, S. Takase and H. Tanaka, J. Chem. Soc., Perkin Trans. 1, 1988, 2387–2395 RSC; J. Lehmann and U. Hartmann, Arch. Pharm., 1989, 322, 451–452 CrossRef CAS; A. R. Katritzky, J. Li and C. V. Stevens, J. Org. Chem., 1995, 60, 3401–3404 CrossRef; D. S. Black, A. J. Ivory and N. Kumar, Tetrahedron, 1995, 51, 11801–11808 CrossRef; J. Tholander and J. Bergman, Tetrahedron, 1999, 55, 6243–6260 CrossRef.
- U. Pindur and J. Müller, Arch. Pharm., 1987, 320, 280–282 CrossRef CAS.
- S. Wakim, B.-R. Aïch, Y. Tao and M. Leclerc, Polym. Rev., 2008, 48, 432–462 CrossRef CAS; T. Janosik, N. Wahlström and J. Bergman, Tetrahedron, 2008, 64, 9159–9180 CrossRef; J. Bergman, T. Janosik and N. Wahlström, Adv. Heterocycl. Chem., 80, 1–71 CrossRef.
- B. Robinson, J. Chem. Soc., 1963, 3097–3099 RSC; L. N. Yudina and J. Bergman, Tetrahedron, 2003, 59, 1265–1275 CrossRef CAS.
- R. Gu, A. Hameurlaine and W. Dehaen, Synlett, 2006, 10, 1535–1538 Search PubMed; R. Gu, K. Van Hecke, L. Van Meervelt, S. Toppet and W. Dehaen, Org. Biomol. Chem., 2006, 4, 3785–3789 Search PubMed; R. Gu, A. Hameurlaine and W. Dehaen, J. Org. Chem., 2007, 72, 2707–2713 CrossRef CAS.
- R. Gu, K. Robeyns, L. Van Meervelt, S. Toppet and W. Dehaen, Org. Biomol. Chem., 2008, 6, 2484–2487 Search PubMed; R. Gu, S. Van Snick, K. Robeyns, L. Van Meervelt and W. Dehaen, Org. Biomol. Chem., 2009, 7, 380–385 Search PubMed; M. Kirkus, J. V. Grazulevicius, S. Grigalevicius, R. Gu, W. Dehaen and V. Jankauskas, Eur. Polym. J., 2009, 45, 410–417 CrossRef CAS; W. Maes, T. H. Ngo, R. Gu, A. S. Starukkin, M. M. Kruk and W. Dehaen, Eur. J. Org. Chem., 2010, 2576–2586 CrossRef.
- F. Yamado, M. Tamura, A. Hasegawa and M. Somei, Chem. Pharm. Bull., 2002, 50, 92–99 CrossRef.
- O. R. Suárez-Castillo, L. Beiza-Granados, M. Meléndeze-Rodriguez, A. Alvarez-Hernández, M. S. Morales-Rios and P. Joseph-Nathan, J. Nat. Prod., 2006, 69, 1596–1600 CrossRef.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; S. P. Stanforth, Tetrahedron, 1998, 54, 263–303 CrossRef; H. Doucet, Eur. J. Org. Chem., 2008, 2013–2030 CrossRef.
- E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 13, 3769–3772 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ob06298k |
| This journal is © The Royal Society of Chemistry 2012 |