Hyaluronic acid modified mesoporous silica nanoparticles for targeted drug delivery to CD44-overexpressing cancer cells
Meihua Yua, Siddharth Jambhrunkara, Peter Thornb, Jiezhong Chenc, Wenyi Gu*a and Chengzhong Yu*a
aARC Centre of Excellence for Functional Nanomaterials and Australian Institute for Bioengineering and Nanotechnology, The University of Queensland, Brisbane, QLD 4072, Australia. E-mail: c.yu@uq.edu.au; w.gu@uq.edu.au; Fax: +61-7-334 63973; Tel: +61-7-334 63283
bSchool of Biomedical Sciences, The University of Queensland, Brisbane, QLD 4072, Australia
cIllawarra Health and Medical Research Institute, University of Wollongong, NSW 2522, Australia
First published on 26th September 2012
Abstract
In this paper, a targeted drug delivery system has been developed based on hyaluronic acid (HA) modified mesoporous silica nanoparticles (MSNs). HA-MSNs possess a specific affinity to CD44 over-expressed on the surface of a specific cancer cell line, HCT-116 (human colon cancer cells). The cellular uptake performance of fluorescently labelled MSNs with and without HA modification has been evaluated by confocal microscopy and fluorescence-activated cell sorter (FACS) analysis. Compared to bare MSNs, HA-MSNs exhibit a higher cellular uptake via HA receptor mediated endocytosis. An anticancer drug, doxorubicin hydrochloride (Dox), has been loaded into MSNs and HA-MSNs as drug delivery vehicles. Dox loaded HA-MSNs show greater cytotoxicity to HCT-116 cells than free Dox and Dox-MSNs due to the enhanced cell internalization behavior of HA-MSNs. It is expected that HA-MSNs have a great potential in targeted delivery of anticancer drugs to CD44 over-expressing tumors.
1. Introduction
In recent decades, the application of nanotechnology in medicine (so called nanomedicine) has attracted much attention.1 Various nanomaterials have been widely used in nanomedicine as potential diagnostic and therapeutic agents for cancer imaging and treatment.2 Compared to traditional organic lipids or polymer-based nanoparticles,3–5 the inorganic counterparts exhibit unique properties such as inertness, stability, and ease of functionalisation.2 Specifically, mesoporous silica nanoparticles (MSNs) with high surface areas, tunable pore structures as well as particle sizes, and controllable surface chemistry have attracted enormous research interest in various bio-applications, including cell imaging,6 diagnosis and bioanalysis,7 and drug/gene delivery.8,9For efficient cancer treatments, it has been well documented that a targeted delivery is vital10 because most anticancer drugs distribute throughout the body and can be harmful to healthy cells. To minimize the side effects, it would be desirable to specifically increase the anticancer drug concentration at the target sites. Nanoparticles with small sizes (1–200 nm) preferably accumulate at tumor sites caused by the enhanced permeability and retention (EPR) effect,11 also called passive targeting. With the advantage of controllable sizes with diameters within 100 nm, MSNs as nanocarriers have the potential to improve cancer treatments. Moreover, there have been considerable efforts devoted to developing MSNs modified with active targeting moieties, including specific ligands (such as folic acid,12–14 mannose15 and arginine–glycine–aspartate (RGD)16,17), peptides,18 and antibodies.19,20 The abundant silanol groups (Si–OH) facilitate the modification of the above active moieties, thereby achieving active targeting of MSNs to specific cancer cells. The active targeting action together with the EPR effect will further enhance the cellular uptake of MSNs in defective cells, leading to a significant improvement in cancer therapy.
Hyaluronic acid (HA) is a biodegradable, biocompatible and non-immunogenic glycosaminoglycan. As a major component of the extracellular matrix, HA is essential for proper cell growth, organ structural stability and tissue organization,21,22 and has been extensively investigated for biomedical and pharmaceutical applications. In particular, researchers focused on the use of HA as a targeting moiety for cancer therapy, because many types of tumor cells over-express HA receptors like CD44.10 Since HA has multiple functional groups available for chemical conjugation with anticancer drugs10,23 or nanocarriers of drugs/genes,24,25 HA-attached drugs/nanocarriers have been developed to increase drug/cargo accumulation specifically in CD44 over-expressing cancer cells. In addition to the targeting function, HA modified delivery systems can enter cells more efficiently via the HA receptor medicated endocytosis pathway. Utilizing the CD44–HA specific affinity is therefore an attractive strategy for cancer targeting treatments. However, to the best of our knowledge, there are few reports on HA conjugated MSNs as the delivery system for targeting CD44 over-expressing cancer cells. Very recently, a HA-MSN conjugate was reported by Ma and co-workers.26 After loading with the hydrophobic drug camptothecin (CPT), HA-MSNs showed enhanced cytotoxicity to HeLa cells. However, the improvement in cell toxicity was not significant compared to free CPT. Although it was claimed in this report that CD44 positive HeLa cells were used, there is no evidence or literature to support that the HeLa cells used in this work were CD-44 positive. Moreover, because HA coated at the outer surface of MSNs is very hydrophilic, the nature of the drug may also have an influence on the cytotoxicity. Therefore, it is hypothesized that in order to show the advantage of HA-MSN delivery systems, both the target cell lines and suitable drug molecules should be carefully designed in the study.
In the present work, MSNs with a mean particle size of 70–100 nm have been synthesized as drug nanocarriers, and their surface is further modified with HA as the targeting ligand. The specificity of HA-MSNs to CD44 over-expressing HCT-116 cells has been studied. HA modified MSNs show a higher endocytosis performance, compared to bare MSNs. Moreover, an anticancer drug doxorubicin (Dox) has been encapsulated into the MSNs with or without HA modification. Dox loaded HA-MSNs show a much more potent effect in HCT-116 cells than free Dox or Dox-MSNs. Our results have demonstrated that MSNs conjugated with HA are effective as a targeted drug delivery system and have a great potential in treating CD44 over-expressing tumors with improved efficiency.
2. Experimental section
2.1. Chemicals
Tetraethylorthosilicate (TEOS), cetyltrimethylammonium bromide (CTAB), (3-aminopropyl)triethoxysilane (APTES), fluorescein-5-isothiocyanate (FITC), N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC), fluoroshield with DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride), triethylamine (TEA), paraformaldehyde (PFA), and doxorubicin hydrochloride (Dox) were purchased from Sigma-Aldrich. Sodium hyaluronate (HA) (Mw = 200 kDa) was purchased from Lifecore Biomedical (Chaska, MN). Reagent grade sodium hydroxide (NaOH) was received from ChemSupply. CellTiter-Blue® Reagent and human CD44 FITC conjugate were ordered from Promega and Invitrogen, respectively.2.2. Synthesis of MSNs
MSNs were synthesized according to an approach reported in our previous work27 with slight modifications. In a typical synthesis, 1.0 g CTAB was dissolved in 480 g deionized water under stirring at room temperature followed by the addition of 3.5 mL NaOH (2 M). The temperature of the solution was raised and kept at 80 °C. To this solution, 6.7 mL TEOS was added. The mixture was continuously stirred for an additional 2 h. The resultant products were collected by filtration and dried at room temperature. The templates were removed by calcination at 550 °C for 5 h.2.3. Synthesis of HA conjugated MSNs (HA-MSNs)
First, 1.5 g calcined MSNs was added to 60 mL toluene and stirred for 6 h before adding 1.0 mL APTES. After stirring at room temperature for 24 h, the particles were extensively washed with toluene and dried in a fume-hood at room temperature (denoted NH2-MSNs). Next, in order to graft HA, the carboxyl group of HA was activated with NHS, using EDC as a coupling agent. The activated carboxyl group of HA then combined with the amine end of the NH2-MSNs. Specifically, 1 g of the prepared NH2-MSNs powder was dispersed in 100 mL deionized water. In another reaction vessel, 20 mL of an aqueous solution containing NHS (0.37 g) and EDC (0.2 g) was mixed with 60 mL HA (113 mg) deionized water solution. Finally the two solutions were mixed and the pH was adjusted to 9.0 using triethylamine as a catalyst. The mixture was stirred at 38 °C overnight. The HA modified MSNs (HA-MSNs) were obtained by centrifugation, washing three times with deionized water, and freeze-drying using Christ Alpha 2-4 LDplus (John Morris Scientific).2.4. FITC modification of MSNs and HA-MSNs
Not all amino groups in NH2-MSNs reacted with HA, thus the remaining free NH2 moieties were utilized for labelling with FITC. The functional group of FITC, thiocyanate, is highly aminoreactive, therefore the prepared NH2-MSNs and HA-MSNs can be conjugated with FITC. Specifically, 20 mg powdered HA-MSNs or NH2-MSNs were dissolved in 3 mL deionized water, and mixed with 5 mL FITC ethanol solution (0.3 mg mL−1). After stirring in the dark for 6 h, the nanoparticles were centrifuged and washed with ethanol three times until the supernatants were colorless. The FITC labelled nanoparticles were used for confocal microscopy observations and flow cytometry analysis after applying in HCT-116 cell uptake experiments.2.5. Characterisation
Transmission electron microscopy (TEM) images were obtained with a JEOL 1010 operated at 100 kV. For TEM measurements, the samples were prepared by dispersing the powder samples in ethanol, after which they were dispersed and dried on carbon film on a Cu grid. An X-ray diffraction (XRD) pattern was recorded on a Rigaku Miniflex X-ray diffractometer with Fe-filtered Co radiation. Nitrogen adsorption–desorption isotherms were measured at 77 K using a Micromeritics ASAP Tristar II 3020 system. The samples were degassed at 473 K overnight on a vacuum line. The pore size distribution curve was derived from the adsorption branch of the isotherm using the Barrett–Joyner–Halanda (BJH) method. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas. The total pore volume was calculated from the amount adsorbed at a maximum relative pressure (P/P0) of 0.99. Zeta potential measurements were carried out on a Malvern NanoZS zetasizer at 25 °C in phosphate buffered saline (PBS) solution. Fourier transform infrared (FTIR) spectra were collected on a ThermoNicolet Nexus 6700 FTIR spectrometer equipped with a Diamond ATR (attenuated total reflection) Crystal. For each spectrum, 32 scans were collected at a resolution of 4 cm−1 over the range 400–4000 cm−1. Before FTIR measurements, all the samples were freeze dried.13C NMR spectra were measured by a solid state Bruker Avance III spectrometer with a 7T (300 MHz for 1 h) magnet and a zirconia rotor, 4 mm, rotated at 7 kHz.2.6. Loading doxorubicin (Dox)
60 μL PBS solution containing 150 μg MSNs or HA-MSNs was mixed with 60 μL Dox–PBS solution (1 mg mL−1). After shaking for 10 minutes, the mixtures were kept in a 4 °C fridge overnight under dark conditions. Then the Dox-HA-MSNs or Dox-MSNs were centrifuged and washed with 120 μL PBS. The Dox-HA-MSNs and Dox-MSNs samples were used for a subsequent cytotoxicity assay against HCT-116 cells. To evaluate the Dox-loading efficiency, the supernatant and washed solutions were collected and the residual Dox content was measured by using a UV-2450 (UV-Vis spectrophotometer, Shimadzu) at a wavelength of 233 nm. The loading amount of Dox can be calculated based on the original and residual Dox concentrations and volumes. Finally, the amount of loaded Dox was 1.81 and 40.25 μg for HA-MSNs and MSNs respectively. The weight percentage of loaded Dox to HA-MSNs and MSNs is 1.2% and 26.8% respectively.2.7. Cell culture and uptake
HCT-116 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented with fetal calf serum (10%, Sigma, MO), L-glutamine (2%), penicillin (1%) and streptomycin (1%) in 5% CO2 at 37 °C. The medium was routinely changed every 2 days and the cells were separated by trypsinisation before reaching confluency. HCT-116 cells were seeded in a 6-well plate (5 × 105 cells per well) and incubated for 24 h. After washing twice with PBS, the cells were incubated with 25 μg mL−1 FITC labeled MSNs or HA-MSNs in 2 mL of the serum-free DMEM medium for 4 h. Subsequently, the cells were washed twice with PBS to remove the remaining nanoparticles and dead cells. For fixed cell imaging, the cells were treated with 500 μL 4% PFA solution for 30 min at 4 °C, and their nuclei were stained with DAPI for 10 min. Finally, the cells were viewed under a confocal microscope (LSM Zeiss 710).For competitive inhibition studies, the medium was replaced with 2 mL serum-free culture medium containing HA polymer (10 mg mL−1) and then FITC labeled HA-MSNs (25 μg mL−1) in HCT-116 cells, followed by the same treatment as described above.
2.8. Fluorescence-activated cell sorter (FACS) analysis
A quantitative determination of the cellular uptake of nanoparticles by FACS analysis was performed as follows. HCT-116 cells were seeded in a 6-well plate (5 × 105 cells per well) and were cultured as described above. After removal of the free FITC–MSNs or FITC–HA-MSNs and dead cells, the cells were trypsinized, and then centrifuged and washed twice with PBS. After that, the cells were suspended in 1 mL 2% PFA–PBS solution. Then the intracellular delivery of the nanoparticles was analyzed using a FACSAria Cell Sorter (Becton Dickinson BD). All the data of the mean fluorescence were obtained from a population of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells after the gating of single cells. Cells incubated in the absence of particles were used as the control.
000 cells after the gating of single cells. Cells incubated in the absence of particles were used as the control.2.9. Measurements of CD44 receptor levels in cultured cells
5 × 105 HCT-116 cells were suspended in 2 mL 2% bovine serum albumin (BSA) in PBS solution and incubated at 37 °C for 30 min. After centrifugation, the cells were suspended in 0.5 mL 2% BSA solution, followed by the addition of 2.5 μL FITC labeled CD44-antibody. After incubation at 37 °C for 45 min, the cells were washed twice with 2% BSA solution, then resuspended in 1 mL 2% PFA solution and incubated for 30 min at 4 °C. The binding efficiency of the CD-44 antibody was tested by FACS analysis.2.10. Cytotoxicity of Dox-HA-MSNs and Dox-MSNs against HCT-116 cells
HCT-116 cells were seeded in a 96-well plate at a density of 2 × 104 cells per well and cultured in 5% CO2 at 37 °C for 24 h. Then, free Dox, Dox-HA-MSNs and Dox-MSNs were added to the cells in DMEM medium at different Dox concentrations of 0.25, 0.125, 0.05 μg mL−1 respectively, and the cells were further incubated in 5% CO2 at 37 °C for 24 h. Subsequently, 15 μL CellTiter-Blue Reagent was added to each well, shaken for 10 seconds and then incubated at 37 °C for 4 h. Then fluorescence readings were measured with an excitation wavelength of 560 nm and an emission wavelength of 590 nm using a microplate reader (SpectraMax M5, Bio-Strategy, Ltd). Cells incubated in the absence of particles were used as the control. All the experiments were performed in triplicate for each group. The statistical significance between the two groups was analyzed by an unpaired Student's t-test using the software in GraphPad Prism. The differences were considered to be significant if p < 0.05.3. Results and discussion
3.1. Synthesis of MSNs and HA-MSNs
The XRD pattern (Fig. 1a) of the MSNs shows three typical well-resolved diffraction peaks that can be assigned to the 10, 11 and 20 reflections of a highly ordered two dimensional (2D) hexagonal mesostructure, similar to previous reports.27,28 The nitrogen adsorption–desorption plot of the MSNs (Fig. 1b) exhibits a typical type IV isotherm and a steep capillary condensation step occurring at a relative pressure (P/P0) of ∼0.3, corresponding to a narrow pore size distribution centred at 2.13 nm (inset of Fig. 1b). The surface area and pore volume are calculated to be 918.5 m2 g−1 and 0.67 cm3 g−1 respectively. From the typical TEM image shown in Fig. 1c, it can be seen that the MSNs are nanoparticles with well-ordered mesopores. The mean particle sizes are estimated to be 70–100 nm. Fig. 1d shows the representative TEM image of HA-MSNs. A vague thin layer as indicated by a white arrow can be seen on the surface of the HA-MSNs, indicating the existence of the HA polymer grafted on the MSNs.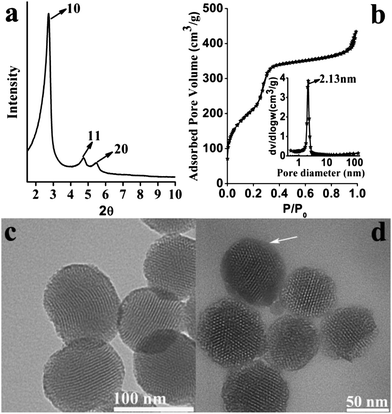 | ||
| Fig. 1 (a) XRD pattern; (b) N2 adsorption–desorption isotherm and the corresponding pore size distribution (inset of (b)) of calcined MSNs; TEM images of (c) MSNs and (d) HA-MSNs. | ||
The surface modification of MSNs has been studied by zeta potential, FTIR and 13C NMR analysis (Fig. 2a–c). From Fig. 2a, it can be seen that the zeta potential of MSNs, NH2-MSNs and HA-MSNs is −23.3, +11.3, and −18.8 mV respectively, consistent with the modification of amine groups (NH2-MSNs) on the negatively charged silica surface (MSNs) and subsequent conjugation of HA on the positively charged NH2-MSNs. Because the carboxyl groups of HA are ionized at pH ∼7.0 (ref. 29), HA-MSNs are negatively charged. As indicated in Fig. 2b, pure HA exhibits a broad O–H stretching bond at 3200–3650 cm−1, a shoulder peak at around 2900 cm−1 associated with –CH2, a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bond at 1100–1300 cm−1, and a CO–NH (amide) bond at 1630–1680 cm−1 (ref. 30). HA-MSNs exhibit typical bonds at 2920 cm−1 (C–H stretching) and 3380 cm−1 (O–H) from HA. The characteristic peaks at 810 and 1060 cm−1 for HA-MSNs can be indexed to Si–O–Si stretching.31 In addition, a shoulder peak at 960 cm−1 can also be observed in HA-MSNs, which can be attributed to the stretching mode of Si–O in the Si–OH group. The band at 1630 cm−1 with a very low intensity is observed in HA-MSNs and MSNs, which is assigned to the bending mode of physisorbed water.31 The 13C NMR solid-state spectra of NH2-MSNs and HA-MSNs are presented in Fig. 2c. Three sharp peaks observed at about 43, 22 and 10 ppm in the spectrum of NH2-MSNs can be assigned to three types of methylene carbons from the amino-silica source (see the inset chemical structure in Fig. 2c). The above three peaks are also observed in the spectrum of HA-MSNs. The extra peaks in the range 70–180 ppm can be attributed to C1 anomeric carbons, C2–C5 carbons and the carboxylate and/or carbonyl acetamide carbons of the HA polymer (103, 75, and 175 ppm, respectively, see the inset structure of HA and indexing).32,33 The results have shown that HA has been successfully conjugated on HA-MSNs.
O stretching bond at 1100–1300 cm−1, and a CO–NH (amide) bond at 1630–1680 cm−1 (ref. 30). HA-MSNs exhibit typical bonds at 2920 cm−1 (C–H stretching) and 3380 cm−1 (O–H) from HA. The characteristic peaks at 810 and 1060 cm−1 for HA-MSNs can be indexed to Si–O–Si stretching.31 In addition, a shoulder peak at 960 cm−1 can also be observed in HA-MSNs, which can be attributed to the stretching mode of Si–O in the Si–OH group. The band at 1630 cm−1 with a very low intensity is observed in HA-MSNs and MSNs, which is assigned to the bending mode of physisorbed water.31 The 13C NMR solid-state spectra of NH2-MSNs and HA-MSNs are presented in Fig. 2c. Three sharp peaks observed at about 43, 22 and 10 ppm in the spectrum of NH2-MSNs can be assigned to three types of methylene carbons from the amino-silica source (see the inset chemical structure in Fig. 2c). The above three peaks are also observed in the spectrum of HA-MSNs. The extra peaks in the range 70–180 ppm can be attributed to C1 anomeric carbons, C2–C5 carbons and the carboxylate and/or carbonyl acetamide carbons of the HA polymer (103, 75, and 175 ppm, respectively, see the inset structure of HA and indexing).32,33 The results have shown that HA has been successfully conjugated on HA-MSNs.
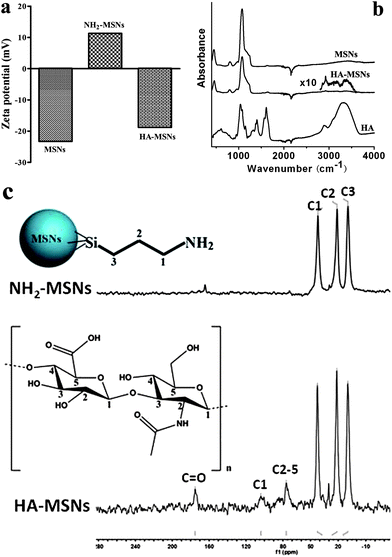 | ||
| Fig. 2 (a) Zeta potential analysis of MSNs, NH2-MSNs and HA-MSNs in PBS solution; (b) FTIR spectra of freeze dried MSNs, HA-MSNs and HA polymer; (c) 13C NMR spectra of NH2-MSNs and HA-MSNs. | ||
3.2. Cellular uptake of MSNs and HA-MSNs
To visualize the cellular uptake of silica nanoparticles, both MSNs and HA-MSNs were labelled with FITC and studied by confocal microscopy. As shown in Fig. 3, when no nanoparticles are used to treat the cells (control group), only the nuclei are observed in blue color after staining with DAPI. When the cells are incubated with FITC labelled HA-MSNs, strong green fluorescent signals originating from FITC are detected inside the cells, indicating that HA-MSNs are readily taken up by HCT-116 cancer cells (Fig. 3). In contrast, when FITC labelled MSNs without HA modification are incubated with HCT-116, a weaker signal is observed, suggesting a decreased amount of MSNs internalized by HCT-116 cells. Furthermore, when HCT-116 cells are pretreated with free HA (10 mg mL−1) to block CD44 prior to FITC labelled HA-MSNs treatment, the green intracellular fluorescence is also weaker compared to the HA-MSN group.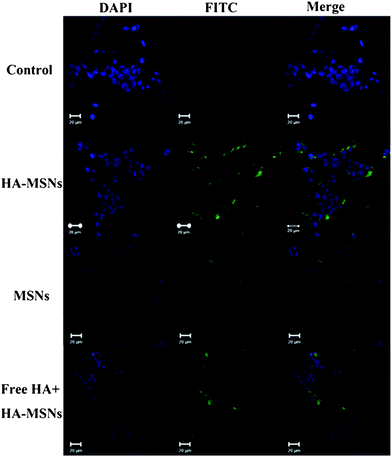 | ||
| Fig. 3 Confocal microscopy images of HCT-116 cells without any treatment as a control (first row), with the treatment FITC labelled HA-MSNs (second row), MSNs (third row) and free HA (10 mg mL−1) together with FITC labelled HA-MSNs (last row). Green fluorescence arises from FITC dyes that are conjugated to silica nanoparticles and nuclei are stained with DAPI, showing blue fluorescence. | ||
To provide a quantitative comparison, the cellular uptake performance of MSNs and HA-MSNs in HCT-116 is further studied by FACS analysis. Before studying the specific affinity of HA-MSNs, the CD44 receptor level of HCT-116 cancer cells has been evaluated using the human FITC–CD44 antibody and FACS analysis. As can be seen from Fig. 4a, compared to the control cells without any treatment, the cells incubated with the FITC–CD44-antibody show a much higher intensity of FITC, indicating the existence of a high level of CD44 on the surface of HCT-116 cells, consistent with a previous description.24
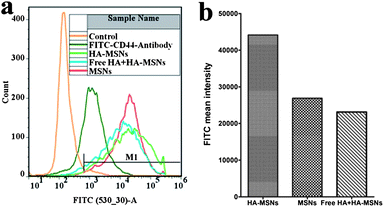 | ||
| Fig. 4 (a) Fluorescence activated cell sorter (FACS) analysis of the FITC intensity of the cells treated with an FITC labeled CD44 antibody, MSNs, and HA-MSNs in the absence or in the presence of free HA (10 mg mL−1); (b) bar chart of FITC mean intensity of the gated positive cells (M1 indicated in Fig. 4a) treated with FITC modified silica particles. | ||
After treating HCT-116 cells with FITC labelled HA-MSNs, the FITC signal emitted from the cells is very strong (Fig. 4b), indicating a high cellular uptake of HA-MSNs. In contrast, the mean intensity of FITC from the cells incubated with FITC labelled MSNs without HA modification is only 61% of that of the FITC labelled HA-MSNs group. To further confirm the specific interaction of HA-MSNs with HCT-16 cells, free HA (10 mg mL−1) is added prior to the addition of FITC labelled HA-MSNs in the cell culture medium. The FITC signal intensity decreases by 48% compared to the FITC labelled HA-MSNs group, suggesting that the interaction between CD44 and HA-MSNs and the subsequent HA receptor-mediated endocytosis has been weakened due to the competition of free HA. The above results have confirmed that HA-MSNs can target CD44 over-expressing HCT-16 cancer cells via the HA receptor-mediated endocytosis pathway and show an improved endocytosis performance compared to the unmodified MSNs. The FACS results provide a quantitative comparison, which is consistent with the confocal microscopy observations (Fig. 3).
3.3. In vitro cytotoxicity of Dox-loaded HA-MSNs and MSNs against HCT-116 cells
Finally, the anti-cancer effects of free Dox, Dox loaded HA-MSNs and MSNs were investigated. HCT-116 cells were treated with free Dox, Dox-HA-MSNs or Dox-MSNs with the same concentration of Dox (see details in Table 1). It should be noted that the Dox loading capacity of the MSNs (26.8%) is much higher than that of the HA-MSNs (1.2%). This is because Dox is a water-soluble molecule with a pKa of 8.2 and thus has a positive charge in PBS solution.9 Consequently, the negatively charged HA on the surface of the HA-MSNs may inhibit the penetration of Dox into the pore channels of HA-MSNs and dramatically decrease the Dox loading percentage.| Dox-HA-MSNs | Dox-MSNs | HA-MSNs | MSNs | Free Dox | |
|---|---|---|---|---|---|
| C1 (μg mL−1) | 20.95 | 1.18 | 20.70 | 0.93 | 0.25 |
| C2 (μg mL−1) | 10.48 | 0.59 | 10.35 | 0.47 | 0.125 |
| C3 (μg mL−1) | 4.19 | 0.24 | 4.14 | 0.19 | 0.05 |
The results in Fig. 5 reveal that both HA-MSNs and MSNs show almost no toxicity to HCT-116 at three concentrations (C1–C3, 20.70, 10.35, 4.14 μg mL−1 in HA-MSNs and 0.93, 0.47, 0.19 μg mL−1 in MSNs respectively), indicating both MSNs and HA-MSNs are excellent biocompatible nano-carriers. HCT-116 cells treated with free Dox, Dox-HA-MSNs and Dox-MSNs show a Dox dosage dependent behavior. However, both free Dox and Dox-MSNs have very low cytotoxicity (∼20% inhibition) to the cells even at the highest concentration (C1). There is no significant difference (p = 0.5249) in cell viability between the two groups. In contrast, the cell proliferation is significantly inhibited when treated with Dox-HA-MSNs (51% inhibition). The antiproliferative action between the two groups (Dox-HA-MSNs and free Dox) shows a significant difference with a p value of 0.0151, as indicated in Fig. 5. This enhanced cytotoxicity can be explained by the enhanced cellular uptake of HA-MSNs via HA receptor-mediated endocytosis.
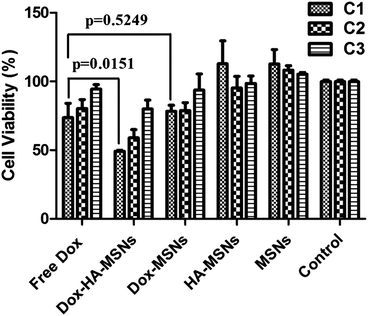 | ||
| Fig. 5 Cytotoxicity of free Dox, Dox-HA-MSNs, Dox-MSNs, HA-MSNs and MSNs against HCT-116 cells at different concentrations C1, C2 and C3 (for details, see Table 1). | ||
As introduced before, when the hydrophobic drug CPT was loaded in HA-MSNs,26 HA-MSNs (∼55% inhibition) showed a slight enhancement in cytotoxicity to CD44 positive cells, compared to free CPT (∼43%). It is possible that hydrophobic anticancer drugs are difficult to release from the mesopores due to the hydrophilic HA polymer layer coated at the outer surface of the MSNs. Thus, HA-MSNs are more feasible for the target delivery of hydrophilic anticancer drugs or some hydrophobic agents, like photosensitizers,34 where drug release is not required. Nevertheless, new approaches should be found to increase the loading amount of such drugs inside HA-MSNs, which can further decrease the dosage needed for efficient cancer treatment.
4. Conclusions
In summary, an anticancer drug delivery system based on HA conjugated MSNs has been developed to specifically target CD44 over-expressing cancer cells. In this study we have demonstrated that HA modified MSNs show enhanced cellular uptake performance via HA receptor mediated endocytosis in CD44 positive HCT-116 cells, compared to that of MSNs without HA modification. Furthermore, HA-MSNs loaded with the hydrophilic anticancer drug Dox has a much better antiproliferative action on HCT-116 cells than free Dox or Dox encapsulated in bare MSNs. This targeted approach based on HA conjugated silica nanoparticles to target CD44 over-expressing cancer cells could have a great potential to improve cancer treatments.Acknowledgements
We thank the Australian Research Council for support, the facilities, and the scientific and technical assistance, of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland, Dr Ekaterina Strounina for the 13C NMR technical help from Centre for Advanced Imaging, the University of Queensland.Notes and references
- B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, N. Engl. J. Med., 2010, 363, 2434–2443 CrossRef CAS.
- H.-C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344–357 CrossRef CAS.
- T. Wang, G. G. M. D'Souza, D. Bedi, O. A. Fagbohun, L. P. Potturi, B. Papahadjopoulos-Sternberg, V. A. Petrenko and V. P. Torchilin, Nanomedicine, 2010, 5, 563–574 CrossRef CAS.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145–160 CrossRef CAS.
- W. C. Hartner, D. D. Verma, T. S. Levchenko, E. A. Bernstein and V. P. Torchilin, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2009, 1, 530–539 CrossRef CAS.
- J. K. Hsiao, C. P. Tsai, T. H. Chung, Y. Hung, M. Yao, H. M. Liu, C. Y. Mou, C. S. Yang, Y. C. Chen and D. M. Huang, Small, 2008, 4, 1445–1452 CrossRef CAS.
- R. J. Tian, H. Zhang, M. L. Ye, X. G. Jiang, L. H. Hu, X. Li, X. H. Bao and H. F. Zou, Angew. Chem., Int. Ed., 2007, 46, 962–965 CrossRef CAS.
- J. Lu, M. Liong, J. I. Zink and F. Tamanoi, Small, 2007, 3, 1341–1346 CrossRef CAS.
- H. A. Meng, M. Liong, T. A. Xia, Z. X. Li, Z. X. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS.
- V. M. Platt and F. C. Szoka, Mol. Pharmaceutics, 2008, 5, 474–486 CrossRef CAS.
- A. S. Hoffman, J. Controlled Release, 2008, 132, 153–163 CrossRef CAS.
- J. Lu, M. Liong, Z. X. Li, J. I. Zink and F. Tamanoi, Small, 2010, 6, 1794–1805 CrossRef CAS.
- Y. F. Zhu, Y. Fang and S. Kaskel, J. Phys. Chem. C, 2010, 114, 16382–16388 CAS.
- L. Pasqua, F. Testa, R. Aiello, S. Cundari and J. B. Nagy, Microporous Mesoporous Mater., 2007, 103, 166–173 CrossRef CAS.
- D. Brevet, M. Gary-Bobo, L. Raehm, S. Richeter, O. Hocine, K. Amro, B. Loock, P. Couleaud, C. Frochot, A. Morere, P. Maillard, M. Garcia and J. O. Durand, Chem. Commun., 2009, 1475–1477 RSC.
- I. J. Fang, I. I. Slowing, C. W. Wu and V. S. Y. Lin, presented in part at The 238th ACS National Meeting, Washington, DC, August 17, 2009 Search PubMed.
- I. J. Fang, I. I. Slowing, K. C. W. Wu, V. S. Y. Lin and B. G. Trewyn, Chem.–Eur. J., 2012, 18, 7787–7792 CrossRef CAS.
- S. H. Cheng, C. H. Lee, M. C. Chen, J. S. Souris, F. G. Tseng, C. S. Yang, C. Y. Mou, C. T. Chen and L. W. Lo, J. Mater. Chem., 2010, 20, 6149–6157 RSC.
- I. I. Slowing, P. A. Kapke, S. Goodison and V. S. Y. Lin, presented in part at The 235th ACS National Meeting, New Orleans, LA, April 8, 2008 Search PubMed.
- C. P. Tsai, C. Y. Chen, Y. Hung, F. H. Chang and C. Y. Mou, J. Mater. Chem., 2009, 19, 5737–5743 RSC.
- Q. Hua, C. B. Knudson and W. Knudson, J. Cell Sci., 1993, 106, 365–375 CAS.
- C. B. Knudson and W. Knudson, FASEB J., 1993, 7, 1233–1241 CAS.
- I. Rivkin, K. Cohen, J. Koffler, D. Melikhov, D. Peer and R. Margalit, Biomaterials, 2010, 31, 7106–7114 CrossRef CAS.
- H. Lee, C. H. Ahn and T. G. Park, Macromol. Biosci., 2009, 9, 336–342 CrossRef CAS.
- M. Y. Lee, S. J. Park, K. Park, K. S. Kim, H. Lee and S. K. Hahn, ACS Nano, 2011, 5, 6138–6147 CrossRef CAS.
- M. Ma, H. R. Chen, Y. Chen, K. Zhang, X. Wang, X. Z. Cui and J. L. Shi, J. Mater. Chem., 2012, 22, 5615–5621 RSC.
- S. Yang, L. Z. Zhao, C. Z. Yu, X. F. Zhou, J. W. Tang, P. Yuan, D. Y. Chen and D. Y. Zhao, J. Am. Chem. Soc., 2006, 128, 10460–10466 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- K. Y. Choi, K. H. Min, J. H. Na, K. Choi, K. Kim, J. H. Park, I. C. Kwon and S. Y. Jeong, J. Mater. Chem., 2009, 19, 4102–4107 RSC.
- E. K. Lim, H. O. Kim, E. Jang, J. Park, K. Lee, J. S. Suh, Y. M. Huh and S. Haam, Biomaterials, 2011, 32, 7941–7950 CrossRef CAS.
- M. Muroya, Colloids Surf., A, 1999, 157, 147–155 CrossRef CAS.
- W. Sicinska, B. Adams and L. Lerner, Carbohydr. Res., 1993, 242, 29–51 CrossRef CAS.
- S. Al-Qadi, A. Grenha and C. Remunan-Lopez, Carbohydr. Polym., 2011, 86, 25–34 CrossRef CAS.
- B. Z. Zhao, J. J. Yin, P. J. Bilski, C. F. Chignell, J. E. Roberts and Y. Y. He, Toxicol. Appl. Pharmacol., 2009, 241, 163–172 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |