Direct electrochemical detection and sizing of silver nanoparticles in seawater media
E. J. E.
Stuart
a,
N. V.
Rees
a,
J. T.
Cullen
b and
R. G.
Compton
*a
aDepartment Of Chemistry, Physical & Theoretical Chemistry Laboratory, Oxford University, South Parks Road, Oxford OX1 3QZ, UK. E-mail: richard.compton@chem.ox.ac.uk; Fax: +44 (0)1865 275410; Tel: +44 (0)1865 275413
bSchool of Earth and Ocean Sciences, University of Victoria, Victoria, BC, Canada V8W 3V6
First published on 19th November 2012
Abstract
We report proof-of-concept measurements relating to the impact of nanoparticles with an electrode potentiostatted at a value corresponding to the diffusion controlled oxidation of silver nanoparticles in authentic seawater media. The charge associated with the oxidation reveals the number of atoms in the nanoparticle and thus its size and state of aggregation.
Introduction
With the number of commercially available products containing silver nanoparticles continuing to rise and the potentially harmful environmental consequences posed by their inevitable release into natural waters1–5 an accurate method capable of directly detecting these widely used nanoparticles in the environment is required.Recently, we have reported the extensive capabilities of a novel nanoparticle detection technique, anodic particle coulometry (APC), involving the observation of Faradaic charge transfer associated with the quantitative destructive oxidation of nanoparticles colliding with an electrode surface (Fig. 1).6 Not only is it possible to utilise this method for nanoparticle detection, it is also possible to determine the identity and concentration of nanoparticles in solution as well as their size and degree of aggregation.6–8 It has previously been shown that this method demonstrates selectivity in mixed nanoparticle systems as the onset of the current–time transients caused by nanoparticle collisions is potential dependent.7 APC also in principle exhibits portability advantages so would be suitable for use in the field and could potentially be used for dissolution studies involving the release of silver nanoparticles into the environment from commercially available products. In this manuscript, we investigate the possible application of APC to environmental testing of silver nanoparticles present in natural waters, in particular seawater.
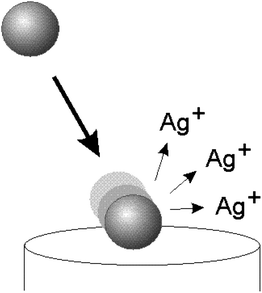 | ||
| Fig. 1 Schematic diagram illustrating the collision of a silver nanoparticle with a potentiostatted electrode. The silver nanoparticle is oxidised upon impact leading to the formation of silver ions. | ||
One of the benefits of APC that is of major importance for environmental monitoring is its ability to determine the persistence of single nanoparticles in aqueous samples via determination of nanoparticle size from the charge transferred during a nanoparticle–electrode impact.8 Indeed, it is well known that silver nanoparticles can have a tendency to aggregate in aqueous solution8 but how does this behaviour change when they are exposed to environmental conditions? With the biocidal nature of silver nanoparticles being particle-size dependent the degree of aggregation has possible implications for their hazardous effect on aquatic ecosystems.9 Hitherto, researchers have combined two or more of the following techniques to detect, size and estimate the degree of silver nanoparticle aggregation in natural waters: dynamic light scattering,5,10–13 scanning electron microscopy,10 atomic force microscopy,11 flow-field flow fractionation,12 UV-vis absorbance measurements,5,11 single particle ICP-MS,14 nanoparticle tracking analysis14 and transmission electron microscopy.5,12,13
Determining the suitability of our APC method to fieldwork testing of this type involved adding a known concentration of synthetic citrate capped silver nanoparticles to seawater samples collected in Saanich Inlet, a fjord located in southern British Columbia, Canada. The results of these proof-of-concept measurements revealed that APC is indeed a suitable method for both the detection and characterisation of silver nanoparticles in natural water samples. This specific method reveals that silver nanoparticles aggregate in seawater.
Experimental
Water column observations and seawater samples from Saanich Inlet, an anoxic fjord in British Columbia, Canada were collected onboard the University of Victoria research vessel M.S.V. John Strickland on June 13, 2012. The temperature, salinity and dissolved oxygen in the inlet were measured using a Sea-Bird 19plus CTD (Conductivity, Temperature and Depth) equipped with a dissolved oxygen sensor (SBE43). Seawater samples were obtained using 5 L Niskin bottles (General Oceanics, Miami, FL, USA) attached to a stainless steel hydrowire that were tripped with Teflon coated messengers. Seawater was collected from the Niskin bottles directly into trace metal clean 250 mL low density polyethylene bottles that were immediately refrigerated and returned to the laboratory for subsequent analyses.NaCl solutions (487 mM and 100 mM, Aldrich) were made using ultrapure water of resistivity ∼18.2 MΩ cm (Millipore). Citrate capped silver nanoparticles were synthesised as described in ref. 15 and were found via SEM imaging to have a mean radius of 14 ± 2 nm. Prior to use, the nanoparticles were ultrasonically dispersed and seawater samples upturned in order to resuspend any settled particulate matter. All experiments were performed inside a faraday cage using a μAutolab II potentiostat and a three electrode configuration: a carbon microelectrode (radius 5.94 μm) or glassy carbon (GC) electrode (diameter 3 mm) was used as the working electrode along with a saturated calomel (SCE) or saturated silver/silver chloride reference electrode and a graphite rod counter electrode. The natural seawater samples and the NaCl solutions were degassed thoroughly with argon (oxygen-free, BOC Gases plc) and an oxygen-free atmosphere was maintained throughout the duration of the experiment. The pH of the seawater sample after purging was found to be 8.28.
The GC electrode was surface modified with silver nanoparticles by depositing 2 μL of the silver nanoparticle solution onto the electrode surface before allowing it to air-dry. OriginPro v.8.5.1 (http://www.OriginLab.com/) was used to analyse the impact spikes and to carry out Gaussian deconvolution of the size distributions.
Results and discussion
The first step was to determine the approximate oxidation potential of silver nanoparticles in seawater. To do this a glassy carbon (GC) electrode (3 mm diameter) that had been surface modified with 1.5 × 10−15 mol silver nanoparticles was scanned anodically using anodic stripping voltammetry in a seawater sample taken at a depth of 205 m. Nanoparticles were deposited on the surface at open current, the electrode was transferred to the cell and the potential scanned linearly from 0 V to +0.4 V (see Fig. 2). The peak potential was +0.03 V (vs. saturated SCE) which was in agreement with previously reported measurements and found to be reproducible to within 5 mV.8 Next, a carbon microelectrode (5.94 μm) was potentiostatted in the same sample at a potential more positive than that required to oxidise the nanoparticles in seawater and no impact spikes were observed. An aliquot of silver nanoparticles was then added so that the concentration of nanoparticles in the cell was 58.9 pM and oxidative impact spikes were observed in the current–time transient, as shown in Fig. 3.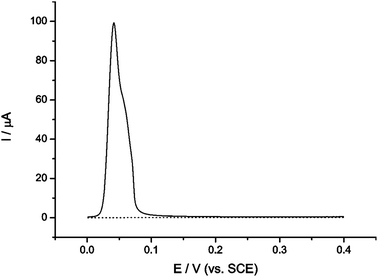 | ||
Fig. 2 Stripping voltammogram of a bare GC electrode (⋯) and a silver nanoparticle modified GC electrode (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) in seawater. ) in seawater. | ||
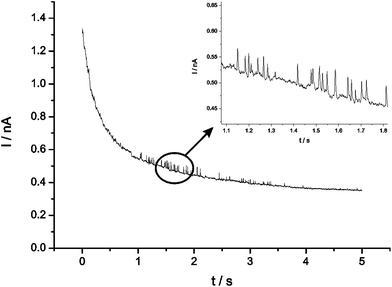 | ||
| Fig. 3 Oxidative current–time transients of citrate capped silver nanoparticles dispersed in seawater at a carbon microelectrode (r = 5.94 μm). | ||
With each spike observed allowing us to detect nanoparticle–electrode collisions, it is also possible to gain quantitative information regarding the number of atoms in each silver nanoparticle (N) and therefore the extent of aggregation by integrating the charge (Q) under each of the current–time transients and using the following equation:6
| Q = eN | (1) |
Fig. 4 shows the size distribution of nanoparticles in terms of the number of atoms making up each nanoparticle inferred from the charge passed during the observed nanoparticle impacts using eqn (1). With the average size of silver nanoparticles having previously been determined via APC to be 13 ± 2 nm in radius,6 it can be estimated that a single silver nanoparticle of this size contains approximately 0.54 × 106 atoms. It is clear therefore that Fig. 4 shows that the nanoparticles are extensively aggregated in solution since most particles show a larger number of atoms. Preliminary deconvolution of Fig. 4 into subdistributions arbitrarily using five Gaussian distributions revealed significant contributions from not only single nanoparticles but also aggregates of 2, 3, 4 and 5 nanoparticles corresponding to 1.08 × 106, 1.62 × 106, 2.16 × 106 and 2.70 × 106 atoms respectively. Therefore, APC distribution data of silver nanoparticles in seawater suggests that over the timescale studied (42 minutes), silver nanoparticles are prone to aggregation. Five size classes were fitted as it is clear that very few impact spikes correspond to colliding nanoparticles beyond the size of five nanoparticle aggregates (2.70 × 106 atoms). The sizes of the nanoparticles and aggregates contributing to the distribution shown were found to be reproducible and further detailed studies will focus on aggregation in depth.
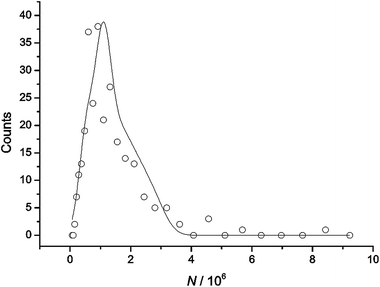 | ||
| Fig. 4 Size distribution of silver nanoparticles in seawater as determined from APC. | ||
It is likely that the behaviour of the silver nanoparticles in seawater is mostly controlled by the major ionic components of seawater which are sodium (469.06 mmol kg−1 at salinity = 35), magnesium (52.82 mmol kg−1), calcium (10.28 mmol kg−1) and potassium (10.21 mmol kg−1) cations and chloride (545.86 mmol kg−1), sulphate (28.24 mmol kg−1) and bicarbonate (1.80 mmol kg−1) anions; sodium and chloride ions are the dominant components.16
From salinity measurements of the seawater at the location and depth where sampling occurred (salinity = 31.3, depth = 205 m), the chloride concentration was determined to be equivalent to 487 mM.16 Therefore, the next step was to perform the nanoparticle collision experiment under exactly the same conditions reported above but this time replacing the seawater sample with 487 mM NaCl. Impact spikes were recorded over a period of 47 minutes.
As before, the size distribution of impacting nanoparticles was plotted (Fig. 5a) and contributions from the same five nanoparticle sizes observed in the seawater sample using arbitrary Gaussian deconvolution also appeared to be prevalent in 487 mM NaCl. With aggregation occurring at a similar extent in this salt concentration we can conclude that the ionic strength of seawater is responsible for the observed silver nanoparticle aggregation in the seawater sample tested.
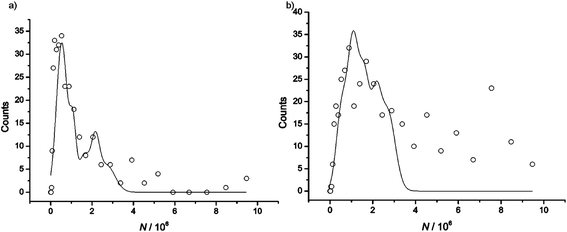 | ||
| Fig. 5 APC distribution of silver nanoparticles in (a) 487 mM NaCl and (b) 100 mM NaCl. | ||
Interestingly, when the same collision experiment was repeated in a lower salt concentration (100 mM NaCl) over a timescale of 42 minutes and a similar APC distribution was plotted (Fig. 5b), it was apparent that the nanoparticles were aggregating at a faster rate at this lower ionic strength. It can be seen that this size distribution can be preliminarily deconvoluted into the same arbitrary sub-distributions for aggregates composed of up to five nanoparticles as before but it is clear that beyond nanoparticle cluster sizes of 2.70 × 106 atoms larger sized aggregates were also formed in solution which were not present at similar timescales in solutions of higher ionic strength i.e. in the seawater and pure 487 mM NaCl samples.
Note that moderately high concentrations of electrolyte are generally thought to encourage aggregation of silver nanoparticles.4,5,17 This is in agreement with the Derjaguin, Landau, Verwey and Overbeek (DLVO) theory for particle interactions which states that an increase in ionic strength will result in the promotion of nanoparticle aggregation as the repulsion between like charged particles will be screened by ions in solution. However, a number of flaws have been identified in traditional DLVO theory, of most relevance here is the disregard for dispersion interactions.18 Specifically Boström et al. have demonstrated the importance of dispersion forces in colloidal systems, noting a significant change in the force between charged interfaces when dispersion forces of counter ions were considered at salt concentrations of 100 mM and an even greater effect was observed when the concentration was increased to 300 mM.18 As a result it has been suggested that classical DLVO theory is inadequate for systems where the salt concentration is 100 mM or higher.18
The slower rate of aggregation of silver nanoparticles observed in the higher ionic strength solutions tested might be explained by the significant ionic dispersion forces that will be dominant at these salt concentrations.
Conclusion
These proof-of-concept measurements reveal the potential possessed by APC for both the detection and characterisation of silver nanoparticles in the environment. This result represents a breakthrough which will be followed by oceanographic applications.Acknowledgements
The authors thank David Janssen for preparation of trace metal clean bottles before sampling and the Leverhulme Trust for financial support.References
- T. M. Benn and P. Westerhoff, Environ. Sci. Technol., 2008, 42, 4133 CrossRef CAS.
- A. Hinther, S. Vawda, R. C. Skirrow, N. Beldhoen, P. Collins, J. T. Cullen, G. van Aggelen and C. C. Helbing, Environ. Sci. Technol., 2010, 44, 8314 CrossRef CAS.
- P. V. Asharani, Y. L. Wu, Z. Gonf and S. Valiyaveettil, Nanotechnology, 2008, 19, 255102 CrossRef CAS.
- L. V. Stebounova, E. Guio and V. H. Grassian, J. Nanopart. Res., 2011, 13, 233 CrossRef CAS.
- A. M. Badaway, T. P. Luxton, R. G. Silver, K. G. Scheckel, M. T. Suidan and T. M. Tolaymate, Environ. Sci. Technol., 2010, 44, 1260 CrossRef.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Angew. Chem., Int. Ed., 2011, 50, 4219 CrossRef CAS.
- E. J. E. Stuart, Y.-G. Zhou, N. V. Rees and R. G. Compton, RSC Adv., 2012, 2, 6879 RSC.
- N. V. Rees, Y.-G. Zhou and R. G. Compton, ChemPhysChem, 2011, 12, 1645 CrossRef CAS.
- J. M. Zook, R. I. MacCuspie, L. E. Locascio, M. D. Halter and J. T. Elliott, Nanotoxicology, 2011, 5(4), 517 CrossRef CAS.
- B. Thio, M. O. Montes, M. A. Mahmoud, D. Lee, D. Zhou and A. A. Keller, Environ. Sci. Technol., 2012, 46, 6985 CrossRef CAS.
- S. L. Chinnapongse, R. I. MacCuspie and V. A. Hackley, Sci. Total Environ., 2011, 409, 2443 CrossRef CAS.
- S. A. Cumberland and J. R. Lead, J. Chromatogr., A, 2009, 1216, 9099 CrossRef CAS.
- A. M. Badaway, K. G. Scheckel, M. Suidan and T. Tolaymat, Sci. Total Environ., 2011, 429, 325 CrossRef.
- J. Farkas, H. Peter, P. Christian, J. A. G. Urrea, M. Hassellöv, J. Tuoriniemi, S. Gustafsson, E. Olsson, K. Hylland and K. V. Thomas, Environ. Int., 2011, 37, 1057 CrossRef CAS.
- A. Pyatenko, M. Yamaguchi and M. Suzuki, J. Phys. Chem. C, 2007, 111, 7910 CAS.
- S. R. Emerson and J. I. Hedges, Chemical Oceanography and the Marine Carbon Cycle, Cambridge University Press, United Kingdom, 4th edn, 2011 Search PubMed.
- M. G. Espinoza, M. L. Hinks, A. M. Mendoza, D. P. Pullman and K. I. Peterson, J. Phys. Chem. C, 2012, 116, 8305 CAS.
- M. Boström, D. R. M. Williams and B. W. Ninham, Phys. Rev. Lett., 2001, 87, 168103 CrossRef.
| This journal is © The Royal Society of Chemistry 2013 |